

Abstract
A critical step in DNA interstrand cross-link repair is the programmed collapse of replication forks that have stalled at an ICL. This event is regulated by the Fanconi anemia pathway, which suppresses bone marrow failure and cancer. In this perspective, we focus on the structure of forks that have stalled at ICLs, how these structures might be incised by endonucleases, and how incision is regulated by the Fanconi anemia pathway.
Introduction
DNA interstrand cross-links (ICLs) are cytotoxic lesions that covalently link the Watson and Crick strands of DNA. From a human health perspective, there are two primary motivations to study ICL repair. First, ICL repair is defective in Fanconi anemia (FA), a human genetic disease caused by biallelic mutations in any one of 16 different FANC genes (Bogliolo et al., 2013; Kashiyama et al., 2013; Kottemann and Smogorzewska, 2013). FA is characterized by congenital abnormalities, bone marrow failure, and cancer predisposition. If ICL repair defects indeed cause FA, as is widely believed, understanding how ICL repair normally occurs and why it fails in patients might point the way to a cure for FA. Second, ICL-inducing agents are widely used in cancer chemotherapy. However, cancers almost invariably become resistant to these agents, in some cases due to up-regulation of repair. Novel inhibitors of ICL repair might augment the efficacy of ICL-inducing agents for chemotherapy, although this might also cause enhanced toxicity.
The major ICL repair pathway operating in proliferating cells is coupled to DNA replication (Akkari et al., 2000; Raschle et al., 2008; Rothfuss and Grompe, 2004; Taniguchi et al., 2002). When forks collide with an ICL, repair is initiated through the excision of the ICL from one parental strand (Figure 1A). This releases or “unhooks” one daughter duplex from the ICL, forming a double-stranded DNA break that must subsequently be repaired. ICL repair is thus a rare instance in which stalled replication forks undergo programmed collapse, and recent evidence suggests this process is dependent on the FANC proteins (Knipscheer et al., 2009). As such, programmed fork collapse can be regarded as a unique event that distinguishes ICL removal from other forms of DNA repair. To shed light on the mechanisms by which forks are processed during ICL repair, we consider here the possible structures of stalled forks prior to collapse and how diverse endonucleases might act on these structures. We also consider the regulation of fork collapse by the FANC proteins.
Figure 1. Possible mechanisms of replication-coupled ICL repair.

Four mechanisms of replication-dependent ICL repair are depicted. The DNA structures acted on by endonucleases in each model are highlighted by a gray box. Incisions are represented by black, blue, red, and green arrowheads. The proposed nuclease(s) that performs each incision is indicated above the arrowhead. (A) The classic ICL repair model, in which a single replication fork collides with the ICL and the leading strand template is incised (Niedernhofer et al., 2005). (B) The classic model, but taking into account the observation that leading strands initially stall 20 nucleotides from the ICL due to the MCM2-7 complex, and that incision occurs on the lagging strand template (Raschle et al., 2008). In models A and B, fork restart would require reloading of the MCM2-7 complex, for which there is no known mechanism. (C) The dual fork convergence model (Raschle et al., 2008). Left inset, 3′ incision substrate if RPA binds the lagging strand template after MCM2-7 removal. Right inset, 3′ incision substrate if parental strands re-anneal after MCM2-7 removal. (D) Traverse model (Huang et al., 2013). The only difference in the incision substrate in the dual fork and traverse models is the location of the 5′ end of the nascent strand on the right side of the ICL (green strand).
Early models of ICL repair
Genetic analysis has identified four major classes of gene products that confer resistance to ICLs. 1) Structure-specific endonucleases, which recognize and incise specific DNA structures. 2) Translesion DNA synthesis (TLS) polymerases, error prone polymerases that are able to tolerate DNA damage in the template strand. (3) DNA recombinases, proteins that mediate strand exchange during homologous recombination. (4) 16 FANC proteins, which are mutated in FA. In the FA “pathway,” eight “group I” FANC proteins assemble into a core complex that mono-ubiquitylates a heterodimer of two “group II” FANC proteins, FANCI and FANCD2 (the “ID” complex)(Alpi et al., 2008; Garcia-Higuera et al., 2001; Smogorzewska et al., 2007). The mono-ubiquitylated ID complex (ID-Ub) is essential for ICL repair (Garcia-Higuera et al., 2001; Knipscheer et al., 2009). The six remaining “group III” FANC proteins fall into the recombinase and nuclease categories. Given the four classes of proteins implicated in ICL repair and the coupling of repair to DNA replication, the following model crystallized several years ago (Niedernhofer et al., 2005; Wang, 2007). Repair is triggered when a DNA replication fork collides with the ICL (Figure 1Ai). This creates a substrate for structure-specific endonucleases, which incise the fork, unhooking the cross-link and generating a double-stranded DNA break (DSB) (Figure 1Aii). The unhooked ICL is bypassed by translesion DNA polymerases (Figure 1Aii). Finally, the fork is restored via homologous recombination (Figure 1Aiii). Although this model accounted for the different gene products implicated in ICL repair and the S phase dependence of repair, it lacked molecular detail. Thus, the precise nature of the DNA intermediates involved remained unclear, making it difficult to understand how the endonucleases and other proteins participate in repair. In addition, it was unknown how the FA pathway promotes repair.
The dual fork convergence model
More recently, replication-dependent ICL repair was recapitulated in Xenopus egg extracts, allowing a more detailed description of repair intermediates(Raschle et al., 2008). When a 6 kb plasmid carrying a single, site-specific ICL is incubated in egg extract, a significant fraction of the lesions is repaired in a replication-dependent manner. Repair begins when two replication forks converge on the ICL (Figure 1Ci and 1Cii). The 3′ ends of both converged leading strands initially stall 20–40 nucleotides from the ICL due to steric hindrance by the MCM2-7 helicase, which translocates along the leading strand template ahead of the polymerase (Fu et al., 2011). Upon collision with the ICL, the 5′ ends of lagging strands are located 50–300 nucleotides from the lesion, and they subsequently undergo resection. Concurrent with MCM2-7 release from the ICL, one leading strand advances to within 1 nucleotide of the ICL (Figure 1Ciii; “Approach”). After Approach, the opposing parental strand is incised on either side of the ICL, leading to unhooking of the ICL and formation of a DSB (Figure 1Civ). In the absence of ID-Ub, incisions are severely impaired and the leading strand remains stuck 1 nucleotide from the lesion (Knipscheer et al., 2009). After incisions, the lesion is bypassed in two steps. First, a nucleotide is inserted across from the damaged base by an unknown translesion DNA polymerase (Figure 1Civ, red arrowhead). The resulting abnormal primer template is then extended by DNA polymerase ζ (Figure 1Civ, blue arrow). Finally, the DSB is repaired via Rad51 dependent strand exchange with the intact sister chromatid (Long et al., 2011). In the Xenopus system, a vestige of the ICL remains attached to one parental strand. This observation implies either that the incisions occur very close together, or that the oligonucleotide between the incisions is processed by a nuclease. Either way, the final adduct is not removed in egg extracts.
