

Abstract
A sp3 – sp2 C-C cross-coupling reaction catalyzed by gold in the absence of a sacrificial oxidant is described. Vital to the success of this method is the implementation of a bimetallic catalyst bearing a bis(phosphino)amine ligand. A mechanistic hypothesis is presented, and observable transmetallation, C-Br oxidative addition, and C-C reductive elimination in a model gold complex are shown. We expect that this method will serve as a platform for the development of novel transformations involving redox-active gold catalysts.
Keywords: Gold Catalysis, C-C Coupling, Oxidative Addition, Bimetallic Catalysis, Allylation
The air- and water-stability of gold catalysts, coupled with their ability to promote complex transformations under mild conditions has attracted considerable interest from the academic community.1 Despite the rapid pace of recent developments, the majority of gold-catalyzed processes rely on a select few reaction manifolds: (i) Lewis acid catalysis, (ii) π-activation, and (iii) the generation of carbenoid intermediates (Scheme 1A).2 While these modes of reactivity have yielded important catalytic methodologies of broad scope and synthetic utility,3 they are typified by catalytic cycles wherein gold maintains a +1 oxidation state, in stark contrast to the 2-electron redox cycles characteristic of late transition metal catalysis.4 Indeed, access to AuIII intermediates under catalytic conditions typically requires strong F+ or I3+ oxidants.5,6
Scheme 1.
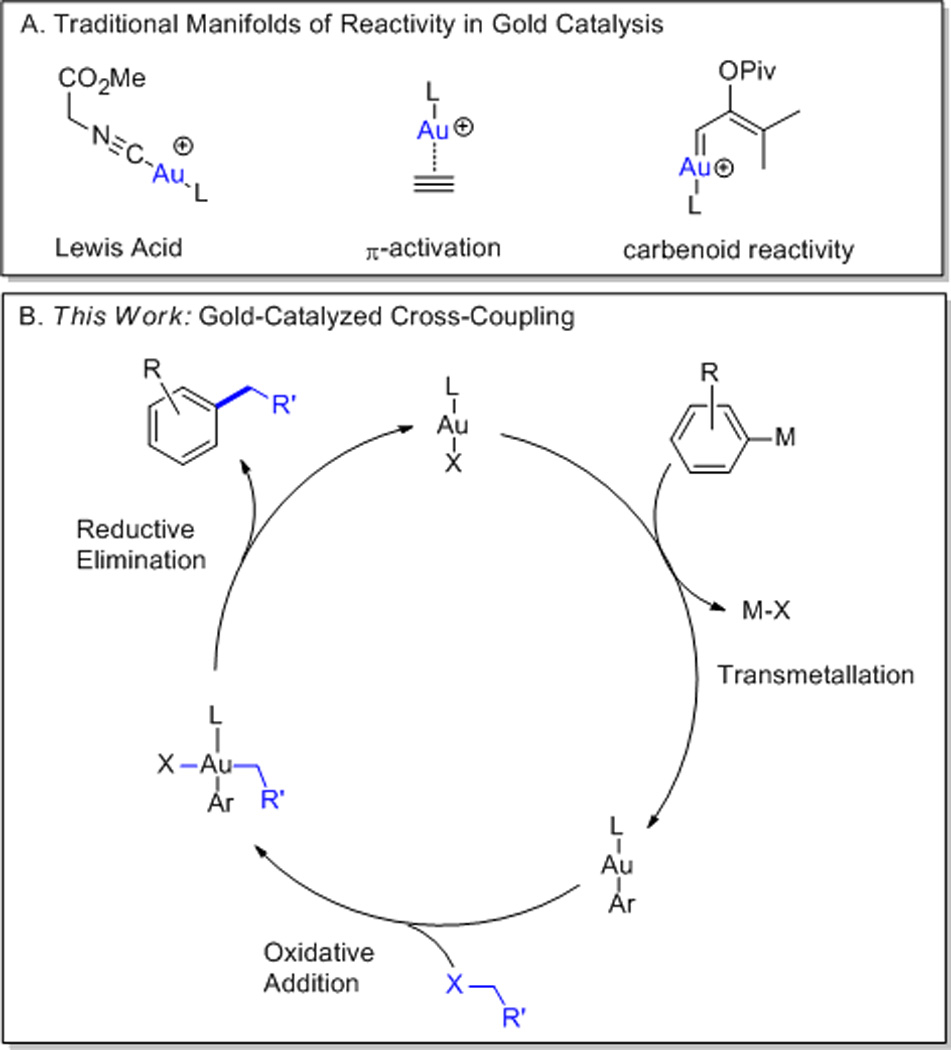
Reactivity in Gold Catalysis
Despite this limitation, seminal work by Kochi and Schmidbaur has shown that AuI complexes oxidatively add alkyl halides, and are further competent to undergo C-C reductive elimination, furnishing formally cross-coupled products.7 However, this mode of reactivity has not previously been realized in a catalytic fashion.
