

Abstract
Aliphatic polycarbonates were discovered a long time ago, with their conventional applications mostly limited to low molecular weight oligomeric intermediates for copolymerization with other polymers. Recent developments in polymerization techniques have overcome the difficulty in preparing high molecular weight aliphatic polycarbonates. These in turn, along with new functional monomers, have enabled the preparation of a wide range of aliphatic polycarbonates with diverse chemical compositions and structures. This review summarizes the latest polymerization techniques for preparing well-defined functional aliphatic polycarbonates, as well as the new applications of those aliphatic polycarbonates, esecially in the biomedical field.
INTRODUCTION
Polycarbonates are polymers with backbones containing repeating carbonate (-O-C(O)-O-) linkages. Aliphatic polycarbonates (APCs) refer to the polycarbonates with no aromatic groups between carbonate linkages. Aliphatic polycarbonates were initially prepared in Wallace Carothers’ laboratory at DuPont around 1930s.1 With characteristic low melting points and high susceptibility to hydrolysis, which were considered inferior to the properties displayed by many other polymers (e.g. polyester, polyamide, poly(methyl methacrylate)) developed in that era for fiber applications, APCs were not pursued commercially.1,2 Unlike aromatic polycarbonates, which had garnered immediate commercial attention since the discovery of bisphenol A (BPA)-based polycarbonate in the 1950s and have been tremendously successful as consumer products,1–3, APCs not only remained largely unexplored commercially, but received little attention from the research field as well until the 1990s (Fig. 1). Although APCs have been proposed as alternative materials for films, packaging, and rigid plastics applications, its current industrial applications are still limited as low molecular weight polycarbonate polyols, macromonomers for the production of polyurethanes and other copolymers.
FIGURE 1.

The number of scientific publications related to APCs versus the time period searched from the database of Web of Science (Thomson Reuters) using various search terms.
The earlier study on APCs focused on improving the mechanical properties and thermal stability of the readily available poly(trimethylene carbonate) by blending it with polymers with complementary properties for applications such as engineering thermoplastics, albeit with limited commercial success. Increasing concerns over greenhouse gas pollution by carbon dioxide (CO2) have motivated the incorporation of CO2 into materials as a way to reduce greenhouse gas and as a means to alleviate the shortage in conventional petroleum fuel supplies. Polycarbonates have received significant renewed attention in this regard.4–8 Increasing demands for more versatile degradable biomaterials have also revived the interest in APCs for biomedical applications,9,10 for which the degradability, low glass transition temperatures and elasticity of APCs, used to be perceived as their major drawbacks, have turned into their competitive advantages over many other polymers in a U-turn.9,10
Indeed, there is a surge of literature on APCs in the past 2 decades (Fig. 2) resulting from new progress on polymerization techniques,7,11–16 functional monomer syntheses17–30 and the many new applications being explored.31–36 This review will first update the latest progress on APC polymerization techniques or new insights on traditional techniques applied to APC preparation, then discuss recent biomedical applications of APC-based hydrogels and drug delivery carriers. The information used in this review mainly comes from the available scientific publications in the past 15 years, along with a few patents.
FIGURE 2.
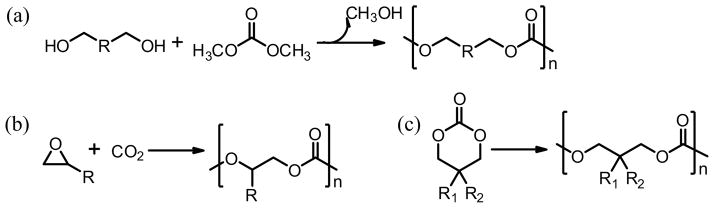
Common polymerization techniques for the preparation of APCs. (a) polycondensation between polyols with dimethyl carbonate; (2) copolymerization of carbon dioxide with epoxides; (3) ring-opening polymerization of cyclic carbonate monomers.
