

Abstract
Adipocyte fatty-acid-binding protein, aP2 (FABP4) is expressed in adipocytes and macrophages, and integrates inflammatory and metabolic responses. Studies in aP2-deficient mice have shown that this lipid chaperone has a significant role in several aspects of metabolic syndrome, including type 2 diabetes and atherosclerosis. Here we demonstrate that an orally active small-molecule inhibitor of aP2 is an effective therapeutic agent against severe atherosclerosis and type 2 diabetes in mouse models. In macrophage and adipocyte cell lines with or without aP2, we also show the target specificity of this chemical intervention and its mechanisms of action on metabolic and inflammatory pathways. Our findings demonstrate that targeting aP2 with small-molecule inhibitors is possible and can lead to a new class of powerful therapeutic agents to prevent and treat metabolic diseases such as type 2 diabetes and atherosclerosis.
Lipids and lipid signals are critical in the integration of metabolic and inflammatory response systems and consequently play significant parts in the pathogenesis of a cluster of chronic metabolic diseases, including type 2 diabetes, fatty liver disease and atherosclerosis1. However, how lipids couple to target signalling pathways or metabolic processes and how their intracellular trafficking is regulated are poorly understood. Cytoplasmic fatty-acid-binding proteins (FABPs) are a family of 14–15-kDa proteins that bind with high affinity to hydrophobic ligands such as saturated and unsaturated long-chain fatty acids and eicosanoids such as hydroxyeicosatetraenoic acid, leukotrienes and prostaglandins2. The adipocyte FABP, aP2 (FABP4), is highly expressed in adipocytes and regulated by peroxisome-proliferator-activated receptor-γ (PPARγ) agonists, insulin and fatty acids2-5.
Studies in aP2-deficient mice have shown that aP2 has a significant role in many aspects of metabolic syndrome. Deficiency of aP2 partially protects mice against the development of insulin resistance associated with genetic or diet-induced obesity6,7. Adipocytes of aP2−/− mice have reduced efficiency of lipid transport in vitro and in vivo, and yet exhibit only minor changes in serum lipids8. Interestingly, recent studies demonstrated that aP2 is also expressed in macrophages and regulated by phorbol 12-myristate 13-acetate, lipopolysaccharide, oxidized low-density lipoproteins and PPARγ ligands9-12. The macrophage is a critical site of FABP action, and total or macrophage-specific aP2-deficiency leads to a marked protection against early and advanced atherosclerosis in apolipoprotein E-deficient (Apoe−/−) mice9,13.
These findings indicate an important role for aP2 in the development of major components of metabolic syndrome through its distinct actions in adipocytes and macrophages of integrating metabolic and inflammatory responses. Hence, pharmacological agents that modify FABP function may offer therapeutic opportunities for many components of metabolic syndrome, such as insulin resistance, type 2 diabetes, and atherosclerosis. Here, we demonstrate the first evidence of the efficacy of a novel chemical aP2 inhibitor in experimental models.
Inhibition of aP2 in cellular models
BMS309403 (Fig. 1a) is a rationally designed, potent, and selective inhibitor of aP2 that interacts with the fatty-acid-binding pocket within the interior of the protein and competitively inhibits the binding of endogenous fatty acids. In a fluorescent 1,8-anilino-8-naphthalene sulphonate (ANS) binding displacement assay, BMS309403 exhibited Ki values <2nM for both mouse and human aP2, compared with 250nM for muscle FABP (FABP3) and 350nM for mal1 (FABP5)14. In this assay, the endogenous fatty acids palmitic acid and oleic acid exhibited aP2 Ki values of 336 and 185 nM, respectively. Results of X-ray crystallography studies suggested the specific interactions of BMS309403 with key residues in the fatty-acid- binding pocket are the basis of its high in vitro binding affinity and selectivity for aP2 over other FABPs14.
Figure 1. Target-specific effects of aP2 inhibition on MCP-1 production in macrophages.
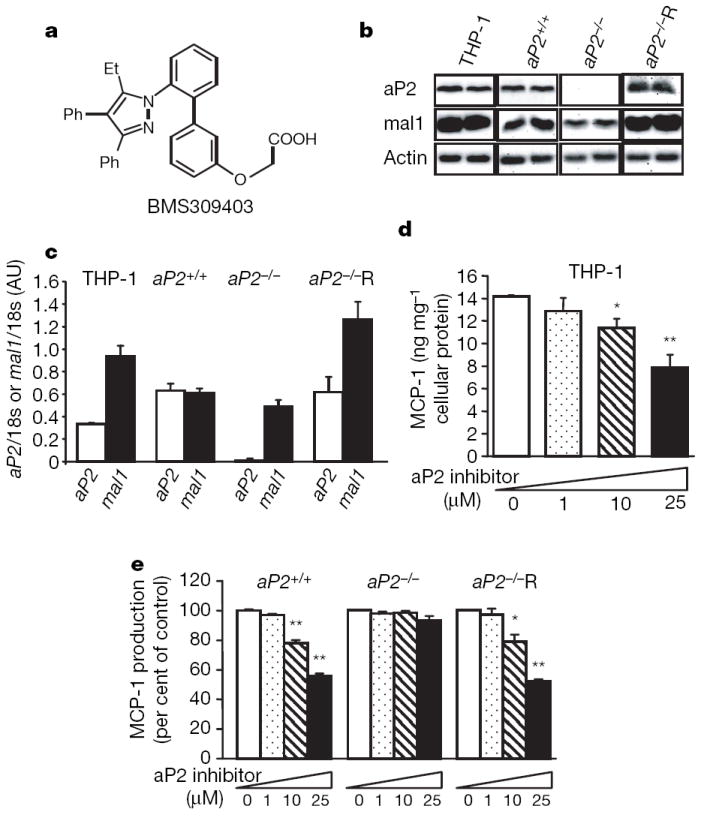
a, Structure of the compound, BMS309403. b, Protein levels of aP2 and mal1 in human THP-1 macrophages and mouse macrophage cell lines, aP2+/+, aP2−/− and aP2−/−R. c, aP2 and mal1 mRNA levels analysed by quantitative real-time PCR. d, MCP-1 production in human THP-1 macrophages treated with aP2 inhibitor at the indicated concentrations for 24 h. e, MCP-1 production in mouse cell lines treated with the aP2 inhibitor at the indicated concentrations for 24 h. Data are shown as the mean ± s.e.m. *P < 0.05, **P < 0.01 compared with the control (each untreated cell line). AU, arbitrary units.
To test the specificity of aP2 inhibition by BMS309403, we developed and used a cellular system with aP2+/+ and aP2−/− macrophage cell lines9,15. In addition, we reconstituted aP2 expression in the aP2−/− cells (aP2−/−R). As shown in Fig. 1b, aP2 protein was expressed in the THP-1 (a human monocytic leukaemia cell line), aP2+/+, and aP2−/−R macrophages but was not detected in the aP2−/− macrophages. In all of the cell lines, mal1 was present (Fig. 1b, c). Similarly, aP2 messenger RNA was readily detectable in THP-1, aP2+/+ and aP2−/−R but not in the aP2−/− macrophages (Fig. 1c). In this system, we examined the impact of aP2 inhibition on production of monocyte chemoattractant protein (MCP)-1 (also known as CCL2), an important aP2-regulated atherogenic product9,15, in macrophages.
Treatment with BMS309403 significantly decreased MCP-1 production from THP-1 macrophages in a dose- and time-dependent manner (Fig. 1d, Supplementary Fig. 1). To address whether this effect is specific, we next investigated MCP-1 production using aP2+/+, aP2−/− or aP2−/−R mouse macrophage cell lines. In a similar way to THP-1 cells, production of MCP-1 from macrophages was decreased in aP2+/+ cells in a dose-dependent fashion. In contrast, BMS309403 had no effect on MCP-1 production in aP2−/− cells at any dose tested. However, re-expression of aP2 (aP2−/−R) rendered the aP2−/− cells responsive to BMS309403 treatment, resulting in a dose-dependent reduction in MCP-1 production and demonstrating the target specificity of this compound (Fig. 1e).