A possible barrier to the implementation of such a redox cycle is the slow rate at which alkyl-alkyl reductive elimination occurs.8 Nevertheless, we were encouraged by our own recent observations that in contrast, aryl-aryl reductive elimination from AuIII is remarkably fast.9 As such, we hypothesized that a process involving oxidative addition to a gold aryl species followed by sp2–sp3 reductive elimination might prove achievable under the influence of a gold catalyst (Scheme 1B).
After examining several classes of aryl nucleophiles and alkyl electrophiles, we found that allyl bromide and phenylboronic acid produced allylbenzene and biphenyl as products when Ph3PAuCl was used as a catalyst (Table 1, entry 1). However, we were unable to substantially improve the yield by implementing other traditional gold catalysts or by increasing catalyst loading (entries 2 – 6, 11).
Table 1.
Catalyst Optimization
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Yield 3a |
Yield 4a |
Entry | Catalyst | Yield 3a |
Yield 4a |
| 1 | Ph3PAuCl | 36% | 10% | 8 | 1 | 66% | 9% |
| 2 | IPrAuCl | 1% | 0% | 9 | 2 | 16% | 7% |
| 3 | tBu3PAuCl | 5% | 6% | 10a | 2 | 17% | 9% |
| 4 | (Johnphos)AuCl | 11% | 3% | 11a | Ph3PAuCl | 41% | 15% |
| 5 | (PhO)3PAuCl | 1% | 0% | 12 | IPrAuOH | 5% | 0% |
| 6 | (pOMePh)3PAuCl | 31% | 6% | 13 | None | 0% | 0% |
| 7 | dppm(AuCl)2 | 24% | 14% | 14b | 1 | 0% | 0% |
Conditions: 4 equiv halide, 3 equiv base, 0.2 M, 18 hrs, Calibrated GC Yields vs. PhCO2Et as an internal standard.
10 mol% catalyst
No allyl bromide added;
IPr = 1,3-bis(2,6-diisopropylphenyl)-1H-imidazol-2(3H)-ylidene, Johnphos = 2-dicyclohexylphosphinobiphenyl

In seeking to improve the reaction, we were drawn to the observation that closely linked bimetallic gold complexes undergo accelerated oxidative addition, due to the formation of AuII-AuII species (rather than discrete AuIII) upon oxidation (Scheme 2).10 While dppm(AuCl)2 showed considerable instability under the reaction conditions, the bimetallic complex 1 produced the desired product in an improved 66% yield.11
Scheme 2.

Intriguingly, the analogous monometallic aminophosphine complex 2 afforded substantially lower yield (even at 10% loading), suggesting that the bimetallic catalyst architecture is responsible for the activity of 1, rather than the electronic character of the aminophosphine ligand.12 However, because monometallic complexes are capable of catalyzing this transformation (albeit with lower efficiency), the influence of the bimetallic catalyst remains to be fully elucidated.
In the absence of allyl bromide (entry 14), neither product was observed, signifying that allyl bromide serves as the oxidant in the homo-coupling process. Notably, the reaction proceeded with identical efficiency in the presence of air and water.
Scheme 3 illustrates the scope of the boronic acid component. While highly basic or nucleophilic functionality was not tolerated, heteroaromatic boronic acids (3j, 3k) were coupled smoothly. Of note, substrates bearing aryl halide moieties reacted with complete chemoselectivity for the external allylic halide (3m), showcasing the discrimination inherent in the SN2-type oxidative addition typically observed with AuI.7 Interestingly, sterically encumbering substituents were found to facilitate the reaction (compare 3f, 3g, 3h). This effect ostensibly arises because the ortho substituents block the formation of homo-coupling side-products.
Scheme 3.
