

Abstract
Medical genetics typically entails the detailed characterization of a patient’s phenotypes followed by genotyping to discover the responsible gene or mutation. Here, we propose that the systematic discovery of genetic variants associated with complex diseases such as autism are progressing to a point where a reverse strategy may be fruitful in assigning the pathogenic effects of many different genes and in determining whether particular genotypes manifest as clinically recognizable phenotypes. This “genotype-first” approach for complex disease necessitates the development of large, highly integrated networks of researchers, clinicians, and patient families, with the promise of improved therapies for subsets of patients.
The genetic study of complex disease has historically been difficult, met with limited success and often even fewer therapeutic advances in patient care. Unlike Mendelian disorders, complex disease is defined as a phenotype that is not caused by a single gene mutation but, rather, by many individual gene events, with a significant contribution from environmental factors. The nature of complex genetic disease makes patient care difficult, as a clinician may never see two individuals with the same gene mutation and, therefore, the same underlying genetic etiology. Classical approaches to the study of complex disease have identified patients with similar phenotypes and have attempted to identify the common causative mutation for this phenotype using association studies. Though there have been numerous loci reported over the last 10 years, in most cases, much of the heritability of complex disease remains unresolved (Manolio et al., 2009). The number of success stories for complex neurocognitive and neurobehavioral disease are even fewer, with enormous numbers of patients (>30,000) being required to discover a small fraction of the genetic risk using genome-wide association study (GWAS) approaches (McCarroll and Hyman, 2013). Complex neurodevelopmental disorders, such as autism, schizophrenia, bipolar disorder, intellectual disability (ID), and developmental delay (DD), require better approaches to link genotype to phenotype. In this Essay, we focus on autism spectrum disorder (ASD)—a highly complex neurodevelopmental disease with a range of phenotypes and a large patient base—and propose a gene-centric methodology to model a streamlined approach for subtyping autism starting with the genotype (Schulze and McMahon, 2004).
The explosion of data from recent exome studies (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012b; Sanders et al., 2012) and earlier work on large copy number variants (CNVs) (de Vries et al., 2005; Sebat et al., 2007; Sharp et al., 2006) have emphasized the importance of sporadic truncating mutations in ASD, revealing a surprising level of genetic heterogeneity among patients. From these data, it has been estimated that ~1,000 distinct loci may be related to disease etiology in ASD, assuming a model of sporadic protein-encoding mutations. Interestingly, more than two decades ago, Percy postulated that a “very wide variety of autistic syndromes depending on underlying etiology” may exist based on his observation that a significant fraction of individuals with fragile X, Rett, and tuberous sclerosis syndromes could be classified as having autistic features (Percy et al., 1990). Whereas traditional genetics approaches were entirely underpowered to detect small subpopulations of autism with a common mutant gene, the advent of next-generation sequencing technology has made it possible to begin to systematically classify genetic subtypes of ASD and, further, to ask whether these define specific clinical subtypes of ASD. For the purpose of this Essay, we will define a “genetic subtype” as a gene in which recurrent mutations show an excess of burden in patients versus controls. This is distinguished from a “molecular subtype” that constitutes a group of genetic subtypes that are linked together in a common pathway (coexpression, protein-protein interaction network, etc.) (O’Roak et al., 2012b).
The extreme genetic heterogeneity exemplified by autism, we believe, requires a shift in the approach to studying the genetics of complex neurological disease. Instead of comprehensive and exhaustive phenotyping as the first step to reducing genetic heterogeneity, we propose to leverage technology to genetically classify subtypes of disease among patients in whom clinical recontact is possible. We outline three logical steps in characterizing genetic subtypes from the perspective of autism: (1) candidate discovery and determination of pathogenicity, (2) comprehensive clinical phenotyping, and (3) resolution of genetic background effects (Figure 1). There are alternative approaches to identifying subtypes of ASD, including the modeling of existing behavioral data sets or the analysis of clinical records to derive clusters of patients with ASD (Bitsika et al., 2008; Doshi-Velez et al., 2014; Sacco et al., 2012), that will continue to contribute to subtype identification. However, we have now been able to identify recurrent rare disruptive mutations in the same gene to the point of statistical significance in ASD patients (O’Roak et al., 2012a), and this is just the beginning. As more patients are analyzed by this approach, we will begin to identify the true scope of genetic subtypes in ASD. The reasonable next step will be to ask whether these genetic subtypes define distinct clinical entities. If so, this clears a path for future functional studies and therapeutic development, as well as patient support groups based on common subtypes of ASD.