The impact of aP2 inhibition on atherosclerosis
To address whether inhibition of aP2 can alter the development of vascular lesions, we performed early and late intervention studies in the Apoe−/− mouse model of atherosclerosis on a western diet. In the early intervention study, a western diet and the aP2 inhibitor BMS309403 were started simultaneously in 5-week-old mice (Supplementary Fig. 2a). In the late intervention paradigm, the aP2 inhibitor was administered after 8 weeks of a western diet (at 12 weeks of age), when significant atherosclerosis has developed (Fig. 2a). Analysis of the en face aorta demonstrated marked reductions in atherosclerotic lesion area in the aP2-inhibitor-treated group compared with the vehicle group in both the early (52.6%, Supplementary Fig. 2a, b) and late (51.0%, Fig. 2a, b) intervention studies. Staining of cross-sections of the proximal aorta with Oil Red O revealed fatty streak lesions (Fig. 2c and Supplementary Fig. 2c). These were almost exclusively macrophage-derived foam cells, as determined by immunohistochemical staining with MOMA-2 (Fig. 2d and Supplementary Fig. 2d). Macrophages were located predominantly on the luminal surface of the lesions. The extent of atherosclerotic lesion area in the proximal aorta was significantly reduced in the aP2-inhibitor-treated group compared with vehicle-treated controls in both the early (Supplementary Fig. 2e) and late (Fig. 2e) intervention studies.
Figure 2. Atherosclerosis in Apoe−/− mice treated with the aP2 inhibitor.
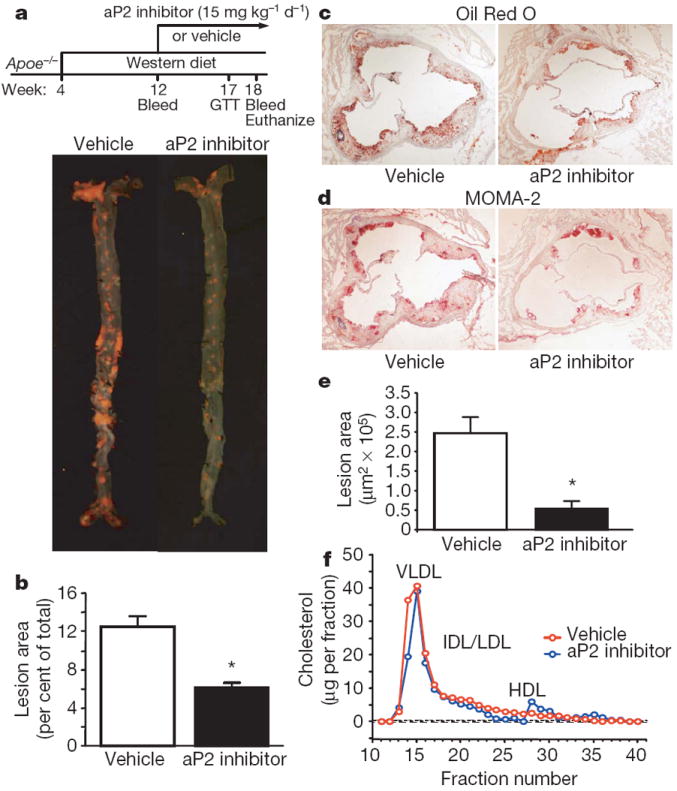
a, Experimental design of the late intervention study and en face aortas stained with Sudan IV. b, Quantitative analyses of the atherosclerotic lesion areas (per cent of total aorta surface area) in the vehicle (n = 16) and aP2 inhibitor (n = 15) groups. c, d, Oil Red O (c) and MOMA-2 (d) stainings of atherosclerotic lesions in the aortic root at the level of the aortic valves. Magnification, ×40. e, Quantitative analyses of the proximal aorta atherosclerotic lesion areas in the vehicle (n = 11) and aP2 inhibitor (n = 6) groups. f, Lipoprotein profile in Apoe−/− mice treated with vehicle (red) and aP2 inhibitor (blue) in the late intervention study. Data are presented as an average (n = 3) per cent distribution of total cholesterol for each group. Data are expressed as the mean ± s.e.m. *P < 0.01. VLDL, very low density lipoprotein; IDL, intermediate density lipoprotein; LDL, low density lipoprotein; HDL, high density lipoprotein. GTT, glucose tolerance test.
The aP2 inhibitor did not influence body weight, systemic glucose or lipid metabolism in Apoe−/− mice (Supplementary Table 1). Examination of the distribution of cholesterol among the serum lipoprotein fractions by size-exclusion chromatography revealed similar lipoprotein profiles between the groups with a large peak in the very low density lipoprotein fractions and a reduced high density lipoprotein peak that was due to APOE-deficiency in both the early (Supplementary Fig. 2f) and late (Fig. 2f) intervention studies. No significant difference in glucose levels during glucose tolerance tests was observed between the vehicle and aP2 inhibitor groups (Supplementary Fig. 3a, b). These results are consistent with previous observations made in mice with genetic deficiency of aP2 in the Apoe−/− background9,13.
Cholesterol and inflammatory responses in macrophages
Macrophage foam cell formation has a critical role in the pathogenesis of atherosclerosis and is a process regulated by FABPs9. Transformation of THP-1 macrophage to foam cells was significantly reduced in the presence of aP2 inhibitor (25 μM) (Fig. 3a). The aP2-inhibitor-treated THP-1 macrophages exhibited 44% reduction in cholesterol ester accumulation compared with vehicle-treated macrophages (Fig. 3b). In aP2+/+ and aP2−/−R macrophages treated with the aP2 inhibitor, total cellular cholesterol ester content was significantly lower than in macrophages treated with vehicle (Fig. 3c). The aP2 inhibitor did not affect cholesterol ester content in the aP2−/− macrophages, again demonstrating the specificity of aP2 inhibitor action in this context.
Figure 3. Effects of aP2 inhibitor on lipid accumulation, cholesterol efflux and inflammatory responses in macrophages.
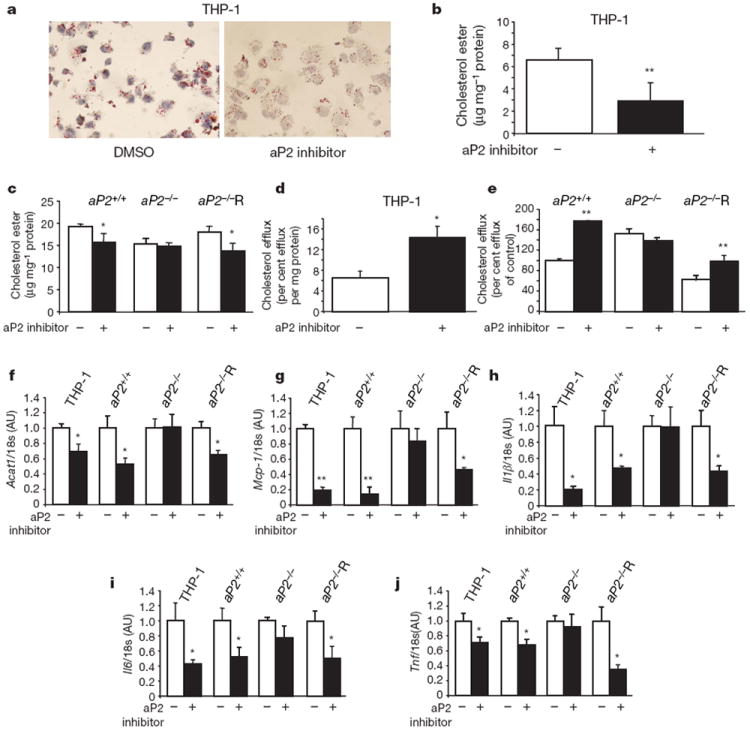
a, Oil Red O staining of THP-1 macrophage foam cells loaded with acetylated low density lipoprotein (50 μg ml−1) in the absence or presence of aP2 inhibitor (25 μM). Magnification, ×400. b, c, Cholesterol ester levels normalized to cellular protein content in human THP-1 macrophages (b) and mouse macrophage cell lines, aP2+/+, aP2−/− and aP2−/−R (c). d, e, APOA1-specific cholesterol efflux in THP-1 macrophages (d) and mouse cell lines (e) in the absence or presence of aP2 inhibitor (25 μM). f–j, Expression of Acat1 (f) and chemoattractant and inflammatory cytokines, Mcp-1 (g), Il1β (h), Il6 (i), and Tnf (j) in macrophages normalized to 18s rRNA levels. Data are normalized to untreated cells and expressed as the mean ± s.e.m. *P < 0.05, **P < 0.01 compared with the control (each untreated cell line). DMSO, dimethyl sulphoxide.