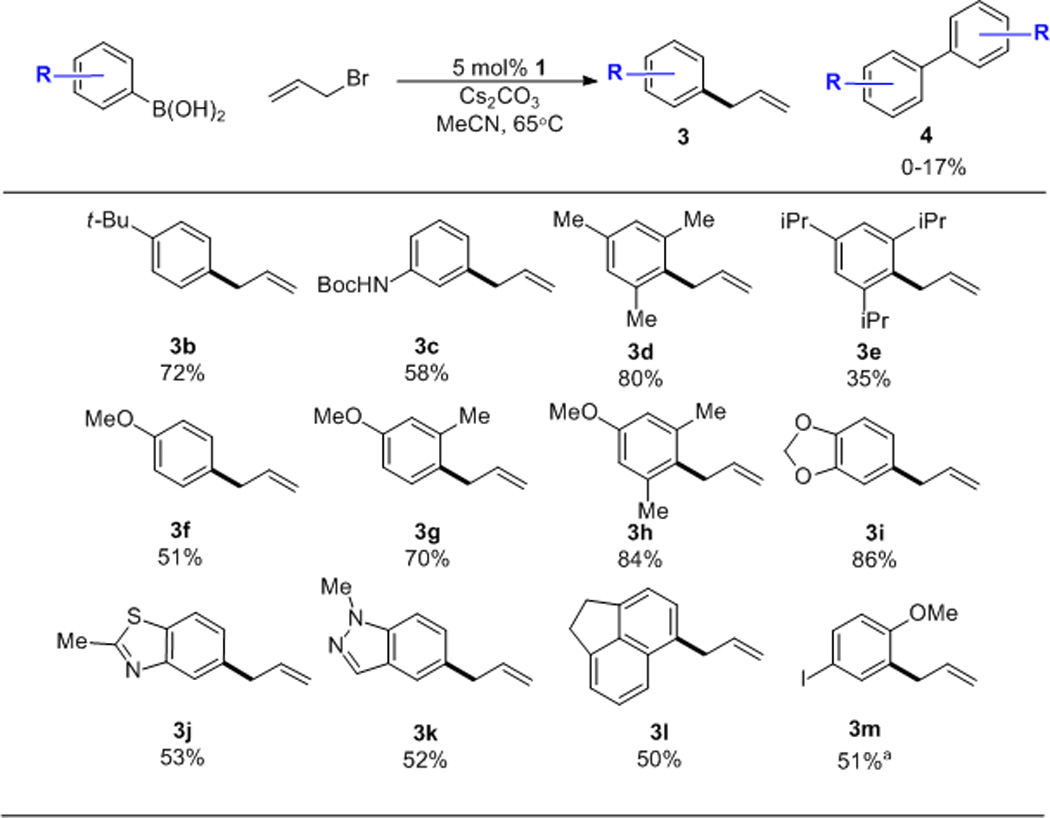
Arylboronic acid scope. Conditions: 4 equiv. halide, 3 equiv base, 0.2 M, 18 hrs. Isolated yields. [a] 10 mol% catalyst
The beneficial effect of sterics led us to examine the scope of the allylic electrophile with mesityl boronic acid, an otherwise challenging cross-coupling substrate (Scheme 4). This effect is further exhibited in products 5f–5i. In all cases, linear products were observed.13
Scheme 4.
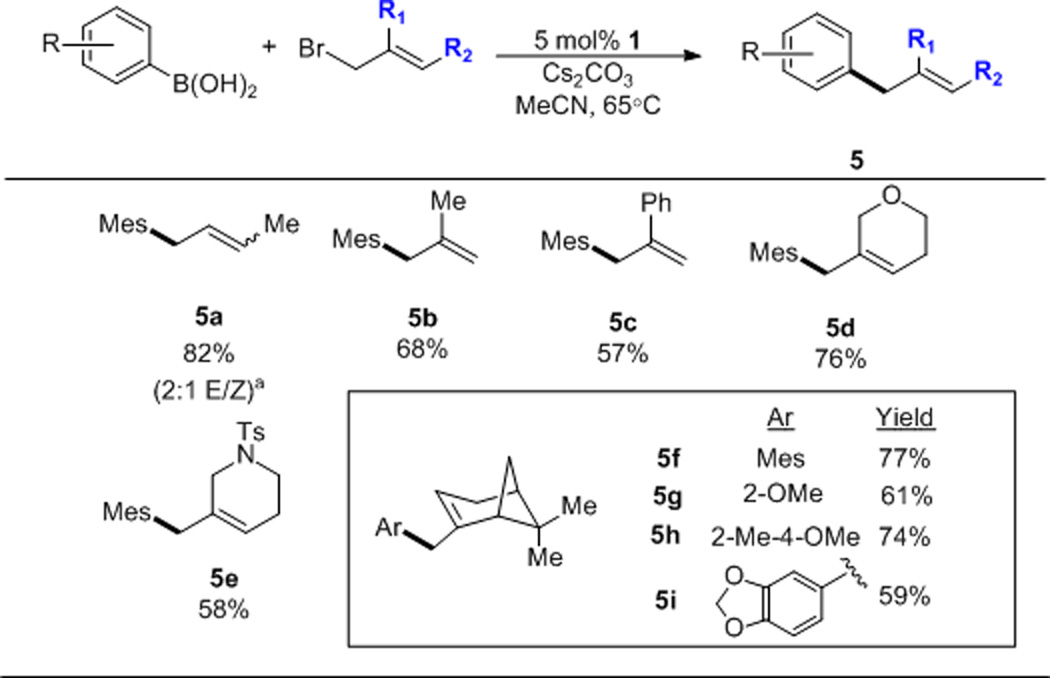
Allylic Bromide Scope. Conditions: 4 equiv. halide, 3 equiv. base, 0.2M, 18 hrs, isolated yields. Mes = 2,4,6-trimethylphenyl. No biaryl detected. [a] Starting from 5:1 E:Z crotyl bromide
The orthogonality of this method to traditional cross-coupling reactions allows the chemoselective preparation of polyfunctionalized products (Scheme 5). While gold and palladium catalysts are both capable of producing 5j, our gold-catalyzed protocol provided higher efficiency and chemoselectivity, allowing access to bifunctionalized products such as 6.14 Furthermore, 3n can be prepared without competitive cyclization or oligomerization.15
Scheme 5.