2. PROGRESS ON APC POLYMERIZATION TECHNIQUES
The sharp difference in mechanical and thermal properties between the most studied aromatic BPA-based polycarbonate and poly(trimethylene carbonate) underscores the importance of the chemical composition of the main chains. Molecular weight (Mn) and polydispersity (PDI) also has significant impact on APCs’ mechanical properties and degradation profiles. By altering these parameters, APCs with a wide range of properties have been reported using one the three major polymerization techniques: (1) polycondensation between aliphatic polyol with dialkyl carbonate (Fig. 2a); (b) copolymerization of carbon dioxide with epoxides (Fig. 2b); (3) ring-opening polymerization (ROP) of cyclic carbonate monomers (Fig. 2c). Significant progress has been made to improve each method over the last 2 decades.
2.1 Polycondenstation
APCs were initially prepared by the polycondensation method1 that involved toxic phosgene or its derivatives and aliphatic diols, and the resulting polymers usually suffered from poor controls over the molecular weight and were characterized with broad molecular weight distributions. Adopting the non-phosgene aromatic polycarbonate preparation technique, APCs were later prepared using dialkyl carbonates instead of phosgene.2,3,37 High molecular weight APCs were obtained via the polycondensation of dialkyl carbonates and aliphatic diols in the melt state, a two-step process involving initial condensation and subsequent chain growth enabled by transesterification between the –OH and –OC(O)R end-groups, using transesterification catalysts.37–39 The choice of catalysts, reaction temperature and the ratio of dialky carbonate to diol are all known to impact the polymerization outcome. Using a novel TiO2/SiO2- poly-(vinylpyrrolidone)-based catalyst (TSP-44), three APCs including Poly(butylene carbonate), poly(pentamethylene carbonate) and poly(hexamethylene carbonate) with high molecular weight (Mw ≥ 166,000 g mol−1) and narrow polydispersity (PDI ≤1.86) were synthesized in > 85% yield by this method.38 A recent study by Bun Yeoul Lee etal.39 revealed that forming the intermediate oligomers with the [–OCH3]/[–OH] ratio of ~1.0 in the first step is the prerequisite for obtaining high molecular weight APCs using this method.
The polycondensation techniques could also be catalyzed by enzyme catalysts.40,41 Enzymes provide distinct advantages over conventional catalysts for the preparation of functional polymers due to milder reaction conditions, high tolerance for functional groups, and higher selectivity that provides control over branching. However, enzyme-catalyzed polycondensations usually require high catalyst loading and long reaction time, and the obtained polycarbonates suffer from relative low molecular weight and broad polydispersity.
A unique advantage of the polycondensation method over the other two APC preparation techniques is that it enables straightforward preparation of APCs with different aliphatic linkages between the carbonates by simply using diols of different lengths during the polymerization.39,42 High-molecular-weight aliphatic copolymers (Mw 90,000–210,000) incorporating multiple aliphatic linkages has been prepared by the two-step melt polycondensation method using a mixture of 1,4-butanediol, 1,6-hexanediol and cyclohexane-1,4-dimethanol.39
2. 2. Alternating copolymerization of carbon dioxide and epoxy
Carbon dioxide (CO2) is one of the most abundant and renewable carbon resources, and the selective transformation of carbon dioxide and epoxides into degradable polycarbonates has been regarded as a promising green and sustainable route to polycarbonates.4,6,7,11,43–47 Since its discovery by Inoue and co-workers in 1969,11 this copolymerization method has become one of the most well-studied and innovative technologies for large-scale utilization of carbon dioxide in chemical synthesis. The search for highly efficient and selective catalysts for this process has been the focus. Besides the earlier zinc-containing heterogeneous catalysts utilized by Inoue et.al,11 a number of active homogeneous metal catalysts have also been reported, including the aluminum-porphyrin complex,48 zinc-phenoxide derivatives49 and β-diiminate-zinc catalysts,50–52 chromium-salen derivatives53 and cobalt salen catalysts.54–56 Studies of these well-defined transition metal coordination complexes as catalysts have revealed much of the mechanisms underlying the alternating copolymerization of carbon dioxide and epoxy.