To determine the potential mechanism for the reduction in cholesterol ester accumulation on inhibition of aP2, we examined APOA1-mediated cholesterol efflux in these cells. Cholesterol efflux from human THP-1 macrophages was significantly increased on treatment with the aP2 inhibitor (Fig. 3d). There was a significant increase in both mRNA and protein levels of the ATP-binding cassette A1 (ABCA1) protein, a critical mediator of cholesterol efflux in macrophages, in the aP2-inhibitor-treated THP-1 cells compared with vehicle-treated controls (Supplementary Fig. 4a, b). Consistent with earlier observations15, cholesterol efflux in aP2−/− macrophages was substantially higher than that of the aP2+/+ cells (Fig. 3e) and was completely abrogated with reconstitution of aP2 expression. Similar to genetic deficiency, treatment with the aP2 inhibitor significantly increased cholesterol efflux in the aP2+/+ and aP2−/−R macrophages but not in the aP2−/− cells. We also examined the impact of aP2 inhibition on principal target molecules that regulate cellular cholesterol ester synthesis and hydrolysis in macrophages. There was a modest reduction in acyl-coenzyme A: cholesterol-acyltransferase 1 (ACAT1), a key enzyme of cholesterol esterification, in the aP2-expressing macrophages (Fig. 3f) but no effect of aP2 inhibition on the expression of hormone-sensitive lipase, which acts as the neutral cholesterol esterase, in macrophages (Supplementary Fig. 5).
Macrophages participate in the pathogenesis of atherosclerosis not only through the formation of foam cells but also by the production of inflammatory mediators. Hence, we determined the impact of aP2 inhibition on several critical chemoattractant and inflammatory cytokines, including MCP-1, interleukin (IL)1β, IL6 and tumour necrosis factor (TNF) in macrophages. Expression of these cytokines was significantly reduced in the aP2-expressing macrophages treated with the aP2 inhibitor compared with those treated with vehicle (Fig. 3g–j). No regulation was evident in aP2−/− cells on treatment with the inhibitor, demonstrating the target specificity of the aP2 inhibitor.
Inhibition of aP2 in adipocytes
The main site of aP2 expression is the adipocyte and although this site does not play a major part in atherosclerosis, it does significantly contribute to systemic insulin resistance and type 2 diabetes6,7,9. To begin to address the specific action of aP2 inhibition in adipocytes, we generated wild-type (aP2+/+mal1+/+) and FABP-deficient (aP2−/− mal1−/−) pre-adipocyte cell lines as well as FABP-deficient cells reconstituted with exogenous aP2 or with control empty vector. These cell lines fully differentiate into adipocytes and in all properties tested, behave in a similar way to commonly used 3T3-L1 or 3T3-F442A adipocytes (Fig. 4a). In genetic aP2-deficiency, the principal alteration observed in adipocytes thus far is a reduction in fatty acid transport8. Hence, we asked whether the chemical inhibition of aP2 could mimic this action in adipocytes and do so in an aP2-dependent fashion. Treatment with the aP2 inhibitor resulted in a dose-dependent decrease in fatty acid uptake in wild-type adipocytes (Fig. 4b). In contrast, there was no action of this inhibitor at any dose in FABP-deficient adipocytes. However, when aP2-deficient cells were reconstituted with aP2, they were rendered responsive to aP2 inhibition in a dose-dependent manner. Hence, the action of the synthetic aP2 inhibitor of regulating lipid transport in adipocytes was specific to its target, aP2.
Figure 4. Metabolic studies in aP2-inhibitor-treated adipocytes and ob/ob mice.
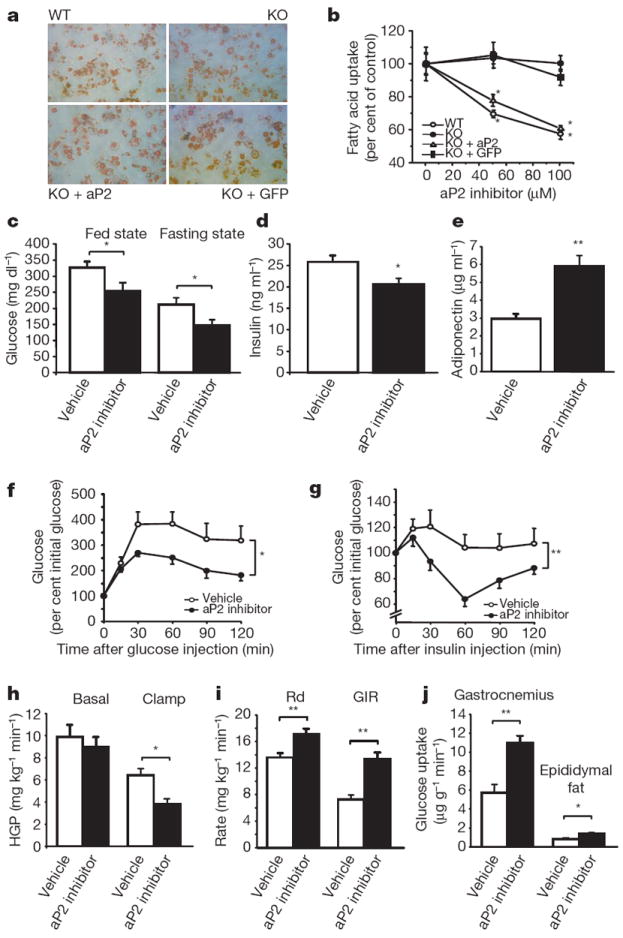
a, Oil Red O staining of wild-type (WT), FABP-deficient (KO), FABP-deficient reconstituted with aP2 (KO + aP2), and FABP-deficient with vector (KO + GFP) adipocyte cell lines. b, Fatty acid uptake using 3H-stearate in adipocyte cell lines. c, Blood glucose levels in ob/ob mice treated with vehicle (n = 6) or aP2 inhibitor (n = 6) at the fed state after 2 weeks of treatment and at the fasting state after 6 weeks of treatment. d, e, Plasma levels of insulin (d) and adiponectin (e) in ob/ob mice treated with vehicle (n = 6) or aP2 inhibitor (n = 6) for 6 weeks. f, Glucose tolerance tests performed after 4 weeks of treatment in ob/ob mice with vehicle (open circle, n = 6) or aP2 inhibitor (closed circle, n = 6). g, Insulin tolerance tests performed after 5 weeks of treatment in ob/ob mice with vehicle (open circle, n = 6) or aP2 inhibitor (closed circle, n = 6). h–j, Hyperinsulinaemic–euglycaemic clamp studies performed in ob/ob mice treated with vehicle (n = 7) or aP2 inhibitor (n = 9) for 4 weeks. Basal and clamp hepatic glucose production (HGP) (h), glucose disposal rate (Rd) and glucose infusion rate (GIR) (i), and tissue glucose uptake in gastrocnemius muscle and epididymal fat (j). Data are shown as the mean ± s.e.m. *P < 0.05, **P < 0.01.
Inhibition of aP2 in obese and diabetic mice
Having established the target specificity of BMS309403 in adipocytes, we administered the compound into a genetic model of obesity and insulin resistance, the leptin-deficient ob/ob (also known as Lepob/ob) mouse, and investigated insulin sensitivity and glucose metabolism. During the course of the 6-week treatment, there was no significant difference in body weight between animals receiving vehicle or aP2 inhibitor (Supplementary Fig. 6a). Similarly, per cent body fat, rates of oxygen consumption and carbon dioxide production, food intake, and physical activity were not different between the vehicle and aP2 inhibitor treatment groups (Supplementary Fig. 6b–f). In contrast, blood glucose levels in both the fed and fasted state were decreased after treatment with the aP2 inhibitor (Fig. 4c). Similar to genetic aP2-deficiency on the ob/ob background7, free fatty acid levels showed a trend towards an increase after treatment with the aP2 inhibitor (Supplementary Table 2, P = 0.07). The aP2 inhibitor decreased insulin and triglyceride levels and increased adiponectin concentration (Fig. 4d, e, and Supplementary Table 2), suggesting a potential increase in systemic insulin sensitivity. In fact, glucose tolerance tests revealed a significant improvement in glucose metabolismin the aP2-inhibitor-treated group (Fig. 4f). Similarly, insulin tolerance tests showed significantly increased insulin sensitivity in the ob/ob mice treated with the aP2 inhibitor (Fig. 4g). At the end of the treatment period, we analysed islet morphology in the pancreas. There was no difference in the pancreatic morphology such as the size, shape, and organization of the non-β-cell mantle between the vehicle- and aP2-inhibitor-treated ob/ob mice (Supplementary Fig. 7).