Figure 1. Schematic of Genotype-First Approach for ASD.

(A–C) Following complex neurodevelopmental disease diagnosis in the clinic, step one is to apply next-generation sequencing (exome or whole-genome) to identify high-impact rare or de novo variants that exist(s) in an individual. Through screening of many individuals, recurrent mutations in a gene or locus are identified with a general diagnosis of ASD or DD. These candidates are selected for targeted resequencing using high-throughput, cost-effective technologies. Molecular inversion probe (MIP) technology, for example, applied to thousands of individuals with ASD or DD identifies genes with an excess mutational burden in probands when compared to controls. Such genes are most likely to contribute to disease etiology and represent future targets for therapeutic intervention. Families with these gene mutations are recontacted and brought back to the clinic for more comprehensive phenotyping (step two). There will be those genes that have a common, strong, single clinical phenotype (A); however, these will likely be rare. There may be those individual genotypes that all affect the same functional pathway (molecular subtypes) and result in similar or potentially opposing phenotypes (e.g., macrocephaly versus microcephaly) (B). Some mutations even within the same gene, however, may have multiple associated clinical phenotypes (C), suggesting high variability in the type of mutation relative to its gene function and/or incomplete penetrance. The latter especially will require more in-depth study for genetic background effects (step three). This approach will group patients foremost based on genotypes or sets of mutated genes. Larger groups of patients with the same presumptive genetic etiology are re-examined to identify specific clinical phenotypes, with the goal of improving diagnosis, patient care, and management.
Gene Discovery and Pathogenicity
The most important step is to identify the genes and the most highly penetrant mutations first. For genetically heterogeneous diseases such as autism, this requires an unprecedented scale of coordination and sample collection. Exome sequencing of patients with ASD is well underway, with the exomes of >10,000 patients expected to be completed in the next year (Buxbaum et al., 2012). Recent work has already demonstrated the unequivocal importance of putative loss-of-function mutations, at least in the case of ID and simplex autism (Veltman and Brunner, 2012). Nevertheless, the biological significance of most de novo mutations discovered as part of exome and genome sequencing projects is uncertain. The locus heterogeneity necessitates resequencing a much larger cohort to prove pathogenicity. If CNV studies are to be a guide, tens of thousands of patients will be required to achieve statistical significance for specific loci. Patients with ID and DD represent a rich resource for the discovery and validation of additional de novo ASD mutations, as more than an estimated 50% of individuals with clinically defined ASD are also intellectually impaired (La Malfa et al., 2004; Matson and Shoemaker, 2009). Indeed, more than half of the recurrent truncating mutations discovered in the Simons Simplex Collection (SSC) occurred among individuals with an IQ of <70 (O’Roak et al., 2012a). Similarly, the majority of the most strongly associated “autism” CNVs (e.g., 16p11.2) are also prevalent among children with ID and DD (Girirajan et al., 2013; Sanders et al., 2011), highlighting the shared genetic etiology underlying these neurodevelopmental disorders.
Dealing with such a large number of patients poses significant hurdles. First, it requires a high-throughput, cost-effective resequencing strategy. Rare and private variants such as these cannot be readily imputed using GWAS approaches but, rather, must be directly detected—as is the case for CNVs associated with ASD. Molecular inversion probes (MIPs) provide one such approach to rapidly resequence candidate genes with high sensitivity and specificity (Turner et al., 2009). Current estimates indicate that, with limited starting material, it is possible to resequence 50 genes for less than $1 per gene/sample (O’Roak et al., 2012a). The second major hurdle is the samples themselves. Most individual sample collections are inadequate and therefore too underpowered to achieve statistical significance. This realization has led to the formation of consortia such as the Autism Sequencing Consortium (ASC) (Buxbaum et al., 2012) and the Autism Spectrum/Intellectual Disability network (ASID), representing groups of researchers and clinicians that have agreed in principle to share samples, technology, and sequencing results to pinpoint genetic risk factors much more quickly (Figure 2). Though this collaboration is an important step forward, simply merging sample collections or sequencing results in a case-control design remains insufficient.
Figure 2. The Autism Spectrum/Intellectual Disability Network.