The copolymerization was initiated by epoxide ring opening by the metal catalyst, followed by CO2 insertion into the metal-oxygen bond generated. Two side reactions that are detrimental to the desired alternating copolymer formation are (1) consecutive epoxide ring opening to form a polyether backbone, and (2) backbiting reactions leading to cyclic carbonate productions. Detailed mechanism on the metal-catalyzed copolymerization of carbon dioxide and epoxides can be found in several excellent reviews.43,47,57 Catalysts with high reactivity towards polymerization while capable of completely suppressing the two side reactions are highly desired. The most active catalysts reported to date are the ‘single component’ cobalt salen complexes bearing ammonium or nucleophilic substituents on pendant arms,55 and the binary systems consisting of simple (salen)Co(III)X and a nucleophilic cocatalyst.58 They exhibited high reactivity under mild conditions (e.g. 0.1MPa of CO2 pressure) and gave rise to copolymers with >99% carbonate linkages and a high regiochemical control (~95% head-to-tail content). The mechanism of the copolymerization of cyclohexene oxide and CO2 was studied quite extensively. The resulting poly(cyclohexylene carbonate), with a Tg of 115°C, however, had inferior mechanical and physical properties compared to BPA-based aromatic polycarbonate,59 and thus did not find practical applications as initially expected. Another widely studied system is the copolymerization between propylene oxide and CO2 to generate poly(propylene carbonate), which has found application as toughening agents for epoxy resins and sacrificial binders for ceramics due to its low Tg (40 °C), sharp and clean decomposition above 200 °C, and the biodegradability.60
Unlike the expanding spectrum of catalysts, epoxides that can copolymerize with CO2 to give truly alternating copolymers remain quite limited. Besides cyclohexene oxide and propylene oxide, styrene oxide, limonene oxide, indene oxide, and epichlorohydrin have been reported for successful copolymerization with CO2. Polycarbonates with built-in side chain functionalities were rarely prepared by this method.61,62, Lukaszczyk et al. copolymerized CO2 with allyl glycidyl ether in the presence of ZnEt2/pyrogallol catalysts, which gave poly(epoxycarbonate) after oxidation.61 The epoxy-functionalized polycarbonate provides potential a functionalization handle for covalent attachment of drugs, thus may be explored as biodegradable drug carriers. Recently, Frey’s and Grinstaff’s research groups independently reported the preparation of poly(1,2-glycerol carbonate) with hydroxylated side chains. Both reports employed a two-step method involving copolymerization of protected epoxides with CO2 followed by selective deprotection under mild conditions.63,64 In Frey’s report,63 the protected epoxide monomer was ethoxy ethyl glycidyl ether (EEGE) or benzyl glycidyl ether (BGE) (Fig. 3a), and the copolymerization of epoxide and CO2 was carried out at room temperature for 72 h in dioxane, in the presence of a heterogeneous catalytic system based on ZnEt2 and pyrogallol at a molar ratio of 2:1, and a CO2 pressure of ~20 bar. Alternating copolymer without ether linkage (Mn = 5,000 to 25,200 g/mol, PDI = 1.24–2.33) was obtained. The protecting groups were removed via acid cleavage and hydrogenation for EEGE and BGE, respectively, with little (EEGE) and no (BGE) backbone degradation. In Grinstaff’s report,64 the atactic and isotactic linear poly(benzyl 1,2-glycerol carbonates) were first synthesized via copolymerization of rac-/(R)-benzyl glycidyl ether (BGE) with CO2 at 22 °C for 4 h using a series of Co-salen complexes and a CO2 pressure of 220 psi (15.2 bar). High molecular weight rac-/R-poly(benzyl 1,2-glycerol carbonate) (Mn = 32,200 –48,100 g/mol) with >97% carbonate linkage selectivity and narrow polydispersity (PDI < 1.2) was obtained. Deprotection using hydrogenation afforded poly(1,2-glycerol carbonate).
FIGURE 3.

Synthetic strategies for preparing functional APCs by the copolymerization of CO2 and epoxides. (a) Preparation of poly(1,2-glycerol carbonate), adapted from Ref. [63], with permission from Wiley; (b) Synthesis of poly((isopropylidene glyceryl glycidyl ether)-co-(glycidyl methyl ether) carbonate) copolymers and subsequent deprotection, adapted from Ref. [65], with permission from Wiley; (c) Preparation of polycarbonates with reactive double bonds and subsequent functionalizations via thiol-ene coupling, adapted from Ref. [66], with permission from Wiley.