We also investigated the effect of aP2 inhibition in a diet-induced obesity model using both wild-type and FABP-deficient mice. The aP2-inhibitor-treated wild-type mice showed a significant decrease in glucose levels during glucose tolerance tests compared with vehicle-treated animals, but there was no change in glucose levels between the vehicle- and aP2-inhibitor-treated FABP-deficient mice on a high-fat diet (Supplementary Fig. 8). These results demonstrate that the insulin-sensitizing effects of the aP2 inhibitor in vivo are target-specific and effective in two independent models of obesity and insulin resistance.
Furthermore, we performed hyperinsulinaemic–euglycaemic clamp studies in ob/ob mice after 4 weeks of treatment. There was no significant difference in basal hepatic glucose production between the vehicle and aP2-inhibitor groups, but clamp hepatic glucose production was significantly suppressed in the aP2-inhibitor-treated ob/ob mice compared with vehicle-treated controls (Fig. 4h). Both whole-body glucose disposal and glucose infusion rates were also significantly increased after treatment with the aP2 inhibitor (Fig. 4i). These data demonstrate that the aP2 inhibitor improves whole-body insulin sensitivity through the suppression of hepatic glucose production and enhancement of insulin-stimulated glucose disposal in peripheral tissues. To explore this further, we determined the rate of glucose uptake in gastrocnemius muscle and epididymal fat during the clamp procedure. In the aP2-inhibitor-treated mice, glucose uptake in muscle and adipose tissues was significantly increased compared with that in the vehicle-treated controls (Fig. 4j).
Effects of aP2 inhibition on adipose tissue in ob/ob mice
Adipocyte size in ob/ob mice treated with the vehicle or aP2 inhibitor was comparable, but macrophage infiltration in adipose tissue was more severe in the vehicle-treated group (Fig. 5a). Expression of two macrophage markers, F4/80 (Emr1) and Cd68, was significantly reduced in the aP2-inhibitor-treated mice compared with vehicle-treated controls (Fig. 5b, c). Obesity leads to increased production of several chemoattractant and inflammatory cytokines, which have a critical role in obesity-associated inflammation and metabolic pathologies. Expression of Mcp-1, Il1β, Il6 and Tnf in adipose tissue was significantly reduced in ob/ob mice treated with the aP2 inhibitor compared with those treated with the vehicle (Fig. 5d–g).
Figure 5. Effects of aP2 inhibitor in adipose tissue of ob/ob mice.
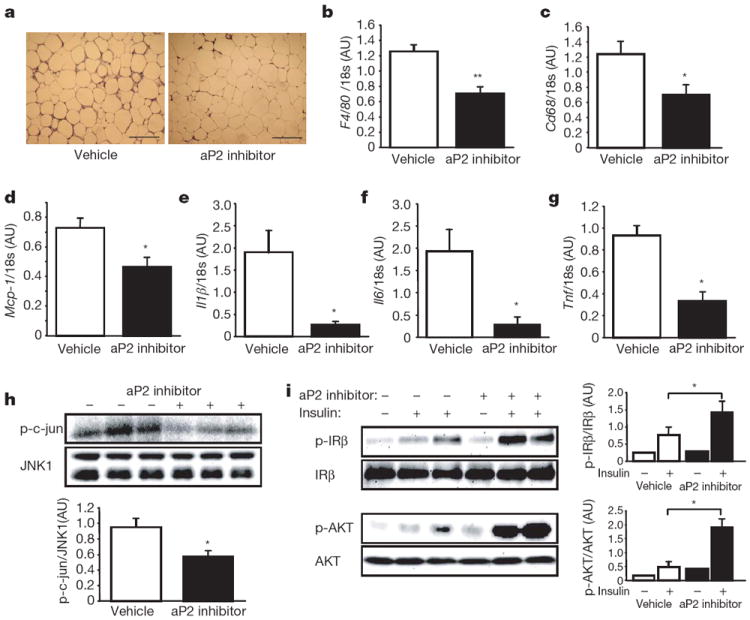
a, Haematoxylin and eosin staining of the adipose tissue in ob/ob mice treated with vehicle or aP2 inhibitor. Scale bar, 200 μm. b–g, Expression of F4/80 (b), Cd68 (c), Mcp-1 (d), Il1β (e), Il6 (f), and Tnf (g) in the adipose tissue of ob/ob mice treated with vehicle (n = 6) or aP2 inhibitor (n = 6). h, JNK1 activity in the adipose tissue of ob/ob mice. Quantification is shown in the graph below. i, Insulin-stimulated IRβ tyrosine 1162/1163 and AKT serine 473 phosphorylation (p) in the adipose tissues of ob/ob mice. The graphs on the right of each blot show the quantification. Data are shown as the mean ± s.e.m. *P < 0.05, **P < 0.01.
Obesity-induced Jun N-terminal kinase (JNK) 1 activity is critical in the generation of inflammatory responses and inhibition of insulin action16,17. To examine whether aP2 inhibition modifies the inflammatory profile and insulin action by this mechanism, we determined JNK1 activity in the adipose tissue of vehicle- and aP2-inhibitor-treated ob/ob mice. There was a significant attenuation (40%) of obesity-induced adipose tissue JNK1 activity in mice treated with aP2 inhibitor compared with vehicle-treated controls (Fig. 5h).
We next examined whether inhibition of aP2 and the alterations seen in inflammatory mediators and JNK activity in adipose tissue resulted in enhanced insulin action at this site. Insulin receptor signalling capacity was examined biochemically in intact mice following insulin administration. Insulin-stimulated tyrosine 1162/1163 phosphorylation of insulin receptor β subunit (IRβ or INSRβ subunit) and serine 473 phosphorylation of AKT were significantly increased in the adipose tissue of aP2-inhibitor-treated ob/ob mice compared with that of vehicle-treated controls (Fig. 5i). These results demonstrate that aP2 inhibition reduced inflammation and increased insulin sensitivity in the adipose tissues of ob/ob mice.
Effects of aP2 inhibition on liver in ob/ob mice
In genetic aP2-deficiency, there is a striking molecular compensation through increased expression of mal1 in adipose tissue6. Owing to this compensation, the phenotype of aP2-deficiency is much milder than aP2–mal1 combined deficiency18. We show here that the compensatory increase in mal1 expression of adipose tissue in the genetic absence of aP2 also occurs in the ob/ob background (Fig. 6a). Because the aP2-inhibitor-treated animals exhibit a significant protection against metabolic disease, we asked whether the compensatory increase in mal1 expression was present or not under these circumstances. Interestingly, after 6 weeks of the aP2-inhibitor treatment, there was no change in levels of aP2 or mal1 protein in the adipose tissue (Fig. 6a). This is a critical observation contributing to the efficacy of chemical inhibition of aP2 action in adult animals. For example, in genetic aP2-deficiency, there is no protection against fatty liver disease but a profound protection is seen in aP2–mal1 combined deficiency18,19. In the aP2-inhibitor-treated ob/ob mice, fatty infiltration of the liver was attenuated (Fig. 6b) with a significant reduction in total liver triglyceride content (Fig. 6c). This reduction in fatty liver disease on aP2 inhibition was associated with diminished expression of key lipogenic enzymes in liver, including the stearoyl-CoA desaturase 1 (Scd1), fatty acid synthase (Fasn), and acetyl-CoA carboxylase 1 (Acaca) (Fig. 6d–f). This phenotype is reminiscent of aP2–mal1 combined deficiency rather than isolated aP2-deficiency18,19.