Orthogonal reactivity of [Au] and [Pd]
Having developed this method, we sought to better understand the mechanism of the overall transformation. In initial stoichiometric experiments (Scheme 6) we found that while 1 underwent halide metathesis upon reaction with allyl bromide, no oxidized species were detected.10f However, the gold aryl complex 9 was formed cleanly via transmetallation from the boronic acid under the reaction conditions.16 Futhermore, 9 underwent facile conversion in reaction with allyl bromide to give the dibromide 8, affording allylbenzene and biphenyl. These experiments suggest a mechanism for the catalytic process in which transmetallation to gold precedes oxidative addition.17,18
Scheme 6.
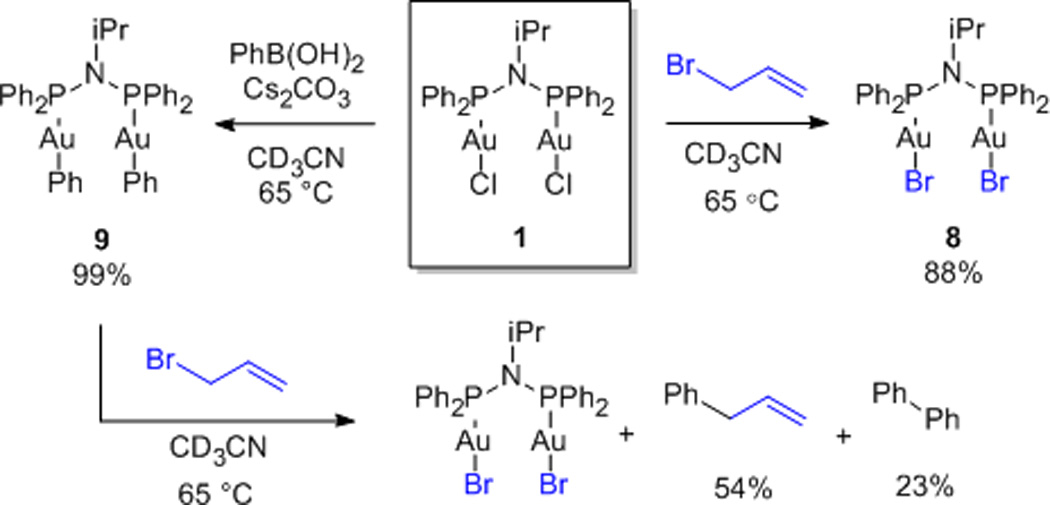
Stoichiometric reactivity of 1
While a number of mechanisms can be proposed for the formation of the desired allylbenzene product from the gold aryl 9, fewer mechanisms can account for the formation of biaryl. Because alternatives to the oxidative addition/reductive elimination process almost invariably necessitate distinct pathways to cross- and homo-coupled products, examination of potential homo-coupling processes can be used to discern between possible mechanistic scenarios (Scheme 7A).19
Scheme 7.