Certain features of the dual fork mechanism likely apply to the single fork model (Figure 1B). Thus, when a single fork strikes an ICL, the leading strand probably also stalls 20 nucleotides from the lesion due to the MCM2-7 footprint (Figure 1Bi), and incisions are likely to require eviction of the MCM2-7 complex (Figure 1Bii). Whether incisions occur in the leading (Figure 1Aii) or lagging strand template (Figure 1Biii) of a single stalled fork remains unclear. However we favor the latter scenario because this allows extension past the lesion using the existing replication apparatus (Figure 1Biii). In addition, as discussed below, it is not obvious which endonuclease would cleave the leading strand template.
Merits of the single versus dual fork collision models
Which of the two collision models applies in cells is subject to lively debate (Kratz et al., 2010; MacKay et al., 2010; Raschle et al., 2008). The single fork mechanism has at least one potential disadvantage. Unlike bacteria (Heller and Marians, 2006) and budding yeast (Anand et al., 2013), metazoans do not contain a known pathway for replicative DNA helicase loading in S phase. Therefore, the restored fork is unlikely to resume synthesis and would have to await the arrival of a converging fork, as seen after HU-induced replication fork collapse (Petermann et al., 2010). Such a helicase-deficient, stationary fork might be unstable and cause genomic rearrangements. The major criticism of the dual fork model is that it was observed in the context of a small plasmid where two forks inevitably converge on the ICL. In vivo, where origins are spaced 100 kb apart and forks move at a rate of 1.5 kb/minute (Duderstadt et al., 2013), most of the time one fork is expected to strike an ICL well before a converging fork arrives. Nevertheless, two forks will converge on an ICL when the lesion is located midway between two origins that fire contemporaneously. Moreover, given that fork processing requires MCM2-7 dissociation, leading strand approach to the ICL, and nuclease recruitment, which together takes at least 30–40 minutes (Fu et al., 2011; Klein Douwel et al., submitted; Raschle et al., 2008), there will exist a significant temporal window during which a second fork can arrive before incisions take place. Thus, based on first principles, it seems likely that both single and dual fork collisions will occur at significant frequencies, necessitating pathways to resolve both structures.
The traverse model
The single versus dual fork debate recently took an unexpected turn when Seidman and colleagues investigated the encounter of DNA replication forks with fluorescently tagged ICLs in vivo using DNA combing (Huang et al., 2013). The work showed that single and dual fork collisions each comprise 15–20% of ICL encounters. Surprisingly, in ~60% of cases, DNA replication forks bypass or “traverse” ICLs without unhooking them (Figure 1Dii). An earlier study inferred that forks generally stall at ICLs, but in this case, the location of ICLs relative to DNA replication tracts was not determined (Vare et al., 2012). Traverse requires the translocase activity of FANCM but not other FANC proteins and is very rapid, taking only a few minutes. When FANCM is defective, the frequency of traverse events goes down while single fork collisions go up, indicating that single fork collisions are followed by traverse. It is unclear whether MCM2-7 jumps over the ICL during traverse, a new MCM2-7 molecule is recruited on the other side of the ICL (unlikely given the absence of known S phase MCM2-7 loading mechanisms), or a different DNA helicase loads distal to the ICL. Interestingly, FANCM has been shown to recruit RPA to an ICL in duplex DNA, suggesting it helps to melt DNA in the vicinity of these structures (Huang et al., 2010), which might facilitate traverse. In summary, it appears that the most common substrate for incisions during ICL repair is an X-shaped DNA molecule, which is generated by some combination of fork traverse and fork convergence events.
Can a single fork trigger ICL repair?
The apparent preponderance of X-shaped structures at ICLs in vivo raises the important question of whether a single fork that has not traversed an ICL can trigger repair. Replication of a psoralen ICL in Xenopus egg extracts suggested that sometimes, a single fork can trigger incisions, but it was unclear whether the observed incisions led to a productive repair outcome (Le Breton et al., 2011). In mammalian cells, repair of an ICL flanked by a replication roadblock suggested that a single fork can trigger repair (Nakanishi et al., 2011). However, the possibility of traverse was not considered and the efficiency of the roadblock was not determined, leaving open the possibility that an X-shaped structure was the substrate for repair. In the future, it will be critical to directly compare the efficiency of processing and repair of single stalled forks, converged forks, and traversed forks.
What DNA structures are formed at ICL-stalled forks?