Figure 6. Effects of aP2 inhibitor in liver of ob/ob mice.
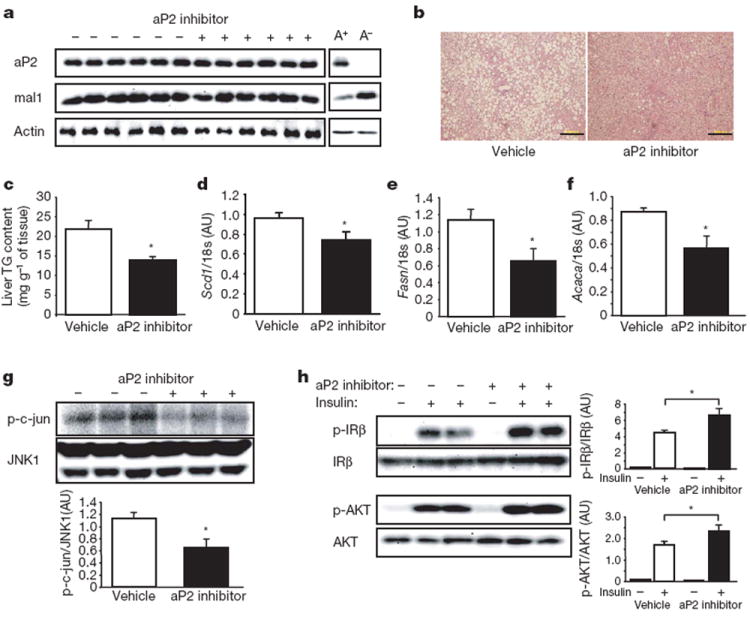
a, aP2 and mal1 protein in the adipose tissue of ob/ob mice treated with vehicle or aP2 inhibitor. For control, the adipose tissue of ob/ob;aP2+/+ (A+) and ob/ob;aP2−/− (A−) mice was used. b, Haematoxylin and eosin staining of the liver of ob/ob mice treated with vehicle or aP2 inhibitor. Scale bar, 200 μm. c–f, Triglyceride (TG) content (c) and mRNA expression of Scd1 (d), Fasn (e), and Acaca (f) in the liver of ob/ob mice treated with vehicle (n = 6) or aP2 inhibitor (n = 6). g, JNK1 activity in the liver of ob/ob mice treated with vehicle or aP2 inhibitor. The graph below the blot shows quantification. h, Insulin-stimulated IRβ tyrosine 1162/1163 and AKT serine 473 phosphorylation in the liver tissues of ob/ob mice treated with vehicle or aP2 inhibitor. The graphs demonstrate the quantification of phosphorylation of each molecule. Data are shown as the mean ± s.e.m. *P < 0.05.
In a similar way to adipose tissue, total JNK1 activity in the liver tissue of aP2-inhibitor-treated ob/ob mice was significantly reduced (43%) compared with that of vehicle-treated control mice (Fig. 6g). Suppression of fatty liver infiltration and inflammatory responses in aP2-inhibitor-treated ob/ob mice also resulted in enhanced insulin action in the liver. Insulin-stimulated tyrosine 1162/1163 phosphorylation of IRβ and serine 473 phosphorylation of AKT were significantly increased in the liver tissue of aP2-inhibitor-treated ob/ob mice compared with vehicle-treated controls (Fig. 6h).
Discussion
A principal mechanistic core of obesity, type 2 diabetes and atherosclerosis resides at the interface of metabolic and inflammatory pathways1. However, this mechanistic platform has not yet been exploited for the development of effective therapeutic strategies.
The locus for the adipocyte/macrophage FABP aP2 is critical in the regulation and dysregulation of metabolic and inflammatory responses as they relate to metabolic diseases6-9,13,15. In addition to cell-autonomous effects in macrophages and adipocytes, aP2 also acts to coordinate the functional interactions between these two critical cell types in adipose tissue. Genetic deletion of the aP2 gene in mice has demonstrated a strong role for this molecule in several chronic metabolic diseases, most notably, atherosclerosis and type 2 diabetes, and has raised the possibility of using aP2 as a potential drug target. Here, we have provided a critical proof of principle in mice that aP2 could be successfully targeted by an orally active, small-molecule inhibitor to generate a profile reminiscent of genetic deficiency in vitro and in vivo.
There are several indications that FABPs might be involved in metabolic homeostasis in a similar fashion in humans. First, the expression and regulation patterns of aP2 are very similar in both adipocytes and macrophages between mice and humans2,6,9. Expression of aP2 is highly regulated on macrophage activation and, interestingly, suppressed by a statin in vitro20. In both mouse and humanmacrophages, aP2 expression modulates inflammatory responses, foam cell formation and cholesterol efflux9,15. Atherosclerotic lesions express high levels of aP2 in both mice and humans9,13,21. Finally, aP2 expression is increased in obesity22. Hence, it is possible that aP2 function may be similar in humans as well. In fact, in a recent study, we produced genetic support for this concept in humans23. A rare genetic variant was identified at the promoter region of the human aP2 orthologue FABP4, coinciding with the binding site for C/EBP. This particular mutation alters C/EBP binding and significantly reduces the transcriptional activity of the human aP2 promoter and the expression level of aP2 in the tissues of the carriers. In a large population sampling, individuals with the aP2 variant had lower triglyceride levels, exhibited reduced cardiovascular disease risk and were protected from obesity-induced type 2 diabetes. This study offers a critical insight and indicates that the metabolic function of aP2 in humans may be similar to that observed in mouse models. It is therefore possible that chemical inhibition of aP2 in humans might also show beneficial effects against diabetes and cardiovascular disease.
METHODS
Biochemical reagents
All biochemical reagents were purchased from Sigma-Aldrich (Saint Louis) unless indicated.
The compound of aP2 inhibitor BMS309403
A synthetic agent, BMS309403, was developed and provided by Bristol–Myers Squibb Pharmaceutical Research Institute. This agent is a selective, high affinity inhibitor of aP2 with the following chemical properties: 2-(2′-(5-ethyl-3,4-diphenyl-1H-pyrazol-1-yl)biphenyl-3-yloxy) acetic acid (BMS309403): m.p: 174–176 °C; 1H-NMR (500 MHz, CDCl3): δ 9.20 p.p.m. (br s, 1H), 7.65 (d, J = 7.1 Hz, 1H), 7.6–7.5 (m, 3H), 7.4 (m, 2H), 7.3–7.1 (m, 5H), 7.04 (d, J = 16.6 Hz, 2H), 6.90 (d, J = 7.7 Hz, 1H), 6.87 (dd, J = 2.2, 8.2 Hz, 1H), 6.66 (s, 1H), 4.36 (s, 2H), 2.05 (br s, 2H), 0.58 (t, J = 7.2 Hz); 13C-NMR (125 MHz, CDCl3): δ172.2, 157.36, 149.1, 145.1, 139.5, 139.0, 137.2, 133.6, 132.7, 130.3, 130.2, 129.7, 129.5, 128.6, 128.4, 128.1, 128.0, 127.5, 126.8, 122.0, 119.0, 114.9, 113.6,64.6, 17.6, 13.0; IR (KBr): 1710 cm−1; analysis (% calculated, % found for C31H26N2O3): C (78.46, 78.30), H (5.52, 5.51), N (5.90, 5.69). Additional information on synthesis and chemical properties has recently been reported14.
Cells
Human monocytic leukaemia THP-1 cells were obtained from ATCC and cultured in Gibco RPMI 1640 medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone), 50 U ml−1 penicillin and 50 μg ml−1 streptomycin (Invitrogen) at 37 °C in 5% CO2. THP-1 monocytes were differentiated into macrophages with 100nM phorbol 12-myristate 13-acetate for 24 h. Immortalized aP2+/+ and aP2−/− mouse macrophage cell lines were generated in our laboratory by a modification of a described procedure (refs 9, 27). Reconstitution of aP2 expression into aP2−/− macrophages to produce the aP2−/−R cells was performed as described15. The levels of aP2 protein, as assessed by western blot, were similar in the aP2−/−R cell line as compared with the aP2+/+ macrophage line. Human THP-1 macrophages and mouse macrophage cell lines, aP2+/+, aP2−/− and aP2−/−R, were incubated in RPMI 1640 supplemented with heat-inactivated 10% FBS or 5% lipoprotein-deficient serum (Biomedical Technologies) in the absence or presence of aP2 inhibitor dissolved in dimethyl sulphoxide (DMSO) at the indicated concentrations. The incubation periods varied according to the experimental protocol. Each experiment was done in at least triplicate.