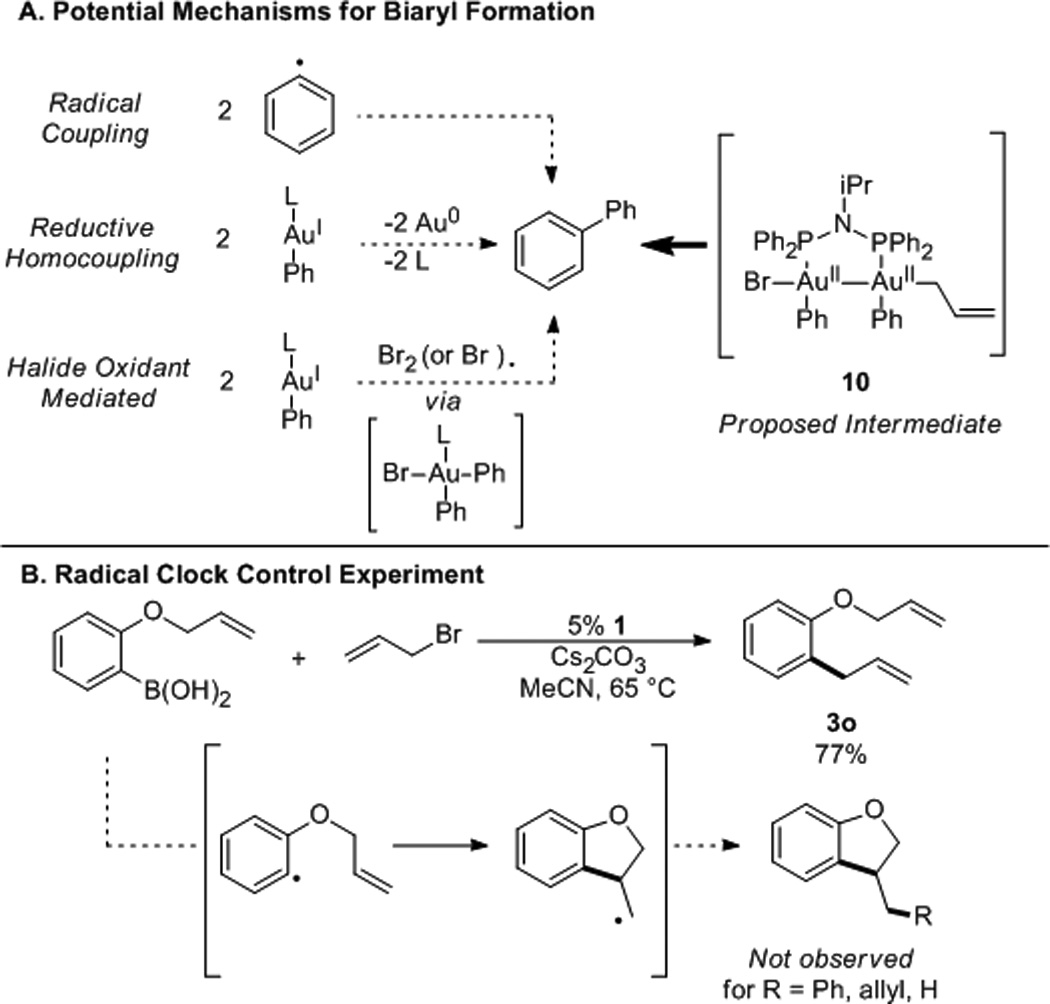
Mechanisms for Biaryl Formation
Of the likely mechanisms, radical clock experiments (Scheme 7B) argue against the implication of radicals, while the stability of 9 to high temperatures argues against reductive homocoupling processes (cf. Table 1, entry 12).14 Finally, halide scavenger experiments argue against trace bromine (or bromine atom) oxidants as agents for the production of biaryl.14,20,21
Combined, these experiments ultimately lead us to implicate the AuII-AuII intermediate 10 as the most likely source of biaryl. Reductive elimination from 10 can presumably also lead to alkyl-aryl bond formation, immediately suggesting a parsimonious mechanism for the overall transformation. Despite this evidence, attempts to isolate or detect the AuII-AuII intermediate directly have so far proven fruitless, likely due to the rapid rate of reductive elimination.9
In light of these difficulties, we turned to the tethered substrate 11 as a mechanistic probe, expecting that the resulting aurocyclic product (e.g. 13) would exhibit hampered reductive elimination, allowing direct observation of reaction intermediates.22 Although transmetallation of 11 to phosphine supported gold complexes such as 1 was accompanied by hydrolysis of the allylic bromide moiety, it was found that clean transmetallation could be accomplished by employing IPrAuOH.23 Although 12 does not react further in benzene, oxidative addition could be initiated upon gentle heating in acetonitrile to yield the isolable AuIII species 13 (Scheme 8).24 Finally, halide abstraction results in reductive elimination to give the exomethylene cyclobutene 14.
Scheme 8.