The Autism Spectrum/Intellectual Disability network (ASID), composed of 21 basic research and clinical laboratories from around the world, has assembled >15,000 patients for gene resequencing. The network is broader than the Autism Sequencing Consortium (ASC) in that it considers patients with ASD, ID, epilepsy, or DD due to their comorbidity. It emphasizes collections where parental DNA is available and where patient recontact is possible to accurately resolve phenotype-genotype correlations. The network includes 10 clinical research labs across the world (blue squares) and 11 labs in the USA (red dots) that recruited families as part of the SSC, where patient recontact has now been made possible after extensive IRB review. Subsets of patients with mutations in a common gene are being reassessed to determine whether the mutation defines a clinical subtype.
As sequencing becomes much more routine, more integrated patient-clinician-researcher networks dedicated to a specific molecular lesion should be envisioned, similar to what has already begun to occur with specific CNVs through the Simons Variation in Individuals (VIP) Project (Consortium, 2012) and Unique (http://www.rarechromo.org/html/home.asp) models. The Simons VIP focuses on the extensive study of individuals with specific recurrent CNVs that increase the risk for developmental disorders. The project includes the development of patient communities through social networking and education built around an online portal. The initial CNV addressed was 16p11.2, and additional CNVs have since been included in the project (e.g., 1q21.1), with more CNVs scheduled. Families engaged in the com munity are also offered the opportunity to participate in research that includes comprehensive behavioral phenotyping, medical examination, and imaging assessments conducted in a clinical context. The combination of an online portal, extensive recruitment, multisite collaboration, and rapid data sharing policies has enhanced the pace of understanding of deletions and duplication at the 16p11.2 locus in a very short amount of time. UNIQUE is an online portal focused on education, awareness, and support for families with rare chromosomal disorders. Through this online presence, families and practitioners are connected so that research findings are translated, education is provided, and families are presented with opportunities for research participation. Both Simons VIP and UNIQUE provide needed resources and education for patients and families while at the same time allowing for the collection of genetically defined populations of patients for advancing scientific understanding.
The final hurdle to establishing pathogenicity is the evaluation of suitable controls. Some of the most interesting mutations may lurk in the general population at low frequency. Well-phenotyped controls, in large numbers, are even more limiting than patient cohorts. The ad hoc use of “population average” controls from exome sequencing projects (e.g., the Exome Sequencing Project [ESP]), as opposed to disease-specific controls in which DNA and individuals can be revisited for additional analyses, is problematic.
Comprehensive Clinical Phenotyping
Once specific genes have been identified with recurrent and/or structural mutations, a critical next step is to explore the phenotypes associated with sporadic mutations. This requires a reassessment at the clinical level once the mutations are discovered because the number of genes that are responsible for autism appears to be so vast. Moreover, it is quite likely that exome or genome sequencing will become a routine diagnostic inevitably incorporated into the electronic medical record, and once a case report is established, clinical evaluation can proceed in a more-directed fashion. There should be two objectives: (1) define clinical subtypes of ASD based on a rigorous examination of different cases with the same mutated gene and (2) assess whether the de novo mutations are highly penetrant. Although rare, this approach has identified some CNVs (e.g., 17q21.31 and 15q24.3 deletions) that appear to be necessary and sufficient for disease (i.e., all cases are de novo and no instances of the mutation have been found in large surveys of the general population). If 1,000 genes are responsible for ASD—each a different gene in a specific family—even the specialist focused on ASD would unlikely see the same genetic cause twice in 25 years of practice. Next-generation sequencing provides the requisite sieve, allowing the needles in the haystack to be sorted at the molecular level down to a handful of highly relevant clinical phenotypes. Only through a genotype-first approach is there the unprecedented opportunity to bring together 10–20 patients with a common genetic etiology for rigorous and detailed phenotyping. Thus, in this investigative paradigm, enabled by next-generation sequencing, phenotype assessment becomes secondary to mutation discovery but also more important (Schulze and McMahon, 2004; Hennekam and Biesecker, 2012).