A detailed knowledge of the DNA structures created after forks encounter an ICL is critical to understand the mechanism of incisions. During repair of ICLs in egg extracts, incisions occur only after one leading strand has advanced to within 1 nucleotide of the ICL (−1 position)(Raschle et al., 2008). Thus, on the 5′ side of the ICL (left side in Figure 1C–D), nucleases act on a replication fork whose leading strand abuts the lesion. The structure on the 3′ (right) side is less clear. After MCM2-7 eviction from converged forks (Fu et al., 2011), the ssDNA between the ICL and the 3′ end of the leading strand (located 20–40 nucleotides from the ICL) might not re-anneal due to RPA binding (Figure 2C, left inset). Since RPA’s preferred binding mode involves a 30 nucleotide footprint (Wold, 1997), it is likely to preferentially occupy the lagging strand template where more ssDNA is available. In this case, incisions by a 3′ flap endonuclease would likely occur close to the ICL (Figure 2C, left inset, red arrowhead). Alternatively, after MCM2-7 departure, the parental strands might re-anneal up to the 3′ end of the leading strand (Figure 2C, right inset). In this case, incisions would likely occur near the dsDNA-ssDNA junction (right inset, pink arrowhead), further away from the ICL, but as discussed below, this is not an ideal substrate for 3′ flap endonucleases. The structure generated during traverse should be similar to the structure that results when two forks converge. The only possible difference is that if DNA synthesis re-initiates just beyond the ICL, the 5′ end of the new leading strand after traverse will be closer to the lesion than at a converged fork (compare green strands in Figures 1C and 1D). This would affect which endonucleases are able to cut the structure (see below). For single fork collisions without traverse, the 3′ side of the ICL will be double-stranded (Figure 1A and B), and therefore not recognized by structure specific endonucleases, unless the ICL is distorting, in which case there could be a limited amount of single-stranded DNA 3′ to the ICL. Most likely, a diversity of structures exists at ICL-stalled forks, which might help explain the many endonucleases that have been implicated in ICL repair.
Figure 2. Model incision substrates for endonucleases.

(A–D) Preferred substrates, as determined in biochemical assays, for the indicated endonucleases. The arrowheads indicate the location of incision. Each substrate is shown in two orientations related by a 180 degree rotation. The left column shows where the nuclease would cut on the 5′ side of a converged fork; the right column shows incisions on the 3′ side. In each column, the nucleases that are likely to cut the ICL-associated X-shaped structure shown at the top are highlighted by a grey box. (E) Classic nucleotide excision repair (NER) substrate and locations of XPF and XPG incisions. (F) Location of incisions when XPF is presented with an ICL-containing fork-shaped structure. (G) Action of SNM1A on ICL-containing DNA.
Endonucleases
Six different nucleases (XPF-ERCC1, MUS81-EME1, SLX1-SLX4, FAN1, SNM1A, and SNM1B) have been implicated in ICL repair. For each nuclease, we discuss the evidence linking it to the incision step, its specificity, and how it might act on the structures shown in Figure 1.
XPF-ERCC1
The best candidate for an incision endonuclease is XPF-ERCC1, which is best known for its role in nucleotide excision repair (NER)(Ciccia et al., 2008). XPF contains an excision repair cross-complementation group 4 (ERCC4) endonuclease domain and a Helix-hairpin-Helix (HhH) DNA binding domain. ERCC1 contains the same domains but the endonuclease domain has acquired mutations that render it catalytically inactive. The preferred substrate of XPF-ERCC1 is a “splayed arm” structure, which it cuts at the base of the 3′ arm, a few nucleotides internal to the DNA duplex (Figure 2A). XPF-ERCC1 also cuts the 3′ flap structure depicted in Figure 2B, but less efficiently than splayed arms (de Laat et al., 1998a), and it does not cut the 3′ arm of 5′ flap structures (Figure 2C) (Rodriguez et al., 1996). During NER, XPF-ERCC1 helps remove bulky lesions by cutting on the 5′ side of an open bubble structure surrounding the lesion (Figure 2E). XPF-ERCC1 is unique among NER proteins in conferring cellular resistance to ICLs (De Silva et al., 2000; Kuraoka et al., 2000; Niedernhofer et al., 2004). Mutations in ERCC1 that disrupt its interaction with XPA, another NER factor, prevent NER but not ICL repair, showing that the two functions of this nuclease can be uncoupled (Orelli et al., 2010). Mutations in mouse Ercc1 cause a spectrum of phenotypes reminiscent of Fanconi anemia (Hsia et al., 2003; McWhir et al., 1993; Prasher et al., 2005), and recently, XPF mutations were discovered in Fanconi patients (Bogliolo et al., 2013; Kashiyama et al., 2013). Together, the data strongly suggest that XPF-ERCC1 operates in the FA-dependent ICL repair pathway.
The specific function of XPF in ICL repair has been slow to emerge. In the absence of XPF or ERCC1, ICL-induced DSBs accumulate and persist, leading to the conclusion that XPF is not required for the first incision of ICL unhooking (De Silva et al., 2000; Niedernhofer et al., 2004). In contrast, recent results from the Knipscheer laboratory show that in XPF-depleted Xenopus egg extracts, forks that have converged on an ICL are not incised (Klein Dowel et al., submitted). These apparently contradictory results can be reconciled by the proposal that XPF is responsible for initial ICL incision in both systems, but that in cells, forks persisting in the absence of XPF are aberrantly cleaved by MUS81 (Wang et al., 2011).
Given its preference for the 3′ arm of a splayed arm structure (Figure 2A), XPF-ERCC1 probably cuts on the 3′ side of ICL-associated X-shaped structures (red arrowhead in Figures 1C). XPF’s cleavage of 3′ splayed arms is dramatically enhanced by RPA binding to the 5′ arm (de Laat et al., 1998b), as depicted in Figure 1C (left inset). However, when the leading strand is present at the junction as depicted in Figure 1C (right inset), XPF may not cut efficiently (Rodriguez et al., 1996), requiring another solution (see SNM1A section below). In Xenopus egg extracts, neither of the two incisions required for unhooking of converged forks occur in the absence XPF-ERCC1, or when XPF-depleted extracts are supplemented with catalytically inactive XPF-ERCC1 (Klein Dowel et al., submitted). One explanation for this observation is that the 5′ incision cannot take place without the 3′ incision. A precedent for coupling between dual incisions is observed in NER, where incision by XPG 3′ to a lesion requires prior incision by XPF-ERCC1 on the 5′ side (Staresincic et al., 2009) (Figure 2E). Another possibility is that XPF performs both incisions. Thus, in the context of a splayed arm structure containing an ICL, purified XPF-ERCC1 cuts on the 5′ and 3′ sides of the lesion (Figure 2F; (Fisher et al., 2008; Kuraoka et al., 2000)). It is presently unclear how this activity would be affected by the presence of a fork on the other side of the ICL. In summary, although XPF-ERCC1 is almost certainly the primary 3′ incision endonuclease, the precise mechanism and circumstances of its action remain to be elucidated.