Frey’s group further improved the preparation of hydroxyl-functionalized APCs by employing a more labile epoxide monomer, 1,2-isopropylidene glyceryl glycidyl ether (IGG), for the copolymerization (Fig. 3b).65 A series of poly((isopropylidene glyceryl glycidyl ether)-co -(glycidyl methyl ether) carbonate) random copolymers with different fractions of IGG units were obtained with >99% carbonate linkages in this manner. The deprotection by acid ion exchange resins (10 wt% in MeOH/THF mixture at 40 °C for 4h in) yielded 1,2-diol functionalized copolymers without any degradation in the polycarbonate backbone. In contrast to the poly(1,2-glycerol carbonate) that degraded completely in THF after 2 weeks, the 1,2-diol functionalized copolymers showed no degradation in THF even after 21 days.
More recently, Frey et al. reported another versatile strategy for preparing functional APCs by the copolymerization CO2 and propylene oxide with aliphatic alkene epoxides (Fig. 3c).66 The reactive double bond on the side chains of the resulting copolymers enabled the introduction of a wide range of functional groups through the thiol-ene reaction, which could alter copolymer properties or provide suitable reactive sites for further grafting.
The lack of commercially available or synthetically readily accessible cyclic ethers beyond the 3-membered cyclic epoxides commonly used in the alternating copolymerization of CO2 and epoxides has largely limited the repeating units in the resulting copolymers to 5-carbon in length. Recently, a series of 4-membered oxetane derivatives were successfully copolymerized with CO2 using (salen)CrCl/onium salt catalysts to generate ether-free polycarbonates.13,67–69 The copolymerization was found to proceed via a six-membered cyclic carbonate intermediate formed between the oxetene and CO2 facilitated by the catalyst, and increasingly so with increasing steric hindrance of the substituents on oxetane.69
2.3 Ring-opening polymerization (ROP) of cyclic carbonate monomers
ROP of cyclic carbonates has become the most effective method to fabricate polycarbonates with good reproducibility and high quality (high molecular weight and low polydispersity). ROP of cyclic carbonate monomers to prepare polycarbonate was mentioned as early as 1932 when the monomer trimethylene carbonate (1 in Fig. 4d) was discovered.70 The polymerization was carried out in the melt with potassium carbonate as the catalyst and resulted in polymers with undesired decarboxylation. The ROP techniques have gradually matured with the development of more effective catalysts for industrial manufacturing of polyesters from cyclic ester monomers such as lactones. Almost all catalysts used for ROP of lactones have been screened for ROP of cyclic carbonate monomers due to the structural similarity between these cyclic monomers. Although many of them were also active for ROP of cyclic carbonates, the polymerization kinetics/mechanisms varied due to the intrinsic difference in electrophilicity of the carbonyl carbon in cyclic carbonates vs. in lactones.
FIGURE 4.

Preparation of six-membered cyclic carbonate monomers from (a) 2,2-bis(hydroxymethyl)propionic acid (Bis-MPA); (b) glycerol or trimethylolalkane; (c) pentaerythritol; and (d) representative chemical structures of functional cyclic carbonate monomers (1–18) and APCs-based macromers (19–25) used in the preparation of hydrogels and drug delivery carriers.
The ROP can be conducted in melt or in solutions by varying mechanisms including cationic, anionic, coordination-insertion, monomer activation, monomer and initiator dual activation, and enzymatic activation. Catalysts available for ROP of cyclic carbonates include transition metal catalysts, alkyl halides, basic and acidic organocatalysts, as well as enzyme catalysts. Concerns over the toxic metal residues in the prepared polymers have motivated the development of metal-free organocatalytic ROP, which has seen great progress in the last decade since Hedrick et al. reported the use of 4-(dimethylamino)pyridine (DMAP) as the catalyst for ROP of lactones.71 Basic organocatalysts14,72 tertiary amines, guanidines, amidines, phosphazenes, N-heterocyclic carbine (NHC), and thiourea (TU)/amines, as well as organic acidic catalysts diphenyl phosphate (DPP),73 methanesulfonic acid (MSA),74 and triflic acid(TFA)75,76 have all been found effective in catalyzing the ROP of cyclic carbonates. Lipases as a class of bio-friendly enzyme catalysts have also been explored for ROP.12,77–82 Compared to their metallic and organocatalyt counterparts, however, lipases are generally less efficient and have poorer control over polydispersity.