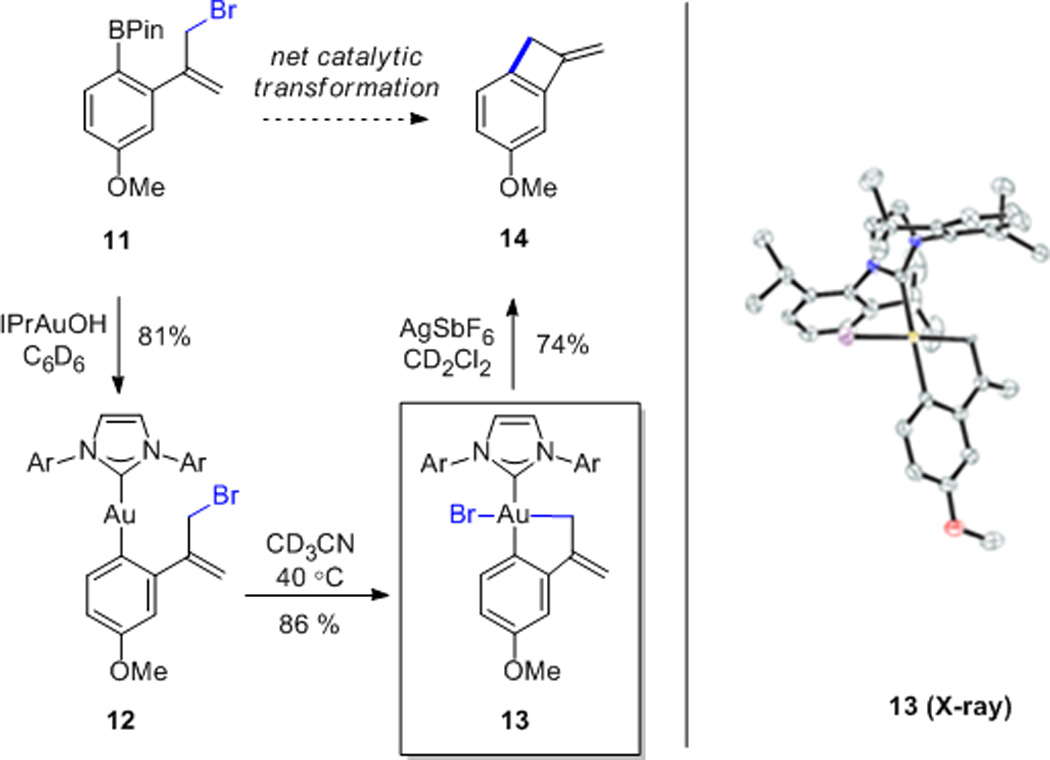
Model Catalytic Cycle and Isolable Product of Oxidative Addition. Ar = 2,6-diisopropylphenyl; asymmetric unit contains two molecules of 13, only one shown; hydrogens omitted for clarity
With the viability of allylic halide oxidative addition to gold aryl complexes demonstrated, we propose the following overall mechanism for this process, following the general outline of Scheme 1B: (i) base-assisted transmetallation of the arylboronic acid to a gold bromide complex, (ii) bimetallic oxidative addition of an allylic halide to the gold aryl species, and (iii) fast C-C reductive elimination to give either allylbenzene or biaryl as product.25,26
In conclusion, we have developed the first example of a net redox-neutral cross-coupling catalyzed by gold.27 The method provides access to sp2 – sp3 coupled products under mild conditions with complete tolerance for air and water. The reaction exhibits unique scope and chemoselectivity, allowing entry to a variety of allylbenzene products. Furthermore, initial experiments suggest an unprecedented mechanism involving oxidative addition to a gold aryl species as a key step. This reaction manifold promises to serve as a powerful strategy for the development of novel gold-catalyzed reactions.
Supplementary Material
Footnotes
We gratefully acknowledge NIHGMS (RO1 GM073932) for financial support. M.D.L thanks the NSF GRFP and ARCS foundation for graduate research fellowships. We gratefully acknowledge Dr. Yi-Ming Wang, Andrew V. Samant, and Dr. David A. Nagib for helpful discussion, and Dr. Antonio DiPasquale for assistance with collecting and analyzing crystallographic data. Prof. Neal P. Mankad is thanked for initial investigations into the chemistry of 1 and 8.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- 1.Nugent WA. Angew. Chem. Int. Ed. 2012;51:8936–8949. doi: 10.1002/anie.201202348. [DOI] [PubMed] [Google Scholar]
- 2.Select reviews on gold catalysis: Shapiro ND, Toste FD. Synlett. 2010:675–691. doi: 10.1055/s-0029-1219369. Fürstner A. Chem. Soc. Rev. 2009;38:3208–3221. doi: 10.1039/b816696j. Jimenez-Nuñez E, Echavarren AM. Chem. Rev. 2008;108:3326–3350. doi: 10.1021/cr0684319. Shen HC. Tetrahedron. 2008;64:7847–7840. Gorin DJ, Sherry B, Toste FD. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. Li Z, Brouwer C, He C. Chem. Rev. 2008;108:3239–3265. doi: 10.1021/cr068434l. Hashmi ASK. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x.