MUS81-EME1
Like XPF, the MUS81-EME1 heterodimer belongs to the XPF/MUS81 nuclease family, with MUS81 contributing the catalytic active ERCC4 domain (Ciccia et al., 2008). MUS81-EME1 greatly prefers 3′ flap structures that contain a 5′ end within 4 nucleotides of the flap junction (Figure 2B) (Bastin-Shanower et al., 2003; Ciccia et al., 2003; Ciccia et al., 2008). Mutations in MUS81-EME1 render cells sensitive to ICLs, but not as sensitive as mutations in XPF-ERCC1 (Crossan et al., 2011; Wang et al., 2011). This suggests that MUS81-EME1 plays a secondary role in ICL repair. While early experiments in mammalian cells suggested that MUS81 is required to generate ICL-induced DSBs (Hanada et al., 2006), implying a role in incisions, more recent evidence indicates that MUS81 generates DSBs when normal fork processing is disrupted (Wang et al., 2011). Consistent with the latter view, depletion of MUS81-EME1 had no effect on the incision of converged forks in Xenopus egg extracts (Klein Dowel et al., submitted). We propose that MUS81 only acts on a subset of the intermediates generated when forks encounter ICLs. For example, if leading strand synthesis resumes immediately downstream of the ICL during fork traverse, a 5′ end is located near the flap junction (Figure 1D; green arrow). The 5′ end disfavors cutting by XPF-ERCC1 (de Laat et al., 1998a; Rodriguez et al., 1996), but creates an ideal substrate for MUS81-EME1. In summary, we propose that the primary role of MUS81-EME1 in ICL repair is to cut on the 3′ side of X-shaped structures in which a 5′ end abuts the ssDNA-dsDNA junction, possibly during a subset of traverse events.
Importantly, the structure generated during a single-fork collision lacks a 5′ end near the junction and therefore should not be cleaved efficiently by MUS81 (Figure 1A–B). It may also not be cut efficiently by XPF, which is inhibited by the presence of a 3′ end at the junction (Rodriguez et al., 1996). Thus, no enzyme implicated in ICL repair is well suited to incise the leading strand template of a single stalled fork (Figure 1A, black arrowhead). These considerations suggest that single forks should be cleaved on the lagging strand template by a 5′ flap endonuclease (Figure 1B, blue arrowhead).
SLX1
SLX1 is a structure-specific endonuclease containing a UvrC-intron-endonuclease domain (URI) and a PHD-type zinc finger domain (Aravind and Koonin, 2000). SLX1’s activity is dramatically stimulated through interaction with a non-catalytic subunit, SLX4. Homozygous deletion of the SLX1 gene in mice causes sensitivity to ICLs similar to that of MUS81, and rescue of this sensitivity requires the catalytic activity of SLX1 (Castor et al., 2013). SLX1-SLX4 is a rather promiscuous endonuclease, in that it cleaves splayed arms, 5′ flaps, 3′ flaps, and holliday junctions (Fricke and Brill, 2003; Wyatt et al., 2013). However, its preferred substrate is a 5′ flap, which it cuts at the ssDNA-dsDNA junction (Figure 2C). In the context of ICL repair, a 5′ flap-like structure is generated on the 5′ side of the ICL when the leading strand of a stalled fork has been extended to the −1 position (Figure 1A–D). Based on this and considerations discussed below, we propose that SLX1 is the primary nuclease that cleaves on the 5′ side of the ICL.
Scaffolding by SLX4
Accumulating evidence indicates that SLX4 serves as a master scaffold for incisions. Deletion of SLX4, the binding partner of SLX1, causes much greater sensitivity to ICLs than deletion of SLX1 or MUS81 (Castor et al., 2013), and it is required for incisions in Xenopus egg extracts (Klein Douwel et al., submitted). Underscoring its importance for ICL repair, SLX4 is a Fanconi gene (Kim et al., 2011; Stoepker et al., 2011). Strikingly, SLX4 co-precipitates with XPF-ERCC1, SLX1, and MUS81-EME1 (Andersen et al., 2009; Fekairi et al., 2009; Munoz et al., 2009; Svendsen et al., 2009). SLX4 binds SLX1 via a helix-turn-helix motif (also referred as SBD: SLX1 binding domain), MUS81 via a SAP motif, and XPF via an MLR motif (Figure 3). The binding of SLX4 to XPF-ERCC1 and SLX1 is critical for the action of these nucleases in ICL repair, probably by recruiting them to sites of damage(Castor et al., 2013; Crossan et al., 2011; Kim et al., 2013; Stoepker et al., 2011). In contrast, the interaction of MUS81 with SLX4 is dispensable for ICL repair (Castor et al., 2013). Together, the data indicate that XPF-ERCC1-SLX4-SLX1 represents a core “XESS” complex that incises ICL-associated X-shaped structures (Figure 3). MUS81 is employed for special scenarios, i.e. when the 3′ side of the ICL contains a true 3′ flap (Figure 1D). Importantly, SLX4 appears to be more than just a recruitment platform, since it dramatically enhances XPF nuclease activity towards splayed arm and ICL-containing structures (Hodskinson et al., submitted). While SLX1 is probably the primary 5′ endonuclease for most situations, the mild ICL-sensitivity of SLX1 mutations suggests its function can be replaced by another endonuclease (next section).
Figure 3. Model for incisions.
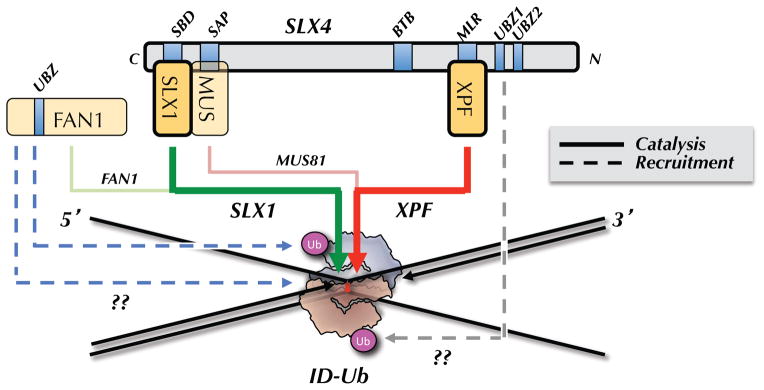
ID-Ub binds directly to an ICL in such a manner as to sequester the bottom parental strand while exposing the top parental strand. ID-Ub also recruits the SLX4-SLX1-MUS81-XPF-ERCC1 complex to the ICL, possibly via a direct interaction between the UBZ domains of SLX4 and the ubiquitin of the ID complex (dashed, grey arrow). XPF is the primary enzyme that incises 3′ to the ICL (thick red arrow), while SLX1 promotes the 5′ incision (green arrow). MUS81 performs a specialty role, cutting on the 3′ side in cases where the 5′ end of the leading strand on the same side abuts the ICL (pink arrow; shown in Figure 1D and 2B). FAN1 is recruited to ICLs via two independent mechanisms, one of which requires ID-Ub (dashed blue arrows). FAN1 cuts on the 5′ side of the ICL (light green arrow), and/or it might function downstream of incisions, during the HR step (not shown).