Numerous cyclic carbonates, mainly six-membered cyclic carbonates with a variety of functional groups (e.g. 1–18 in Fig. 4d), have been prepared and polymerized by means of ROP, as extensively reviewed by Zhang et al.9 and Dove et al.10 Most of these functional monomers were derived from compounds containing 1,3-diols. Among them, 2,2-bis(hydroxymethyl)propionic acid (Fig. 4a),18,23,26 glycerol or trimethyolalkane (Fig. 4b),24,25,81,83–86 and pentaerythritol (Fig. 4c),28,87,88 and their derivatives are the most utilized starting materials for deriving functional cyclic carbonates (1–18 in Fig. 4d). The functionalities could be introduced via either protected monomers that requires post-polymerization deprotection or unmasked monomers when they are compatible with the carbonate structure and the polymerization conditions.
Compared to cyclic ethers and esters, there are greater functional diversity within cyclic carbonates. Combined with the development of ROP techniques with milder reaction conditions, it has enabled the facile preparation of a wide range of functional APCs. The degradability but slow degradation rate of APCs can be exploited to engineer desired degradation profiles of polymers for biomedical applications by virtue incorporation of APCs with other non-degradable or faster degrading polymers.
3. Applications
The advance of the polymerization techniques, especially the CO2-based copolymerization technique, makes it possible to prepare APCs at relatively low cost in the industrial scale. They have been explored for a range of applications as thermoplastics, binders, electronics, coating resins, surfactants and foams and others.89–94,95 The relatively low thermal stability and poor mechanical properties associated with APCs have still limited them to applications that are less demanding in these properties in general. Of particular note, functional APCs with controlled architectures have been increasingly explored for biomedical applications in the last two decades including as tissue engineering scaffolds in the form of electrospun fibers,96,97 biodegradable elastomers,98–103 hydrogels,31,32,104–116, and as drug delivery carriers in the form of micelles,34–36,88,117–128 polymersomes,129–132 and polycomplexes,33,34,111,133–135 etc. Here we review some representative applications of APCs as hydrogels and drug delivery carriers.
3.1 Hydrogels
Hydrogels are three-dimensional polymer networks with intrinsic ability to absorb/hold water,136 and have been widely used in personal care products,137 wound dressing,138,139 protein microchips,140 drug and gene delivery carriers,141 ophthalmic prostheses,142 and tissue engineering scaffolds.143–145 APCs themselves are usually hydrophobic, thus copolymerization with hydrophilic polymers is often required for the preparation of APC-based hydrogels. Both physically and chemically crosslinked APC-based hydrogels with varying hydrophobicity, mechanical properties and degradation profiles have been prepared.
Physically crosslinked hydrogels can be formed from diblock or triblock polycarbonate-containing amphiphilic polymers driven by hydrophobic interactions between the carbonate segments.104–107 High-concentration aqueous solutions of diblock copolymer poly(ethylene glycol)-poly-(trimethylene carbonate) (PEG-PTMC, 20 in Fig. 4d) with relatively short PEG and PTMC segments underwent a sol-to-gel transition as the temperature increased.104 The sol-to-gel transition temperature could be tuned within the range of 20–75 °C by varying the aqueous concentration, molecular weight, and composition of the polymer. Subcutaneous injection of aqueous polymer solutions (30 wt %, 0.5 mL) into rats led to in situ gelation. Whereas the polymer was stable in PBS (pH = 7.4) for over 90 days (e.g. no changes in molecular weight, pH, or gel mass), about 15 wt% mass loss due to the dissolution of lower molecular weight polymers from the gel was detected in vivo within the same time frame. To improve the mechanical properties of hydrogels formed from diblock copolymer with relatively low molecular weight, which had storage modulus of only tens of pascals (Pa), triblock copolymers PTMC-PEG-PTMC (21 in Fig. 4d)with longer hydrophilic and hydrophilic blocks were used for hydrogel preparation.105 These triblock copolymers gelled upon cooling rather than heating, and exhibited storage modulus ranging from 220 to 4700 Pa depending on the composition and concentration of the copolymer.