We generated aP2+/+ mal1+/+(WT) and aP2−/− mal1−/− (KO) pre-adipocytes from mouse models using a previously described protocol24. The KO pre-adipocytes were also reconstituted by lentivirus with exogenous aP2 (KO+aP2) or with control empty vector including green fluorescent protein (KO+GFP). These cell lines were maintained and propagated in Dulbeco’s Modified Eagle’s Media (Invitrogen) with 10% cosmic calf serum (Hyclone), 50 U ml−1 penicillin and 50 μg ml−1 streptomycin at 37 °C in 10% CO2. Differentiation was then initiated (day 0) by incubation in induction medium (1 μM dexamethasone, 0.5 mM isobutylmethyxanthine, 1 μM rosiglitazone and 5 μg ml−1 insulin). Following a 4-day induction period (two 48-h incubations), the medium was changed to a post-induction medium (1 μM rosiglitazone and 5 μg ml−1 insulin) for an additional 2 days. Thereafter, the medium was replaced with a medium supplemented only with 0.5 μg ml−1 of insulin.
Mice
Animal care and experimental procedures were performed with approval from animal care committees of Harvard University. Male Apoe−/− mice in the C57BL/6J background (Jackson Laboratory) were kept on a 12-h light cycle and were fed a high-cholesterol atherogenic western diet (D12079B: 21% fat, 0.21% cholesterol; Research Diets) ad libitum, beginning at 4–5 weeks of age. The mice were treated by oral gavage with vehicle including 10% 1-methyl-2-pyrrolidone and 5% cremophor EL with ethanol in 100 μl of water or 15 mg kg−1 d−1 of the aP2 inhibitor BMS309403 dissolved in the vehicle for 6 weeks starting at 5-(Supplementary Fig. 2a, early intervention) or 12-weeks of age (Fig. 2a, late intervention). Before and after treatment, blood samples were collected from animals that had fasted for 6 h, and the glucose tolerance test was performed by intraperitoneal glucose injection (1.5 g kg−1) on conscious mice after an overnight (15 h) fast at the fifth week of treatment.
Male ob/ob mice (Jackson Laboratory) were treated by oral gavage with 40 mg kg−1d−1 of the aP2 inhibitor or vehicle for 6 weeks beginning at 8-weeks-old. Before and after treatment, blood samples were collected from mice that had fasted for 6 h. Blood glucose levels at the fed state were also examined after 2 weeks of treatment. After 4 weeks of treatment, glucose tolerance test was performed by intraperitoneal glucose injection (0.5 g kg−1) after an overnight fast. After 5 weeks of treatment, insulin tolerance test was performed by intraperitoneal insulin injection (2 IU kg−1) after a 6 h fast.
Mice deficient in both aP2 and mal1 were generated as previously described18. These mice were backcrossed > 12 generations into the C57BL/6J genetic background. Both wild-type and aP2−/− mal1−/− mice were placed on a high-fat diet (D12492: 60% kcal% fat; Research Diets), beginning at 4 weeks of age. In these mice, the aP2 inhibitor (40 mg kg−1 d−1) or vehicle was administered by oral gavage for 4 weeks starting at 20 weeks of age. Glucose tolerance test was performed by intraperitoneal glucose injection (1.5 g kg−1) after an overnight fast.
Assessment of atherosclerosis and immunohistochemistry
Perfusion fixation, preparation of aortas and quantification of atherosclerotic lesions were performed as previously described25. En face pinned-out aortas were stained with Sudan IV. The heart with aorta was embedded in OCT and snap-frozen in liquid nitrogen. Lesions in the proximal aorta from serial 7-μm-thick cytosections were stained with Oil Red O and counterstained with haematoxylin. The images of the aortas were captured with a digital colour camera (DP70; Olympus) mounted on a microscope (VistaVision). Quantitative analysis of lipid-stained lesions was performed using ImageJ software. Macrophages in the arterial lesions were examined by immunohistochemistry using 7-μm cryosections of the proximal aorta fixed in acetone at 4 °C. The sections were immersed in phosphate-buffered saline (PBS) and incubated overnight at 4 °C with monoclonal rat anti-MOMA-2 (Accurate Chemical & Scientific). The sections were treated with rabbit biotinylated antibodies to rat IgG for 1 h at room temperature followed by incubation with avidin–biotin complex labelled with alkaline phosphatase (Vectastain ABC-AP) and visualized with red alkaline phosphatase substrate (Vector Laboratories). Slides were counterstained with haematoxylin.
Plasma measurements
Blood glucose concentration was determined with 3-μl whole blood using Ascensia BREEZE blood glucose meter (Bayer Company). Plasma insulin was measured with a commercially available ultra sensitive ELISA kit (Crystal chemicals). Total cholesterol, free fatty acids, glycerol and triglycerides were determined with colorimetric assay systems (Wako chemicals and Sigma-Aldrich) adapted for microtitre plate assay. Plasma adiponectin was measured with an ELISA kit (Linco Research). Particle size distribution of the lipoproteins was determined by fast-performance liquid chromatography (FPLC), using pooled samples of plasma as previously described25. In brief, plasma from mice was subjected to FPLC analysis using a Superose 6 column (Pharmacia Biotech) on an HPLC system model 600 (Waters Chromatography). A 100-μl aliquot of plasma was injected onto the column and separated with a buffer containing 0.15 M NaCl, 0.01 M Na2HPO4, 0.1 mM EDTA (pH 7.5), at a flow rate of 0.5 ml min−1. Forty 0.5-ml fractions were collected, and tubes 11–40 were analysed for cholesterol. Fractions 14–17 contain VLDL and chylomicra; fractions 18–24 contain LDL and IDL; fractions 25–29 contain HDL; and fractions 30–40 contain non-lipoprotein-associated proteins.
Examination of macrophage MCP-1 production
Supernatants from macrophages incubated in RPMI 1640 supplemented with 5% LPDS in the presence of aP2 inhibitor (0–25 μM) for 24 h were evaluated for MCP-1 production with ELISA (OptEIA; BD Biosciences). MCP-1 levels were normalized to cellular protein content in THP-1 macrophages. In the macrophage cell lines, aP2+/+, aP2−/− and aP2−/−R, data were also normalized to those in the absence of aP2 inhibitor.
Intracellular lipid analysis in macrophages
THP-1 macrophages were incubated for 24 h in RPMI 1640 supplemented with 5% LPDS and 50 μg ml−1 of acLDL (Biomedical Technologies), followed by RPMI 1640 supplementation with 10% FBS with the vehicle or aP2 inhibitor (25 μM) dissolved in DMSO. After 3 days, the macrophage foam cells were fixed in 10% formalin for 90 min, washed thoroughly with PBS, and incubated with a working solution of Oil Red O for 3 h. Intracellular lipids in macrophages were extracted with hexane/isopropanol (3:2) after 4 days incubation with the vehicle or aP2 inhibitor (25 μM), evaporated and dissolved in isopropanol containing 10% Triton X-100 for preparation of a sample solution. Free cholesterol and total cholesterol were determined by commercial assay systems (Wako chemicals). Cholesterol ester was estimated by subtracting free cholesterol from total cholesterol. Data are normalized to cellular protein content.
Cholesterol efflux assays in macrophages
APOA1-specific cholesterol efflux to the medium was determined as described15. Macrophages were incubated for 24 h in RPMI 1640 supplemented with 5% LPDS, 1 μCi ml−1 of 3H-cholesterol (PerkinElmer Life Sciences) and 50 μg ml−1 of acLDL in the absence or presence of aP2 inhibitor (25 μM) for 24 h. To equilibrate cholesterol pools, cells were washed twice with PBS and incubated for 24 h in RPMI 1640 containing 0.2% low endotoxin fatty-acid-free bovine serum albumin with no lipoproteins in the absence or presence of aP2 inhibitor (25 μM). Cells were washed again with PBS and incubated in RPMI containing 0.2% BSA in the absence or presence of APOA1 (20 μg ml−1; Calbiochem) for 4 h. Radioactivity of culture supernatants and cellular lysate was measured separately by liquid scintillation. Results are normalized to cellular protein content and expressed as the percentage of 3H-cholesterol in the medium divided by the total 3H-cholesterol in the cells and medium per milligram of protein. In the mouse macrophage cell lines, data are also normalized to those in the untreated aP2+/+ macrophages.