FAN1
Fanconi Associated Nuclease 1 (FAN1) contains a UBZ4-type ubiquitin binding domain, a SAP DNA binding domain, and a PD-D/E(X)K nuclease motif, placing it in the same nuclease superfamily as XPF and MUS81 (Kratz et al., 2010). Like SLX1, FAN1 prefers 5′ flap structures. However, unlike SLX1, which cleaves at the ssDNA-dsDNA junction, FAN1 cleaves four nucleotides 3′ to the branch point (Figure 2D)(Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010). FAN1 also exhibits robust 5′→3′ exonuclease activity of 5′ recessed DNA ends, nicks, or gaps. FAN1 gene knockdown or knock-out selectively sensitizes cells to ICL-inducing agents, although not to the same extent as disruption of the FA pathway (Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010; Trujillo et al., 2012; Yoshikiyo et al., 2010; Zhou et al., 2012). FAN1 and FA pathway mutations are not epistatic, suggesting distinct roles in ICL repair (Yoshikiyo et al., 2010; Zhou et al., 2012). Indeed, FANC mutations cause much greater damage-induced chromosomal instability than FAN1 mutations (Trujillo et al., 2012; Yoshikiyo et al., 2010; Zhou et al., 2000). Consistent with these observations, FAN1 and FANC mutations cause distinct clinical phenotypes, the former being associated with karyomegalic interstitial nephritis, a form of chronic kidney disease instead of FA (Trujillo et al., 2012; Zhou et al., 2012). Together, the data indicate that FAN1 participate in ICL repair, but via a distinct and secondary role relative to the FA pathway.
The function of FAN1 in ICL repair remains enigmatic. ICL-induced γH2AX foci still form in the absence of FAN1, leading to the proposal that FAN1 might function in the HR step of ICL repair, downstream of incisions (MacKay et al., 2010). However, γH2AX foci are caused by DNA damage other than dsDNA breaks (Petermann et al., 2010), leaving open the possibility that FAN1 is required for incisions. When FAN1 is depleted from Xenopus egg extracts, there is no detectable repair or incision defect (Klein Dowel et al., submitted), but this might be due to redundancy with other nucleases such as SLX1. In the future, SLX1-FAN1 redundancy should be examined using biochemical and genetic ICL repair assays. A reasonable working hypothesis is that FAN1 plays a secondary role in cleaving the 5′ side of ICL-associated X-shaped structures (Figure 3; light green line).
SNM1 family nucleases
SNM1 nucleases (SNM1A, SNM1B, and SNM1C) belong to the β-CASP subfamily of metallo-beta-lactamases, which are DNA processing enzymes (Cattell et al., 2010). The single SNM1 family member in yeast, Pso2, exhibits singular sensitivity to ICLs but not other damaging agents. In vertebrates, SNM1A and SNM1B mutants are selectively sensitive to ICLs, with SNM1A showing the greatest sensitivity (Cattell et al., 2010; Ishiai et al., 2004). In contrast, SNM1C/Artemis mutants are not sensitive to ICLs but to ionizing radiation, consistent with this protein’s role in non-homologous end-joining. While SNM1A and B are reported to exhibit non-epistasis with homologous recombination, translesion DNA synthesis, and FANC genes in chicken cells (Ishiai et al., 2004), there are hints of an epistatic relationship with XPF in mammalian cells (Wang et al., 2011). Thus, although SNM1A and B appear to participate in ICL repair, whether they function in the FA-dependent pathway remains uncertain.
Given its greater sensitivity to ICLs, functional complementation of yeast pso2 mutants (Hazrati et al., 2007), and more robust nuclease activity (Sengerova et al., 2012), we will focus on SNM1A. Unlike the endonucleases discussed above, SNM1A functions as a 5′-> 3′ exonuclease with no obvious role in incisions (Hazrati et al., 2007; Hejna et al., 2007). Consistent with this, pso2 mutants in yeast still form ICL-induced dsDNA breaks(Grossmann et al., 2000; Magana-Schwencke et al., 1982). Interestingly, SNM1A can use its 5′ → 3′ exonuclease activity to digest one DNA strand a few nucleotides past an ICL (Figure 2G; (Wang et al., 2011)). This suggests that as long as an incision is made on the 5′ side of an ICL (e.g. by SLX1 or FAN1), SNM1A might be able to complete the unhooking reaction without the need for 3′ endonucleolytic cleavage by XPF or MUS81. Exonucleolytic degradation by SNM1A therefore might replace the 3′ incision in cases where the 3′ side of the ICL is not ideal for incision by XPF or MUS81 (as in Figure 1C, right insert). Another possible role for SNM1A is that after 5′ and 3′ incisions have occurred, SNM1A reduces the oligonucleotide between the incision points to a mono-adduct that can be bypassed by translesion DNA polymerases.