Cyclic carbonate monomers can be copolymerized with other hydrophobic monomers to improve the gelling characteristics. Compared to gels formed from triblock copolymer poly(caprolactone-b-ethylene glycol-b-caprolactone) without APC segments, the poly(caprolactone-co-trimethylene carbonate)-poly(ethylene glycol)-poly(caprolactone-co-trimethylene carbonate) (PCTC-PEG-PCTC) triblock copolymer (PCTC-PEG-PCTC) hydrogel achieved better sol stability while maintaining the thermogelling property within a physiologically relevant temperature range of 10–50 °C.106 Subcutaneous implantation of the hydrogel in rats revealed substantial degradation, although the hydrogel was quite stable upon incubation in PBS(pH=7.4) for >50 days.106
Physically crosslinked hydrogels could be delivered in an injectable form due to the dynamic physical crosslinking over time. David Putnama’s group had prepared injectable hydrogels from diblock copolymer consisting of monomethoxy-poly(ethylene glycol) (MPEG) and poly(2-oxypropylene carbonate) (MPEG-pDHA, 22 in Fig. 4d).107 The copolymer was prepared by MPEG-initiated ROP of 2,2-dimethoxypropylene carbonate (2 in Fig. 6) that derived from metabolic intermediate dihydroxyacetone, followed by deprotection under acidic conditions. The poly(2-oxypropylene carbonate) is hydrophilic even though insoluble in water. These injectable hydrogel were used for the prevention/alleviation of seroma (benign pocket of body fluids), a common postoperative complication following ablative and reconstructive surgeries. The MPEG-pDHA hydrogels were thixotropic, exhibiting decreasing viscosities with increasing shear rates, thus allowing the hydrogels to be delivered to (potential) sites of seroma by injection. The in vitro degradation rate of the hydrogel in PBS (pH 7.4) was surprisingly rapid, achieving complete degradation in 24 h, and the degradation rate decreased with increasing pDHA lengths. The in vivo degradation of the hydrogel in a rat mastectomy model was slightly slower than that in vitro, with complete degradation accomplished in less than 3 days. The seroma volumes decreased significantly when MPEG-pDHA was administrated compared to the untreated control group. Moreover, the MPEG-pDHA gel and its degradation products did not adversely impact the early wound healing.
FIGURE 6.

Macromer synthesis, crosslinking and cell encapsulation strategies of a “clickable” APC-based hydrogel system. a) ROP of AzDXO initiated by PEG. b) Synthesis of PEG-(DBCO)x; c) Schematic illustration of cell encapsulation by crosslinking PEG-P(AzDXO)2m and PEG-(DBCO)x via SPAAC “click” reaction; d) A representative demonstration of the rapid gelation of the cell-hydrogel constructs within 1 min of mixing the BMSC cell suspension (106 cells/mL) in a PEG20k-P(AzDXO)4 solution (10 w/v% in BMSC expansion media) and a 4-arm-PEG10k-DBCO solution (10 w/v% in BMSC expansion media). The BMSC expansion media consisted of α-MEM with 20% FBS. (Reprinted from Ref [31], with permission from Wiley)
Hydrogels usually suffer from inadequate mechanical properties without sufficient covalent or physical crosslinking. Covalently crosslinked APC-based hydrogels have been prepared by photo-polymerization of the water-soluble, end-group acrylated PTMC-PEG-PTMC triblock copolymers (23 in Fig. 4d).146 Varghese et al. recently reported a mechanically tough biodegradable hydrogel prepared from APC-containing macromer oligo(trimethylene carbonate)-block-poly(ethylene glycol)-block-oligo(trimethylene carbonate) (OTMC-PEG-OTMC) diacrylate.108 Very tough hydrogel (TMC20), was obtained from photopolymerization of OTMC-PEG-OTMC with appropriate block lengths of the hydrophilic PEG (Mn = 20,000 g/mol) and the hydrophobic OTMC (Mn = 325 g/mol). The critical balance of hydrophilic-hydrophobic moieties resulted in hydrogels with enhanced toughness (215.3±46.4 kJ/m3) and modulus (14.9 ±0.2 kPa) while maintaining good fracture strains (98.2 ±1.3%) compared with the hydrogel without the APC component (PEG20), which exhibited a toughness of 130.2±45.4 kJ/m3, modulus of 7.4±0.8 kPa, and fracture strain of 98.7 ± 3.5% (Fig. 5). Moreover, these APC-containing hydrogels were shown to support the adhesion and spreading of human bone marrow derived mesenchymal stem cells (hMSCs) and primary bovine articular chondrocytes.108,110 When chondrocytes were encapsulated in the TMC20 gel, they underwent spontaneous aggregation in vitro, which was not observed with the cells encapsulated in the PEG20 control. More cartilage matrix (GAG and collagen) syntheses were observed with the aggregated chondrocytes in TMC20 than those encapsulated in PEG20.