Fatty acid uptake in adipocytes
Differentiated adipocytes were incubated in DMEM supplemented with 2% FBS in the absence or presence of aP2 inhibitor at the indicated concentrations for 24 h. Medium was changed to DMEM containing 2% BSA in the absence or presence of aP2 inhibitor. Cells were then incubated with 3H-stearate (PerkinElmer Life Sciences) at 37 °C for 30 min. Radioactivity of cellular lysate was measured by liquid scintillation. Results are normalized to cellular protein content and expressed as the percentage of those in the absence of aP2 inhibitor in adipocytes.
Metabolic studies
Total body fat was assessed by dual energy X-ray absorptiometry (DEXA; PIXImus). For additional metabolic measurements, mice were placed in an indirect open circuit calorimeter (Oxymax System). Oxygen and carbon dioxide concentrations by volume were monitored at the inlet and outlet parts of a partially sealed chamber, through which a known flow of ambient air was forcibly ventilated. The concentration difference measured between the parts was used to compute oxygen consumption (VO2) and carbon dioxide production (VCO2). The consumption and production information were presented in units of ml kg−1 h−1 and normalized to 25 °C and 760mmHg. Food intake was investigated by using the Oxymax Feed Scale Device (Columbus Instruments). The physical activity of the mice was monitored with OPTOM3 Activity Application Device (Columbus Instruments). The movements (other than scratching, grooming, digging and so on) of each animal were determined by infrared beams in x, y and z axes.
Hyperinsulinaemic–euglycaemic clamp studies
Hyperinsulinaemic–euglycaemic clamps were performed in ob/ob mice after 4 weeks of treatment by a modification of a described procedure26. Four days before experiments, ob/ob mice were anaesthetized with an intraperitoneal injection of ketamine (80 mg kg−1) and xylazine (40 mg kg−1), and the right jugular vein was catheterized with a PE-10 polyethylene tube (inside and outside diameters, 0.28 mm and 0.61 mm, respectively; Becton Dickinson) filled with heparin solution (100 U ml−1) (United States Pharmacopeia). After an overnight fast, HPLC purified 3H-glucose (0.05 μCi min−1; Perkin Elmer) was infused during the 2-h basal period, and blood samples were collected at the end to estimate the rate of basal hepatic glucose production. After the basal period, a 120-min hyperinsulinaemic–euglycaemic clamp was conducted with a primed-continuous infusion of human insulin (Humulin R; Eli Lilly) at a rate of 12.5 mU kg−1min−1. Blood samples were collected at 20-min intervals for the immediate measurement of plasma glucose concentration, and 25% glucose was infused at variable rates to maintain plasma glucose at basal concentrations. Insulin-stimulated whole-body glucose disposal was estimated with a continuous infusion of 3H-glucose throughout the clamps (0.1 μCi min−1). All infusions were performed using microdialysis pumps (CMA/Microdialysis). To estimate insulin-stimulated glucose uptake in individual tissues, 2-14C-deoxyglucose (2-14C-DG; Perkin Elmer) was administered as a bolus (10 μCi) 75 min after the start of clamps. Because 2-deoxyglucose is a glucose analogue that is phosphorylated but not metabolized, insulin-stimulated glucose uptake in individual tissues can be estimated by determining the tissue content of 2-deoxyglucose-6-phosphate. Blood samples were collected at 80, 85, 90, 100, 110, and 120 min after the start of clamps for the determination of plasma 3H-glucose, 3H2O, and 2-14C-DG concentrations. At the end of clamps, animals were euthanized. Within 5 min, gastrocnemius muscles from both hindlimbs and epididymal adipose tissue were harvested. Each tissue was frozen immediately using liquid N2 and stored at −80 °C until further analysis.
For the determination of plasma 3H-glucose and 2-14C-DG concentrations, plasma was deproteinized with ZnSO4 and Ba(OH)2, dried to remove 3H2O, resuspended in water and counted in scintillation fluid (Ecoscint H) on channels for 3H and 14C. The plasma concentration of 3H2O was determined by the difference between 3H counts without and with drying. For the determination of tissue 2-14C-DG-6-phosphate content, tissue samples were homogenized, and the supernatants were subjected to an ion-exchange column to separate 2-14C-DG-6-phosphate from 2-14C-DG.
Rates of basal hepatic glucose production and insulin-stimulated whole-body glucose uptake were determined as the ratio of the 3H-glucose infusion rate to the specific activity of plasma glucose at the end of the basal period and during the final 30 min of clamps, respectively. Hepatic glucose production during the hyperinsulinaemic–euglycaemic clamps was determined by subtracting the glucose infusion rate from the whole-body glucose uptake. Glucose uptake in individual tissues was calculated from the plasma 2-14C-DG profile, which was fitted with an exponential curve, and tissue 2-14C-DG-6-phosphate content.
Portal vein insulin infusion and protein extraction from tissue
Following 6 h of food withdrawal, ob/ob mice were anaesthetized with a intraperitoneal injection of tribromoethanol (250 mg kg−1), and insulin (2 IU kg−1) or phosphate buffered saline (PBS) was injected into mice through the portal vein. Three minutes after infusion, tissues were removed and frozen in liquid nitrogen and kept at −80 °C until processing. For protein extraction, tissues were placed in a cold lysis buffer containing 50 mM Tris-HCl (pH 7.0), 2 mM EGTA, 5 mM EDTA, 30 mM NaF, 10 mM Na3VO4, 10 mM Na4P2O7, 40 mM β-glycerophosphate, 0.5% NP-40 and 1% protease inhibitor cocktail. After homogenization on ice, the tissue lysates were centrifuged, and the supernatants were used for western blot analysis or JNK kinase assay.
Western blot analysis
Total protein content of the samples was assessed by microplate protein assay (Bio-Rad), and equal amounts of protein per sample and known molecular weight markers were subjected to SDS–polyacrylamide gel electrophoresis. Proteins were electrophoretically transferred onto PVDF membranes (Whatman) and incubated for 1 h at room temperature with blocking solution (2% BSA or 5% nonfat milk in TBS buffer containing 0.1% Tween 20). The blocked membranes were incubated with anti-aP2, anti-mal1, anti-ABCA1 (Novus Biologicals), anti-IRβ-pTyr 1162/1163 (Calbiochem), anti-IRβ (Santa Cruz Biotechnology), anti-AKT-pSer 473 (Santa Cruz Biotechnology), anti-AKT (Santa Cruz Biotechnology), anti-JNK1 (Santa Cruz Biotechnology), anti-tubulin (Santa Cruz Biotechnology), and anti-β-actin (Cell Signaling Technology) for overnight at 4 °C, and washed three times with TBS buffer containing 0.1% Tween 20. The membranes were incubated with the secondary antibody conjugated with horseradish peroxidase (Amersham Biosciences) for 1 h at room temperature and washed. Immunodetection analyses were accomplished using the enhanced chemiluminescence kit (Roche Diagnostics).
c-Jun N-terminal kinase (JNK) kinase assay
Tissue lysates containing 500–1,000 μg of protein were mixed with 1 μg of JNK1 antibody (Santa Cruz Biotechnology) and 20 μl of protein A sepharose beads. The mixture was agitated at 4 °C overnight, pelleted by centrifugation and washed twice with lysis buffer followed by 3 additional washes with JNK kinase assay buffer containing 25 mM Hepes (pH 7.4), 20 mM MgCl2, 20 mM β-glycerophosphate, 0.5 mM EGTA, 0.5 mM NaF, 0.5 mM Na3VO4 and 1 mM PMSF for equilibration. After washing with kinase buffer, the beads were incubated in 25 μl kinase buffer containing 10 μCi 32P γATP (PerkinElmer Life Sciences), 10 μM ATP and 5 μg of c-jun fusion protein at 30 °C for 20 min. The reaction was terminated by addition of Laemmli buffer.