Recontact and longitudinal assessment via large consortia is key to this dissection. Ironically, most existing cohorts, even those that are well phenotyped and supported for future work (e.g., the SSC), are not collected in such a way that patient recontact is immediately possible. Recontact with consortia participants can be established but requires significant time and coordination with institutional review boards (IRBs) in order to ensure that appropriate attention to research subjects’ wishes regarding privacy and contact are maintained (Figure 2). Nevertheless, even with limited phenotype data, important genotypephenotype data are beginning to emerge. For example, the recent characterization of patients with recurrent disruptive mutations in the β-catenin/Wnt-signaling pathway identified a subset with macrocephaly (e.g., PTEN) and a subset with microcephaly (e.g., DYRK1A) (Figure 3). Although these results are preliminary, owing to the small number of individuals and the need to control for additional covariates, the data suggest that the extreme genetic heterogeneity (i.e., 1,000 different genes) may be reduced to much smaller subsets of biologically related networks (coexpression or protein networks) with restricted phenotypic presentation (i.e., molecular subtypes).This pathway and the genes associated with it may be an important first step in identifying molecular and clinical subtypes of ASD arising from defects in neural progenitor cell proliferation and apoptosis.
Figure 3. Potential Genetic Definition of Autism Subtypes.
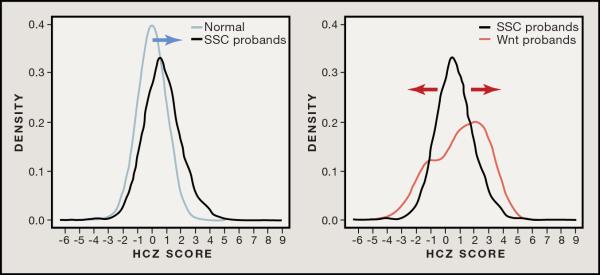
Individuals with autism have been described as having an increased head circumference size (HCZ) distribution (Courchesne et al., 2003). This is observed among ASD probands of the SSC where HCZ is positively skewed (blue arrow) when compared to a normal distribution (left). Subselecting patients (red) with de novo mutations in the β-catenin/Wnt-signaling network (Iossifov et al., 2012; Neale et al., 2012; O’Roak et al., 2012b; Sanders et al., 2012)—defined as those de novo proband events that cluster around the central CTNNB1 node using either STRING or Ingenuity Pathway Analysis enrichment (n = 26 individuals)—further transforms this into a bimodal distribution (right), suggesting reciprocal macrocephaly and microcephaly associated with de novo mutations in this pathway.
As patients and families are recontacted and engaged in follow-up evaluation, the phenotypic workup of patients should be intensive and should include a careful assessment of core behaviors in the domains of ASD; critical evaluation of related symptoms and behaviors such as affective, attentional, or behavioral challenges; and standardized testing of cognitive and adaptive functioning. It should include a standardized assessment of comorbid medical conditions (e.g., intellectual disability, epilepsy, neuropsychiatric disease, cancer risk, etc.), careful review of medical history, brain imaging, and physical examination with a focus on dysmorphology. This approach has been particularly useful in our ongoing examination of 100 cases of the 16p11.2 microdeletion, in which we identified speech sound disorders, motor coordination impairment, and medical concerns such as enuresis and seizures in a significant fraction of cases. More generally, patient recontact has been critical for the identification of new syndromes associated with specific CNVs. Adding to the challenge is that the process needs to be dynamic and iterative. Namely, index cases (the original families used to establish the first version of the working clinical phenotype) will be required to help establish a set of standards and checklists that can evolve in consultation with clinicians, counselors, and families as additional cases are discovered.
The genotype-first approach helps to address the challenges that are inherent in attempting to carve nature at its joints based strictly on a behavioral presentation. A key problem with classifying subtypes by strict clinical criteria is that particular manifestations may depend on a specific point in time in which a patient is evaluated (Charman et al., 2005; Fountain et al., 2012; La Malfa et al., 2004; Lord et al., 2012; McGovern and Sigman, 2005; Pellicano, 2012). This is especially relevant if ASD is regarded as primarily a disorder of neurodevelopment. A 2-year-old with ASD can present remarkably differently just 1 year later as a function of a dynamic developmental trajectory. That is, the same degree or quality of impairment in social communication with peers can appear significantly different for a 3-year-old preschool child compared to a 6-year-old first-grade student based on developmental context. The influence of interactive endogenous and exogenous factors renders multiple assessments conducted in a developmental context essential for the accurate behavioral phenotyping of complex neurodevelopmental disorders. Longitudinal follow-up is thus necessary to define disease trajectories as the patients age. The genetic mutation in a key autism gene provides an objective point of reference to study and compare such developmental progressions.