Regulation of ICL processing
How does the FA pathway regulate incisions (Knipscheer et al., 2009)? Upon exposure of cells to ICLs, the ID-Ub complex forms DNA damage foci that overlap with several DNA repair factors, including Rad51 and BRCA1, suggesting it binds to sites of damage (Taniguchi et al., 2002). Consistent with this idea, during repair of an ICL in egg extracts, ID-Ub localizes to the ICL immediately before incisions take place, as measured by chromatin immunoprecipitation (Klein Douwel et al., submitted). Moreover, FANCI and FANCD2 bind preferentially to a variety of branched DNA structures(Joo et al., 2011; Longerich et al., 2009; Park et al., 2005; Yuan et al., 2009), and the crystal structure of FANCI with DNA suggests that the ID complex should be able to accommodate an ICL-associated X-shaped structure (Figure 3 (Joo et al., 2011)). Thus, ID-Ub likely binds directly to ICLs, allowing it to exert local control over the process of incisions (Figure 3).
Several connections between the ID complex and the incision machinery have now been observed. First, the recruitment of FAN1 to damage foci in cells requires ubiquitylated FANCD2, as well as the UBZ domain of FAN1 (Kratz et al., 2010; Liu et al., 2010; MacKay et al., 2010; Smogorzewska et al., 2010). On the other hand, FAN1 and the FANC genes are not epistatic (Yoshikiyo et al., 2010; Zhou et al., 2012), and a FAN1 mutant disrupting the UBZ domain is functional for ICL resistance (Zhou et al., 2012). These data suggest that although FAN1 can be recruited to ICLs via ID-Ub, this is not essential in some cases, perhaps due to another, independent recruitment pathway (Figure 3, dashed blue arrows). The connection between ID-Ub and XPF is more compelling. Most importantly, in Xenopus egg extracts, the binding of SLX4 and XPF to ICLs during repair requires ubiquitylated FANCD2 (Klein Douwel et al., submitted). This observation is consistent with the earlier finding that SLX4 damage localization in cells depends on the SLX4 UBZ motif and ubiquitylated FANCD2 ((Yamamoto et al., 2011), but see (Kim et al., 2013)). Although the molecular details remain unclear, the data strongly suggest that ID-Ub recruits the XESS complex to sites of damage.
Why has a system as elaborate as the FA pathway evolved to regulate incisions? We propose this regulation arose to deal with the complexity inherent in ICL-associated X-shaped structures. In principle, these structures could be cleaved by structure-specific endonucleases in any one of the four arms that meet at the ICL. While several combinations of cuts are possible, the only one that yields a productive outcome involves incising the same parental strand on either side of the lesion (Figure 3, red and green arrows), or incising on the 5′ side of the ICL and using an exonuclease to degrade past the ICL. We propose that ID-Ub has at least two functions. First, it recruits the XESS complex to the lesion to promote 5′ and 3′ incisions of one parental strand. Second, it embraces the ICL in such a manner as to suppress incisions on the other parental strand. Ultimately, to understand how ID-Ub controls incisions, the reaction will have to be reconstituted with purified components and a crystal structure of the ubiquitylated ID complex with an appropriate X-shaped structure will be needed. However, already one can envision that ID-Ub sequesters the lower strand of the X-shaped structure to protect it from cleavage (Figure 3). It is important to note that the X-shaped DNA structure is not symmetrical, since a 3′ end abuts the ICL on just one side. This would allow the ID complex to interact differentially with the two parental strands.
Outlook
It is now clear that a signature event in ICL repair is the ID-Ub-dependent incision of stalled replication forks. The discovery of the XESS complex lays the foundation to understand how these incisions are carried out and regulated. Going forward, it will be critical to re-examine the specificities of all the relevant endonucleases on DNA substrates that better mimic the various structures predicted to exist at ICL-stalled DNA replication forks (Figure 1A–D), including with distorting and non-distorting ICLs. This should help clarify which endonuclease is employed under which circumstances. A more ambitious goal is to reconstitute ID-Ub-dependent incisions with defined components so that we may precisely define the function of the Fanconi anemia pathway. These biochemical approaches will have to be complemented with incision assays under physiological conditions. Finally, it will be critical to understand why deficiencies in two endonucleases (FAN1 versus XPF-ERCC1-SLX4) that appear to resolve the same type of DNA damage cause such different clinical phenotypes. As we address these important issues, nature will undoubtedly serve up more surprises.
Acknowledgments
We thank KJ Patel and Puck Knipscheer for communicating unpublished results. The work on ICL repair in the Walter laboratory is funded by NIH grant HL098316 and a grant from the Fanconi anemia research foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M. DNA replication is required To elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol Cell Biol. 2000;20:8283–8289. doi: 10.1128/mcb.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpi AF, Pace PE, Babu MM, Patel KJ. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Molecular cell. 2008;32:767–777. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Anand RP, Lovett ST, Haber JE. Break-Induced DNA Replication. Cold Spring Harb Perspect Biol. 2013 doi: 10.1101/cshperspect.a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen SL, Bergstralh DT, Kohl KP, LaRocque JR, Moore CB, Sekelsky J. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Molecular cell. 2009;35:128–135. doi: 10.1016/j.molcel.2009.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. SAP - a putative DNA-binding motif involved in chromosomal organization. Trends Biochem Sci. 2000;25:112–114. doi: 10.1016/s0968-0004(99)01537-6. [DOI] [PubMed] [Google Scholar]
- Bastin-Shanower SA, Fricke WM, Mullen JR, Brill SJ. The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Molecular and cellular biology. 2003;23:3487–3496. doi: 10.1128/MCB.23.10.3487-3496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillon J, Ramirez MJ, Pujol R, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92:800–806. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castor D, Nair N, Declais AC, Lachaud C, Toth R, Macartney TJ, Lilley DM, Arthur JS, Rouse J. Cooperative Control of Holliday Junction Resolution and DNA Repair by the SLX1 and MUS81-EME1 Nucleases. Molecular cell. 2013;52:221–233. doi: 10.1016/j.molcel.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattell E, Sengerova B, McHugh PJ. The SNM1/Pso2 family of ICL repair nucleases: from yeast to man. Environmental and molecular mutagenesis. 2010;51:635–645. doi: 10.1002/em.20556. [DOI] [PubMed] [Google Scholar]