FIGURE 5.

Photographs demonstrating how the TMC20 hydrogels better sustained compression, knot formation and stretching compared to PEG20 control: (a) PEG20 hydrogels deformed under compression and broke into pieces at higher stress. The dotted circle denotes the damaged hydrogel; (b) deformation and recovery of TMC20 hydrogel under compressive stress; (c) knots formed from PEG20 hydrogels (top) were broken into pieces upon stretching (bottom); (d) TMC20 hydrogels knots (top) were able to withstand stretching/tightening (bottom); (e) stress-strain profiles of the hydrogels under uniaxial compression. (Reproduced from Ref. [108], with permission from The Royal Society of Chemistry)
Triblock copolymer diacrylate with a more hydrophobic APC block, oligo(2,2-dimethyltrimethylene carbonate)-block-poly(ethylene glycol)-block-oligo(2,2-dimethyltrimethylene carbonate) diacrylate (DPD-DA, 24 in Fig. 4d), prepared through ROP of dimethyltrimethylene carbonate (3 in Fig. 6), were also photo-crosslinked by Liao et al. to form hydrogels.114 Although the hydrogel prepared from DPD-DA exhibited some good mechanical properties, their toughness was inferior to those of PEG20 and TMC20 reported by Varghese et al.,108 probably due to the relative low degree (~70%) of acrylation of the DPD precursor. A series of methacrylate-functionalized PTMC-PEG-PTMC triblock copolymers were also used for hydrogel preparation through photo-polymerization. In contrast to the hydrogels reported by the Varghese’s group,108 these hydrogels, with similar lengths of PEG and PTMC blocks, only achieved modest mechanical properties with a compressive modulus of < 15 kPa and toughness of 25 kJ/m3.
Multiple functional cyclic carbonates can also be crosslinked into hydrogels directly by ROPs.32,109,115 A pH-responsive APC-based hydrogels formed by covalent crosslinking and strengthened by secondary non-covalent interactions was reported by Mespouille et al.32 Functional cyclic carbonates bearing BOC-protected guanidines (MTC-GuaBOC, 14 in Fig. 4d) and tert-butyl-protected carboxylic acids (MTC-tBAc, 15 in Fig. 4d) were first synthesized from 2,2-bis(hydroxymethyl)propionic acid. A PEG-based trimethylene carbonate crosslinker, poly(ethylene oxide)-α,ω-methylcarboxy trimethylene carbonate (MTC-PEO-MTC, 19 in Fig. 4d), was obtained by esterification of poly(ethylene oxide)- α,ω-hydroxyl by 5-methyl-2-oxo-[1,3]dioxane-5-carboxylic acid. The hydrogel was formed by organocatalytic ROP of the two functional cyclic carbonates, MTC-GuaBOC and MTC-tBAc, at varying ratios with the MTC-PEO-MTC crosslinker in organic solvents. Monolithic and transparent hydrogels were obtained with high gel content (>92%). Selective de-protection of the BOC- and tert-butyl protection groups result in hydrogels with guanidines and carboxylic acid side chains without degradation of the polycarbonate backbone pH-Dependent swelling behavior was observed with the deprotected hydrogels due to the coexistence of the oppositely charged carboxylic acid and guanidine residues. The same group also reported morpholine-functionalized hydrogels through the copolymerization of 2-(morpholin-4-yl) ethyl-functionalized cyclic carbonate monomer (MTC-Morph, 16 in Fig. 4d) with MTC-PEO-MTC.115 The morpholine-containing hydrogels can be exploited for heavy metal ion sequestrations.