Genetic Background Effect and Phenotypic Heterogeneity
Based on experience with large CNVs, we anticipate that there will be multiple categories of highly penetrant mutations that will constitute the genetic subtypes of ASD. First, and perhaps the most satisfying, will be those mutations that are most penetrant for disease and associate with a prescribed set of clinical features irrespective of the genetic background (Figure 1A). Such mutations will direct diagnosis and will drive the definition of new syndromic forms of autism. However, the proof for these mutations will ultimately rest on demonstration of enrichment or exclusivity of this type of mutation in patients with a particular subset of features. Most mutations will, unfortunately, not be this simple. A second category of mutations may be those linked to each other through a common protein interaction pathway or functional network, leading to phenotypic manifestations that are virtually indistinguishable at a clinical level (Figure 1B). For example, there are suggestions that de novo mutations in different genes within the β-catenin/Wnt-signaling pathway will define a subset of microcephalic and macrocephalic individuals with ASD (Figure 3). Although more complex, these mutations may also define syndromic forms of ASD and drive patient care when identified. There will still be mutations in other genes and their associated binding partners that appear, on first blush, hopelessly phenotypically variable and/or incompletely penetrant. Mutations in ARID1B, for example, have been found in patients with Coffin-Siris syndrome, syndromic ID, and ASD (Halgren et al., 2012; Santen et al., 2013). In the end, there will undoubtedly remain many genes whose mutation will lead to either clinically subtle or variable phenotypes, limiting any immediate diagnostic value to the approach that we have described. However, even in such cases, understanding the molecular basis of disease will ultimately drive more precise therapeutic intervention.
Even in the cases of apparent gene pleiotropy, we predict that, as larger and larger numbers of patients are studied, smaller subtypes will emerge, as has begun to be observed with certain variable expressive CNVs, wherein a particularly sensitive genetic background may give way to disease due to additional genetic modifiers that would otherwise be genetically silent. In the case of the 1q21 deletion, for instance, a number of subcategories began to emerge (e.g., multiple cases with cataracts, others with structural heart defects, and still others with cognitive deficits) after a larger number of patients were identified (Mefford et al., 2008). We argue that grouping on the “primary” mutation (e.g., 1q21 deletion) enables a more-directed study on the full extent of variable expressivity for that lesion where subsets of clinical manifestation may become apparent. The genotype-first approach provides the means to explore both pleiotropy and variable expressivity (Figure 1C). For variants such as these, it will be critical to understand the potential effect of modifiers on the phenotype. Although we may never have the statistical power to identify a single genetic modifier at a specific locus, signal may become apparent in the aggregate. For example, a higher burden of CNVs of both known and unknown pathogenic significance in aggregate compound the severity of disease presentation for patients with the 16p12.1 microdeletion (Girirajan et al., 2012). Such information is ultimately critical for practical management of disease and patient counseling.
We stress that a significant association signal and a large effect size are not proof positive of causality. Indeed, there is a disturbing trend of excluding patient DNA from further analysis due to the presence of a putative CNV or loss-of-function mutation in a gene that is presumed to be the primary cause of disease. We argue that such samples should be subjected instead to more intensive genetic scrutiny to understand the genetic basis of the phenotypic variability and the complete spectrum of potentially deleterious alleles. Full-genome sequencing, for example, will lead to the characterization of small CNVs within complex regions of the genome and to the discovery of mutations in conserved regulatory regions. Such mutations may provide the evidence needed to explain phenotypic variability in such cases. Studying such mutations in the context of families—i.e., genomic sequencing of unaffected transmitting parents—may provide some important clues as well. Numerous lines of evidence argue that the genetic background is key for such phenotypic variability and that multiple mutations in different genes may compound to result in a specific outcome (Girirajan and Eichler, 2010; Schaaf et al., 2011). The important point here is that patients grouped by a common genetic lesion represent, once again, an objective starting point to begin to assess the effect of other modifiers. This includes not only genetic but epigenetic and environmental factors that may affect the phenotypic outcome of a particular mutation and autism.
Conclusion
With sequencing becoming increasingly cheaper as well as the preferred frontline diagnostic test (Johnson et al., 2011), researchers are now in the position to effectively break down the umbrella of ASD. We propose that defining molecular subtypes will serve as a superior classifier compared to ever-changing psychiatric nosological definitions (e.g., DSM-5 [American Psychiatric Association, 2013]). Indeed, NIMH has made a dedicated commitment to addressing limitations in diagnostic boundaries defined by consensus through the establishment of the Research Domain Criteria for clinical research (Insel et al., 2010). There is a pressing need to systematically prioritize gene-disruptive events and to rapidly and cost effectively resequence candidates in many thousands of individuals and controls in the context of the families to identify high-impact risk factors. Because there is so much overlap among ASD, ID, and epilepsy, broader consortia that consider patients outside of the strict ASD diagnosis should be envisioned. Patients with common genetic etiology should be revaluated for common clinical features no matter their initial diagnosis (e.g., ASD, ID, epilepsy). This has the benefit of controlling for ascertainment bias and discovering truly disease-specific genes in addition to more broadly defined neurodevelopmental disease genes.