- Ciccia A, Constantinou A, West SC. Identification and characterization of the human mus81-eme1 endonuclease. The Journal of biological chemistry. 2003;278:25172–25178. doi: 10.1074/jbc.M302882200. [DOI] [PubMed] [Google Scholar]
- Ciccia A, McDonald N, West SC. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem. 2008;77:259–287. doi: 10.1146/annurev.biochem.77.070306.102408. [DOI] [PubMed] [Google Scholar]
- Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PH, McIntyre RE, Gallagher F, Kettunen MI, Lewis DY, Brindle K, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet. 2011;43:147–152. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Jaspers NG, Hoeijmakers JH. DNA structural elements required for ERCC1-XPF endonuclease activity. The Journal of biological chemistry. 1998a;273:7835–7842. doi: 10.1074/jbc.273.14.7835. [DOI] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Sugasawa K, Weterings E, Jaspers NG, Hoeijmakers JH. DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes & development. 1998b;12:2598–2609. doi: 10.1101/gad.12.16.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duderstadt KE, Reyes-Lamothe R, van Oijen AM, Sherratt DJ. Replication-Fork Dynamics. Cold Spring Harb Perspect Biol. 2013 doi: 10.1101/cshperspect.a010157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Dong MQ, Ruse C, Yates JR, 3rd, Russell P, et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009;138:78–89. doi: 10.1016/j.cell.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher LA, Bessho M, Bessho T. Processing of a psoralen DNA interstrand cross-link by XPF-ERCC1 complex in vitro. The Journal of biological chemistry. 2008;283:1275–1281. doi: 10.1074/jbc.M708072200. [DOI] [PubMed] [Google Scholar]
- Fricke WM, Brill SJ. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, Walter JC. Selective Bypass of a Lagging Strand Roadblock by the Eukaryotic Replicative DNA Helicase. Cell. 2011;146:931–941. doi: 10.1016/j.cell.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Grossmann KF, Ward AM, Moses RE. Saccharomyces cerevisiae lacking Snm1, Rev3 or Rad51 have a normal S-phase but arrest permanently in G2 after cisplatin treatment. Mutat Res. 2000;461:1–13. doi: 10.1016/s0921-8777(00)00035-5. [DOI] [PubMed] [Google Scholar]
- Hanada K, Budzowska M, Modesti M, Maas A, Wyman C, Essers J, Kanaar R. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. Embo J. 2006;25:4921–4932. doi: 10.1038/sj.emboj.7601344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazrati A, Ramis-Castelltort M, Sarkar S, Barber LJ, Schofield CJ, Hartley JA, McHugh PJ. Human SNM1A suppresses the DNA repair defects of yeast pso2 mutants. DNA Repair (Amst) 2007 doi: 10.1016/j.dnarep.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Hejna J, Philip S, Ott J, Faulkner C, Moses R. The hSNM1 protein is a DNA 5′-exonuclease. Nucleic acids research. 2007;35:6115–6123. doi: 10.1093/nar/gkm530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat Rev Mol Cell Biol. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- Hodskinson MR, Silhan J, Crossan GP, Garaycoechea JI, Mukherjee S, Scharer OD, Patel KJ. Mouse Slx4 is a tumour suppressor that stimulates the activity of the nuclease Xpf-Ercc1 in DNA crosslink repair. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia KT, Millar MR, King S, Selfridge J, Redhead NJ, Melton DW, Saunders PT. DNA repair gene Ercc1 is essential for normal spermatogenesis and oogenesis and for functional integrity of germ cell DNA in the mouse. Development. 2003;130:369–378. doi: 10.1242/dev.00221. [DOI] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM. The DNA Translocase FANCM/MHF Promotes Replication Traverse of DNA Interstrand Crosslinks. Molecular cell. 2013 doi: 10.1016/j.molcel.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Kim JM, Shiotani B, Yang K, Zou L, D’Andrea AD. The FANCM/FAAP24 complex is required for the DNA interstrand crosslink-induced checkpoint response. Molecular cell. 2010;39:259–268. doi: 10.1016/j.molcel.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiai M, Kimura M, Namikoshi K, Yamazoe M, Yamamoto K, Arakawa H, Agematsu K, Matsushita N, Takeda S, Buerstedde JM, et al. DNA cross-link repair protein SNM1A interacts with PIAS1 in nuclear focus formation. Molecular and cellular biology. 2004;24:10733–10741. doi: 10.1128/MCB.24.24.10733-10741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo W, Xu G, Persky NS, Smogorzewska A, Rudge DG, Buzovetsky O, Elledge SJ, Pavletich NP. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. 2011;333:312–316. doi: 10.1126/science.1205805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet. 2013;92:807–819. doi: 10.1016/j.ajhg.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, Smogorzewska A. Mutations of the SLX4 gene in Fanconi anemia. Nat Genet. 2011;43:142–146. doi: 10.1038/ng.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Spitz GS, Veturi U, Lach FP, Auerbach AD, Smogorzewska A. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood. 2013;121:54–63. doi: 10.1182/blood-2012-07-441212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein Douwel D, Boonen RACM, DTL, Szypowska AA, Raschle M, Walter JC, Knipscheer P. XPF-ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. (submitted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–1701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz K, Schopf B, Kaden S, Sendoel A, Eberhard R, Lademann C, Cannavo E, Sartori AA, Hengartner MO, Jiricny J. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell. 2010;142:77–88. doi: 10.1016/j.cell.2010.06.022. [DOI] [PubMed] [Google Scholar]
- Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. The Journal of biological chemistry. 2000;275:26632–26636. doi: 10.1074/jbc.C000337200. [DOI] [PubMed] [Google Scholar]
- Le Breton C, Hennion M, Arimondo PB, Hyrien O. Replication-fork stalling and processing at a single psoralen interstrand crosslink in Xenopus egg extracts. PLoS One. 2011;6:e18554. doi: 10.1371/journal.pone.0018554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ghosal G, Yuan J, Chen J, Huang J. FAN1 Acts with FANCI-FANCD2 to Promote DNA Interstrand Cross-Link Repair. Science. 2010 doi: 10.1126/science.1192656. [DOI] [PubMed] [Google Scholar]