Lipid content in liver
Total lipids in liver were extracted by Bligh-Dyer method (ref. 28). Triglyceride content was determined by the colorimetric method using a commercial kit (Sigma-Aldrich).
Quantitative real-time PCR analysis
Total RNA was isolated using Trizol reagent (Invitrogen). For reverse transcription, 0.5–1.0 μg of the total RNA was converted to first strand complementary DNA in 20 μl reactions using a cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR analysis was performed using SYBR Green in a real-time PCR machine (iCycler; Bio-Rad). The thermal cycling program was 4 min at 95 °C for enzyme activation and 50 cycles of denaturation for 15 s at 95 °C, 30 s annealing at 58 °C and 30 s extension at 72 °C. Primers used in the present study are listed in Supplementary Table 3. To normalize expression data, 18s rRNA was used as an internal control gene. For some experiments, another housekeeping gene, GAPDH, was used, and the experiments produced identical results to those obtained when 18s rRNA was used as a reference.
Statistical analysis
Experimental results were shown as the mean ± s.e.m. After analysis of normality using the Shapiro-Wilk W test, the mean values for biochemical data from each group were compared by Student’s t-test. Data on vascular lesions, which were not normally distributed, were analysed with a non-parametric Mann–Whitney test. Comparisons between time points were analysed using repeated-measures analysis of variance, ANOVA. All statistical tests with P < 0.05 were considered significant.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the NIH and the American Diabetes Association. M.F. is supported by a JSPS Postdoctoral Fellowship for Research Abroad from the Japan Society for the Promotion of Science. G.T. is supported by a fellowship from the Iacocca Foundation.
Footnotes
Author Contributions G.S.H. designed and supervised experiments and analysed data. M.F. designed and performed experiments and analysed data. G.T., C.Z.G., E.V. and K.K. performed experiments. L.M. and G.A. developed cell lines frommice. V.R.B., S.F. and M.F.L. analysed lipoprotein profiles and advised on experiments. R.S., J.A.R. and R.A.P developed the aP2 inhibitor, BMS309403. M.F. and G.S.H wrote the manuscript. All authors discussed the results and commented on the manuscript.
The authors declare competing financial interests: details accompany the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 2.Hertzel AV, Bernlohr DA. The mammalian fatty acid-binding protein multigene family: molecular and genetic insights into function. Trends Endocrinol Metab. 2000;11:175–180. doi: 10.1016/s1043-2760(00)00257-5. [DOI] [PubMed] [Google Scholar]
- 3.Hunt CR, Ro JH, Dobson DE, Min HY, Spiegelman BM. Adipocyte P2 gene: developmental expression and homology of 5′-flanking sequences among fat cell-specific genes. Proc Natl Acad Sci USA. 1986;83:3786–3790. doi: 10.1073/pnas.83.11.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melki SA, Abumrad NA. Expression of the adipocyte fatty acid-binding protein in streptozotocin-diabetes: effects of insulin deficiency and supplementation. J Lipid Res. 1993;34:1527–1534. [PubMed] [Google Scholar]
- 5.Distel RJ, Robinson GS, Spiegelman BM. Fatty acid regulation of gene expression. Transcriptional and post-transcriptional mechanisms. J Biol Chem. 1992;267:5937–5941. [PubMed] [Google Scholar]
- 6.Hotamisligil GS, et al. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 7.Uysal KT, Scheja L, Wiesbrock SM, Bonner-Weir S, Hotamisligil GS. Improved glucose and lipid metabolism in genetically obese mice lacking aP2. Endocrinology. 2000;141:3388–3396. doi: 10.1210/endo.141.9.7637. [DOI] [PubMed] [Google Scholar]
- 8.Scheja L, et al. Altered insulin secretion associated with reduced lipolytic efficiency in aP2−/− mice. Diabetes. 1999;48:1987–1994. doi: 10.2337/diabetes.48.10.1987. [DOI] [PubMed] [Google Scholar]
- 9.Makowski L, et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nature Med. 2001;7:699–705. doi: 10.1038/89076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kazemi MR, McDonald CM, Shigenaga JK, Grunfeld C, Feingold KR. Adipocyte fatty acid-binding protein expression and lipid accumulation are increased during activation of murine macrophages by toll-like receptor agonists. Arterioscler Thromb Vasc Biol. 2005;25:1220–1224. doi: 10.1161/01.ATV.0000159163.52632.1b. [DOI] [PubMed] [Google Scholar]
- 11.Fu Y, Luo N, Lopes-Virella MF. Oxidized LDL induces the expression of ALBP/aP2 mRNA and protein in human THP-1 macrophages. J Lipid Res. 2000;41:2017–2023. [PubMed] [Google Scholar]
- 12.Pelton PD, Zhou L, Demarest KT, Burris TP. PPARγ activation induces the expression of the adipocyte fatty acid binding protein gene in human monocytes. Biochem Biophys Res Commun. 1999;261:456–458. doi: 10.1006/bbrc.1999.1071. [DOI] [PubMed] [Google Scholar]
- 13.Boord JB, et al. Adipocyte fatty acid-binding protein, aP2, alters late atherosclerotic lesion formation in severe hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2002;22:1686–1691. doi: 10.1161/01.atv.0000033090.81345.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sulsky R, et al. Potent and selective biphenyl azole inhibitors of adipocyte fatty acid binding protein (aFABP) Bioorg Med Chem Lett. doi: 10.1016/j.bmcl.2006.12.044. in the press. [DOI] [PubMed] [Google Scholar]
- 15.Makowski L, Brittingham KC, Reynolds JM, Suttles J, Hotamisligil GS. The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor γ and IκB kinase activities. J Biol Chem. 2005;280:12888–12895. doi: 10.1074/jbc.M413788200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirosumi J, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 17.Tuncman G, et al. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc Natl Acad Sci USA. 2006;103:10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maeda K, et al. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 2005;1:107–119. doi: 10.1016/j.cmet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 19.Cao H, et al. Regulation of metabolic responses by adipocyte/macrophage fatty acid-binding proteins in leptin-deficient mice. Diabetes. 2006;55:1915–1922. doi: 10.2337/db05-1496. [DOI] [PubMed] [Google Scholar]
- 20.Llaverias G, et al. Atorvastatin reduces CD68, FABP4, and HBP expression in oxLDL-treated human macrophages. Biochem Biophys Res Commun. 2004;318:265–274. doi: 10.1016/j.bbrc.2004.04.021. [DOI] [PubMed] [Google Scholar]
- 21.Fu Y, Luo N, Lopes-Virella MF, Garvey WT. The adipocyte lipid binding protein (ALBP/aP2) gene facilitates foam cell formation in human THP-1 macrophages. Atherosclerosis. 2002;165:259–269. doi: 10.1016/s0021-9150(02)00305-2. [DOI] [PubMed] [Google Scholar]
- 22.Fisher RM, et al. Fatty acid binding protein expression in different adipose tissue depots from lean and obese individuals. Diabetologia. 2001;44:1268–1273. doi: 10.1007/s001250100643. [DOI] [PubMed] [Google Scholar]
- 23.Tuncman G, et al. A genetic variant at the fatty acid-binding protein aP2 locus reduces the risk for hypertriglyceridemia, type 2 diabetes, and cardiovascular disease. Proc Natl Acad Sci USA. 2006;103:6970–6975. doi: 10.1073/pnas.0602178103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sethi JK, et al. Characterisation of receptor-specific TNFα functions in adipocyte cell lines lacking type 1 and 2 TNF receptors. FEBS Lett. 2000;469:77–82. doi: 10.1016/s0014-5793(00)01250-3. [DOI] [PubMed] [Google Scholar]
- 25.Babaev VR, Patel MB, Semenkovich CF, Fazio S, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J Biol Chem. 2000;275:26293–26299. doi: 10.1074/jbc.M002423200. [DOI] [PubMed] [Google Scholar]
- 26.Kim JK, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest. 2000;105:1791–1797. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blasi E, et al. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature. 1985;318:667–670. doi: 10.1038/318667a0. [DOI] [PubMed] [Google Scholar]
- 28.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.