This genotype-first perspective also offers both short- and long-term benefits to patients. The identification of genetic subtypes of ASD (especially those mutations deemed most penetrant) provides a medium to rapidly network families, researchers, and clinicians. For families, this will translate into better diagnosis, counseling, and the formation of patient-driven support groups through meetings and interactive websites that link families across the world who have children with a common genetic etiology. Such networks, if properly implemented, can effectively drive research and advance clinical understanding of phenotype-genotype correlations and may further spawn the formation of foundations and dedicated research endeavors (e.g., the Simons VIP).
From a longer-term perspective, the genotype-first approach that we propose will lead to genetic classification of ASD subtypes and may be more broadly applied to complex diseases, such as schizophrenia, ID, DD, and bipolar disorder. There is emerging evidence that such seemingly diverse clinical diagnoses may be, in some cases, genetically linked. The discovery of mutations, genes, and pathways across such diverse diseases not only promises to revolutionize our biological understanding but also may lead to the development of therapies focused on the mutation as opposed to a nosological definition. This holds the promise of better clinical intervention and a full realization of precision medicine.
ACKNOWLEDGMENTS
This work was supported, in part, by U.S. National Institute of Mental Health (NIMH) grant MH101221 and by the Simons Foundation Autism Research Initiative (SFARI) 303241 to E.E.E. E.E.E. is an investigator of the Howard Hughes Medical Institute. E.E.E. is on the scientific advisory boards (SABs) of DNAnexus, Inc. and was an SAB member of Pacific Biosciences, Inc. (2009–2013) and SynapDx Corp. (2011–2013).
REFERENCES
- American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. Fifth Edition American Pyschiatric Publishing; Arlington, VA: 2013. [Google Scholar]
- Bitsika V, Sharpley CF, Orapeleng S. An exploratory analysis of the use of cognitive, adaptive and behavioural indices for cluster analysis of ASD subgroups. J. Intellect. Disabil. Res. 2008;52:973–985. doi: 10.1111/j.1365-2788.2008.01123.x. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Daly MJ, Devlin B, Lehner T, Roeder K, State MW, Autism Sequencing Consortium The autism sequencing consortium: large-scale, high-throughput sequencing in autism spectrum disorders. Neuron. 2012;76:1052–1056. doi: 10.1016/j.neuron.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charman T, Taylor E, Drew A, Cockerill H, Brown JA, Baird G. Outcome at 7 years of children diagnosed with autism at age 2: predictive validity of assessments conducted at 2 and 3 years of age and pattern of symptom change over time. J. Child Psychol. Psychiatry. 2005;46:500–513. doi: 10.1111/j.1469-7610.2004.00377.x. [DOI] [PubMed] [Google Scholar]
- Consortium, V. Simons Vip Consortium Simons Variation in Individuals Project (Simons VIP): a genetics-first approach to studying autism spectrum and related neurodevelopmental disorders. Neuron. 2012;73:1063–1067. doi: 10.1016/j.neuron.2012.02.014. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Carper R, Akshoomoff N. Evidence of brain overgrowth in the first year of life in autism. JAMA. 2003;290:337–344. doi: 10.1001/jama.290.3.337. [DOI] [PubMed] [Google Scholar]
- de Vries BB, Pfundt R, Leisink M, Koolen DA, Vissers LE, Janssen IM, Reijmersdal Sv., Nillesen WM, Huys EH, Leeuw Nd., et al. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi-Velez F, Ge Y, Kohane I. Comorbidity clusters in autism spectrum disorders: an electronic health record time-series analysis. Pediatrics. 2014;133:e54–e63. doi: 10.1542/peds.2013-0819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountain C, Winter AS, Bearman PS. Six developmental trajectories characterize children with autism. Pediatrics. 2012;129:e1112–e1120. doi: 10.1542/peds.2011-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Eichler EE. Phenotypic variability and genetic susceptibility to genomic disorders. Hum. Mol. Genet. 2010;19(R2):R176–R187. doi: 10.1093/hmg/ddq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012;367:1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girirajan S, Dennis MY, Baker C, Malig M, Coe BP, Campbell CD, Mark K, Vu TH, Alkan C, Cheng Z, et al. Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am. J. Hum. Genet. 2013;92:221–237. doi: 10.1016/j.ajhg.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halgren C, Kjaergaard S, Bak M, Hansen C, El-Schich Z, Anderson CM, Henriksen KF, Hjalgrim H, Kirchhoff M, Bijlsma EK, et al. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin. Genet. 2012;82:248–255. doi: 10.1111/j.1399-0004.2011.01755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennekam RC, Biesecker LG. Next-generation sequencing demands next-generation phenotyping. Hum. Mutat. 2012;33:884–886. doi: 10.1002/humu.22048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, Sanislow C, Wang P. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am. J. Psychiatry. 2010;167:748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson HM, Gaitanis J, Morrow EM. Genetics in autism diagnosis: adding molecular subtypes to neurobehavioral diagnoses. Med. Health R. I. 2011;94:124–126. [PMC free article] [PubMed] [Google Scholar]
- La Malfa G, Lassi S, Bertelli M, Salvini R, Placidi GF. Autism and intellectual disability: a study of prevalence on a sample of the Italian population. J. Intellect. Disabil. Res. 2004;48:262–267. doi: 10.1111/j.1365-2788.2003.00567.x. [DOI] [PubMed] [Google Scholar]
- Lord C, Luyster R, Guthrie W, Pickles A. Patterns of developmental trajectories in toddlers with autism spectrum disorder. J. Consult. Clin. Psychol. 2012;80:477–489. doi: 10.1037/a0027214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson JL, Shoemaker M. Intellectual disability and its relationship to autism spectrum disorders. Res. Dev. Disabil. 2009;30:1107–1114. doi: 10.1016/j.ridd.2009.06.003. [DOI] [PubMed] [Google Scholar]
- McCarroll SA, Hyman SE. Progress in the genetics of polygenic brain disorders: significant new challenges for neurobiology. Neuron. 2013;80:578–587. doi: 10.1016/j.neuron.2013.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern CW, Sigman M. Continuity and change from early childhood to adolescence in autism. J. Child Psychol. Psychiatry. 2005;46:401–408. doi: 10.1111/j.1469-7610.2004.00361.x. [DOI] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, Huang S, Maloney VK, Crolla JA, Baralle D, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012a;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012b;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicano E. Do autistic symptoms persist across time? Evidence of substantial change in symptomatology over a 3-year period in cognitively able children with autism. Am. J. Intellect. Dev. Disabil. 2012;117:156–166. doi: 10.1352/1944-7558-117.2.156. [DOI] [PubMed] [Google Scholar]
- Percy A, Gillberg C, Hagberg B, Witt-Engerström I. Rett syndrome and the autistic disorders. Neurol. Clin. 1990;8:659–676. [PubMed] [Google Scholar]
- Sacco R, Lenti C, Saccani M, Curatolo P, Manzi B, Bravaccio C, Persico AM. Cluster analysis of autistic patients based on principal pathogenetic components. Autism Res. 2012;5:137–147. doi: 10.1002/aur.1226. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, Moreno-De-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santen GW, Aten E, Vulto-van Silfhout AT, Pottinger C, van Bon BW, van Minderhout IJ, Snowdowne R, van der Lans CA, Boogaard M, Linssen MM, et al. Coffin-Siris consortium Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum. Mutat. 2013;34:1519–1528. doi: 10.1002/humu.22394. [DOI] [PubMed] [Google Scholar]
- Schaaf CP, Sabo A, Sakai Y, Crosby J, Muzny D, Hawes A, Lewis L, Akbar H, Varghese R, Boerwinkle E, et al. Oligogenic heterozygosity in individuals with high-functioning autism spectrum disorders. Hum. Mol. Genet. 2011;20:3366–3375. doi: 10.1093/hmg/ddr243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze TG, McMahon FJ. Defining the phenotype in human genetic studies: forward genetics and reverse phenotyping. Hum. Hered. 2004;58:131–138. doi: 10.1159/000083539. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- Turner EH, Lee C, Ng SB, Nickerson DA, Shendure J. Massively parallel exon capture and library-free resequencing across 16 genomes. Nat. Methods. 2009;6:315–316. doi: 10.1038/nmeth.f.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]