- 3.For an example of AuI catalysed allyl-allyl coupling: Porcel S, Lopez-Carrillo V, Garcia-Yerba C, Echavarren AM. Angew. Chem. Int. Ed. 2008;47:1883–1886. doi: 10.1002/anie.200704500.
- 4.Hartwig JF. Organotransition Metal Chemistry, from Bonding to Catalysis. Sausalito: University Science Books; 2010. [Google Scholar]
- 5.For relativistic influences on the oxidation potential of gold: Gorin DJ, Toste FD. Nature. 2007;446:395–403. doi: 10.1038/nature05592.
- 6.a) Brenzovich WE, Benitez D, Lackner AD, Shunatona HP, Tkatchouk E, Goddard WA, Toste FD. Angew. Chem. Int. Ed. 2010;49:5519–5522. doi: 10.1002/anie.201002739. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tkatchouk E, Mankad NP, Benitez D, Goddard WA, Toste FD. J. Am. Chem. Soc. 2011:133, 14293–14300. doi: 10.1021/ja2012627. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Melhado A, Brenzovich WE, Lackner AD, Toste FD. J. Am. Chem. Soc. 2010;132:8885–8887. doi: 10.1021/ja1034123. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang G, Cui L, Wang Y, Zhang L. J. Am Chem. Soc. 2010;132:1474–1475. doi: 10.1021/ja909555d. [DOI] [PubMed] [Google Scholar]; e) Ball LT, Green M, Lloyd-Jones GC, Russel CA. Org. Lett. 2010;12:4724–4727. doi: 10.1021/ol1019162. [DOI] [PubMed] [Google Scholar]; f) Brenzovich WE, Brazeau JF, Toste FD. Org. Lett. 2010;12:4728–4731. doi: 10.1021/ol102194c. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Ball LT, Lloyd-Jones GC, Russel CA. Chem. Eur. J. 2012;18:2931–2937. doi: 10.1002/chem.201103061. [DOI] [PubMed] [Google Scholar]; h) Hopkinson MN, Tessier A, Salisbury A, Giuffredi GT, Combettes LE, Gee AD, Governeur V. Chem. Eur. J. 2010;16:4739–4743. doi: 10.1002/chem.201000322. [DOI] [PubMed] [Google Scholar]; i) Ball LT, Lloyd-Jones GC, Russel CA. Science. 2012;337:1644–1648. doi: 10.1126/science.1225709. [DOI] [PubMed] [Google Scholar]; j) Zhang G, Luo Y, Wang Y, Zhang L. Angew. Chem. Int. Ed. 2011;50:4450–4454. doi: 10.1002/anie.201100293. [DOI] [PubMed] [Google Scholar]; k) Zhang G, Peng Y, Cui L, Zhang L. Angew. Chem. Int. Ed. 2009;48:3112–3115. doi: 10.1002/anie.200900585. [DOI] [PubMed] [Google Scholar]; l) Qian D, Zhang J. Beilstein J. Org. Chem. 2011;7:808–812. doi: 10.3762/bjoc.7.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For examples of oxidative addition to mononuclear gold(I): Tamaki A, Kochi JK. J. Organomet. Chem. 1974;64:411–425. Shiotani A, Schmidbaur H. J. Organomet. Chem. 1972;37:C24–C26. Tamaki A, Magennis SA, Kochi JK. J. Am. Chem. Soc. 1973;95:6487–6488. Guenther J, Mallet-Ladeira S, Estevez L, Miqueu K, Amgoune A, Bourissou D. J. Am. Chem. Soc. 2014;136:1778–1781. doi: 10.1021/ja412432k.
- 8.a) Tamaki A, Magennis SA, Kochi JK. J. Am. Chem. Soc. 1974;96:6140–6148. [Google Scholar]; b) Komiya S, Albright TA, Hoffmann R, Kochi JK. J. Am. Chem. Soc. 1976;98:7255–7265. [Google Scholar]; c) Komiya S, Kochi JK. J. Am. Chem. Soc. 1976;98:7599–7607. [Google Scholar]; d) Kuch PL, Tobias RS. J. Organomet. Chem. 1976;122:429–446. [Google Scholar]; e) Komiya S, Shibue A. Organometallics. 1985;4:684–687. [Google Scholar]; f) Komiya S, Ozaki S, Shibue A. Chem. Commun. 1986:1555–1556. [Google Scholar]; g) Vicente J, Bermudez MD, Escribano J. Organometallics. 1991;10:3380–3384. [Google Scholar]
- 9.Wolf WJ, Winston MS, Toste FD. Nature Chemistry. 2013;6:159–164. doi: 10.1038/nchem.1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For examples of oxidative addition to bimetallic gold complexes: Fackler JP. Polyhedron. 1997;16:1–17. Murray HH, Fackler JP. Inorg. Chim. Acta. 1986;115:207–209. Laguna A, Laguna M. Coord. Chem. Rev. 1995;193:837–856. Murray HH, Fackler JP, Mazany AM, Porter LC, Shain J, Falvello LR. Inorg. Chim. Acta. 1986;114:171–178. Fackler JP, Basil JD. Organometallics. 1982;1:871–873. f) Halide metathesis via oxidative addition: Fackler JP, Murray HH, Basil JD. Organometallics. 1984;3:821–823. Basil JD, Murray HH, Fackler JP, Tocher J, Mazany AM, Trzcinska-Bancroft B, Knachel H, Dudis D, Delord TJ, Marlier DO. J. Am. Chem. Soc. 1985;107:6908–6915.