- Long DT, Raschle M, Joukov V, Walter JC. Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science. 2011;333:84–87. doi: 10.1126/science.1204258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longerich S, San Filippo J, Liu D, Sung P. FANCI binds branched DNA and is monoubiquitinated by UBE2T-FANCL. J Biol Chem. 2009;284:23182–23186. doi: 10.1074/jbc.C109.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay C, Declais AC, Lundin C, Agostinho A, Deans AJ, MacArtney TJ, Hofmann K, Gartner A, West SC, Helleday T, et al. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell. 2010;142:65–76. doi: 10.1016/j.cell.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magana-Schwencke N, Henriques JA, Chanet R, Moustacchi E. The fate of 8-methoxypsoralen photoinduced crosslinks in nuclear and mitochondrial yeast DNA: comparison of wild-type and repair-deficient strains. Proc Natl Acad Sci U S A. 1982;79:1722–1726. doi: 10.1073/pnas.79.6.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhir J, Selfridge J, Harrison DJ, Squires S, Melton DW. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat Genet. 1993;5:217–224. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- Munoz IM, Hain K, Declais AC, Gardiner M, Toh GW, Sanchez-Pulido L, Heuckmann JM, Toth R, Macartney T, Eppink B, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Molecular cell. 2009;35:116–127. doi: 10.1016/j.molcel.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Cavallo F, Perrouault L, Giovannangeli C, Moynahan ME, Barchi M, Brunet E, Jasin M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nature structural & molecular biology. 2011;18:500–503. doi: 10.1038/nsmb.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Lalai AS, Hoeijmakers JH. Fanconi anemia (cross)linked to DNA repair. Cell. 2005;123:1191–1198. doi: 10.1016/j.cell.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelli B, McClendon TB, Tsodikov OV, Ellenberger T, Niedernhofer LJ, Scharer OD. The XPA-binding domain of ERCC1 is required for nucleotide excision repair but not other DNA repair pathways. The Journal of biological chemistry. 2010;285:3705–3712. doi: 10.1074/jbc.M109.067538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park WH, Margossian S, Horwitz AA, Simons AM, D’Andrea AD, Parvin JD. Direct DNA binding activity of the Fanconi anemia D2 protein. J Biol Chem. 2005;280:23593–23598. doi: 10.1074/jbc.M503730200. [DOI] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasher JM, Lalai AS, Heijmans-Antonissen C, Ploemacher RE, Hoeijmakers JH, Touw IP, Niedernhofer LJ. Reduced hematopoietic reserves in DNA interstrand crosslink repair-deficient Ercc1−/− mice. EMBO J. 2005;24:861–871. doi: 10.1038/sj.emboj.7600542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle M, Knipsheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008;134:969–980. doi: 10.1016/j.cell.2008.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez K, Wang Z, Friedberg EC, Tomkinson AE. Identification of functional domains within the RAD1.RAD10 repair and recombination endonuclease of Saccharomyces cerevisiae. The Journal of biological chemistry. 1996;271:20551–20558. doi: 10.1074/jbc.271.34.20551. [DOI] [PubMed] [Google Scholar]
- Rothfuss A, Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2004;24:123–134. doi: 10.1128/MCB.24.1.123-134.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengerova B, Allerston CK, Abu M, Lee SY, Hartley J, Kiakos K, Schofield CJ, Hartley JA, Gileadi O, McHugh PJ. Characterization of the human SNM1A and SNM1B/Apollo DNA repair exonucleases. J Biol Chem. 2012;287:26254–26267. doi: 10.1074/jbc.M112.367243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiacovo MP, et al. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell. 2010;39:36–47. doi: 10.1016/j.molcel.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, Giglia-Mari G, Clarkson SG, Vermeulen W, Scharer OD. Coordination of dual incision and repair synthesis in human nucleotide excision repair. The EMBO journal. 2009;28:1111–1120. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet. 2011;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- Svendsen JM, Smogorzewska A, Sowa ME, O’Connell BC, Gygi SP, Elledge SJ, Harper JW. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138:63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002;100:2414–2420. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- Trujillo JP, Mina LB, Pujol R, Bogliolo M, Andrieux J, Holder M, Schuster B, Schindler D, Surralles J. On the role of FAN1 in Fanconi anemia. Blood. 2012;120:86–89. doi: 10.1182/blood-2012-04-420604. [DOI] [PubMed] [Google Scholar]
- Vare D, Groth P, Carlsson R, Johansson F, Erixon K, Jenssen D. DNA interstrand crosslinks induce a potent replication block followed by formation and repair of double strand breaks in intact mammalian cells. DNA repair. 2012;11:976–985. doi: 10.1016/j.dnarep.2012.09.010. [DOI] [PubMed] [Google Scholar]
- Wang AT, Sengerova B, Cattell E, Inagawa T, Hartley JM, Kiakos K, Burgess-Brown NA, Swift LP, Enzlin JH, Schofield CJ, et al. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes & development. 2011;25:1859–1870. doi: 10.1101/gad.15699211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66:61–92. doi: 10.1146/annurev.biochem.66.1.61. [DOI] [PubMed] [Google Scholar]
- Wyatt HD, Sarbajna S, Matos J, West SC. Coordinated Actions of SLX1-SLX4 and MUS81-EME1 for Holliday Junction Resolution in Human Cells. Molecular cell. 2013;52:234–247. doi: 10.1016/j.molcel.2013.08.035. [DOI] [PubMed] [Google Scholar]
- Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K, Jiricny J, Takeda S, Hirota K. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc Natl Acad Sci U S A. 2011;108:6492–6496. doi: 10.1073/pnas.1018487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikiyo K, Kratz K, Hirota K, Nishihara K, Takata M, Kurumizaka H, Horimoto S, Takeda S, Jiricny J. KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc Natl Acad Sci U S A. 2010;107:21553–21557. doi: 10.1073/pnas.1011081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, El Hokayem J, Zhou W, Zhang Y. FANCI protein binds to DNA and interacts with FANCD2 to recognize branched structures. J Biol Chem. 2009;284:24443–24452. doi: 10.1074/jbc.M109.016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Chaturvedi P, Spring K, Scott SP, Johanson RA, Mishra R, Mattern MR, Winkler JD, Khanna KK. Caffeine abolishes the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-telangiectasia-mutated kinase activity. J Biol Chem. 2000;275:10342–10348. doi: 10.1074/jbc.275.14.10342. [DOI] [PubMed] [Google Scholar]
- Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, Diaz K, Lach FP, Bennett GR, Gee HY, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat Genet. 2012;44:910–915. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]