- 11.For the first report of complex 1: Chan C, Cheung K, Yam VW. J. Chem. Soc. Dalton Trans. 1996;20:4019–4022.
- 12.In accordance with this hypothesis, our group has previously observed improved reactivity in oxidative gold-catalyzed transformations using bimetallic catalysts. See Ref 5a,5b.
- 13.The origin of this regioselectivity is not known. For regioselectivity in Pd, Mo, W, and Cu catalyzed allylic subsitution: Trost BM, Hung M-H. J. Am. Chem. Soc. 1984;106:6837–6839. Yoshikai N, Nakamura E. Chem. Rev. 2012;112:2339–2372. doi: 10.1021/cr200241f.
- 14.See SI for details.
- 15.For an example of chemoselectivity in Pd-catalyzed allylic substitution: Hussain MM, Walsh PJ. Angew. Chem. Int. Ed. 2010;49:1834–1837. doi: 10.1002/anie.200905399.
- 16.For boronic acid transmetallation to phosphine-supported gold complexes: Partyka DV, Zeller M, Hunter AD, Gray TG. Angew. Chem. Int. Ed. 2006;45:8188–8191. doi: 10.1002/anie.200603350. Hashmi ASK, Ramamurthi TD, Rominger FJ. J. Organomet. Chem. 2009;694:592–597.
- 17.A mechanism involving reversible oxidative addition followed by slow transmetallation is also possible, but likewise does not account for the formation of biphenyl.
- 18.The difference in product ratio between the catalytic and stoichiometric experiments is likely due to the higher concentration of [AuI]-Ar species, which transmetallate rapidly with AuIII (see ref 9). Monoaryl species have been detected by 31PNMR in the stoichiometric reaction of 9 with allyl bromide and are clearly likewise competent to produce allylbenzene products.
- 19.Such mechanistic possibilities for allylbenzene formation include a) electrophilic aromatic substitution (in analogy to protodeauration): Roth KE, Blum SA. Organometallics. 2010;29:1712–1716. and b) carboauration-elimination (in analogy to “dehydrative” allylic substitutions): Mukherjee P, Widenhoefer RA. Angew. Chem. Int. Ed. 2012;51:1405–1407. doi: 10.1002/anie.201107877.
- 20.Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. Sausalito: University Science Books; 2006. [Google Scholar]
- 21.Kochi JK. Organometallic Mechanisms and Catalysis. New York, NY: Academic Press; 1978. [Google Scholar]
- 22.Albrecht M. Chem. Rev. 2010;110:576–623. doi: 10.1021/cr900279a. [DOI] [PubMed] [Google Scholar]
- 23.Gaillard S, Slawin AMZ, Nolan SP. Chem. Commun. 2010;46:2742–2744. doi: 10.1039/c0cc00018c. [DOI] [PubMed] [Google Scholar]
- 24.This solvent effect appears stronger than those previously reported for SN2-like transition metal oxidative addition to alkyl halides, although the rates have not been quantified. For a comprehensive study on this effect see: Chock PW, Halpern J. J. Am. Chem. Soc. 1966;88:3511–3514.
- 25.Reductive elimination to yield biphenyl presumably generates a gold allyl species which can react further with allyl bromide. Correspondingly, 1,5-hexadiene has been detected by 1HNMR under the reaction conditions in varying amounts.
- 26.Based on the analogous aryl-aryl reductive elimination, we expect that isomerization from 10 (or its monoaryl analogue) to a mixed-valent AuIII - AuI species precedes reductive elimination. (see ref. 9).
- 27.For a discussion of putative gold catalyzed Suzuki and Sonogashira reactions, see: Lauterbach T, Livendahl M, Rosellon A, Espinet P, Echavarren AM. Org. Lett. 2010;12:3006–3009. doi: 10.1021/ol101012n.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.