Strategies to directly encapsulate cells in hydrogels with tunable mechanical properties and degradability without harmful gelling conditions are highly desired for regenerative medicine applications. The gelling of most physically crosslinked hydrogels requires substantial changes in environmental conditions (e.g., pH, temperature, ionic strength), which could be detrimental to the in situ encapsulated cells. On the other hand, the cytotoxicity of crosslinking reagents and initiators as well as heat or UV irradiations used for chemically crosslinked hydrogels can negatively impact the viability and long-term fate of the encapsulated cells. A hydrogel system that can be crosslinked under physiological conditions without external perturbations or cross-reactivities with cellular or tissue environment is highly desired. Functional APC-based hydrogel precursors carrying orthogonal reactive groups that can efficiently chemically crosslink to form functional hydrogels under physiological conditions without the use of cytotoxic catalyst, heat or UV irradiations are ideal for addressing such a critical challenge. To enable this strategy, our group recently developed an azido-functionalized six-membered cyclic monomer,28 5,5-bis(azidomethyl)-1,3-dioxan-2-one (AzDXO, 4 in Fig. 4d), and prepared the azido-functionalized APC hydrogel precursors by ROP. Cytocompatile degradable hydrogel was then formed via a bioorthogonal azido-alkyne reaction with another alkynylated hydrogel precursor.31
Specifically, the hydrogel was formed from two orthogonal synthetic macromers, azido-functionalized poly(azido carbonate)-poly(ethylene glycol)-co-poly(azido-carbonate) triblock copolymer, P(AzDXO)m-PEG-P(AzDXO)m, and dibenzocyclooctyne-functionalized poly(ethylene glycol), PEG-(DBCO)2 or PEG-(DBCO)4, through copper-free, strain-promoted azide-alkyne cylcloaddition (SPAAC) “click” chemistry (Fig. 7). The azido-functionalized triblock P(AzDXO)m-PEG-P(AzDXO)m were prepared by organocatalytic ROP of AzDXO under the catalysis of DBU, using PEG-diol (Mn = 6,000, 10,000, and 20,000 g/mol) as initiators, in dichloromethane at room temperature (Fig. 6a). The triblock copolymer macromers with expected molecular weight and narrow polydispersity (PDI < 1.1) were obtained with high monomer conversion (~90%). The solubility of the P(AzDXO)m-PEG-P(AzDXO)m macromers decreased with increasing lengths of the polycarbonate block and was less dependent on the PEG block length. Water soluble triblock copolymers with more than 14 azido groups could be obtained. Robust hydrogels were formed upon mixing the azido-functionalized triblock copolymer macromers with the DBCO-functionalized PEG macromers (Fig. 6b) in aqueous solutions. The gelation time ranging from 20 s to 5 min and the shear modulus ranging from 200 Pa to 10 KPa could be tuned by the polycarbonate block length, macromer concentrations, temperature, and azido/DBCO ratio. The high fidelity and orthogonality of the SPAAC “click” chemistry as well as its high efficiency under physiological conditions present significant advantages over other in situ crosslinking chemistries for biological applications. The gelation could be carried out in water, PBS and even cell culture media without noticeable compromises on the gelling kinetics. Rat bone marrow stromal cells (BMSC) were dispersed in the culture media containing DBCO-and azido-functionalized macromers, which rapidly gelled upon mixing (Figs. 6c & d). The encapsulated cells remained viability after 48 h at a greater percentage than those encapsulated in conventional photo-polymerized PEG hydrogel. These hydrogel formulations are being optimized in terms of mechanical properties and degradation rates, for potential cartilage tissue engineering applications.
FIGURE 7.
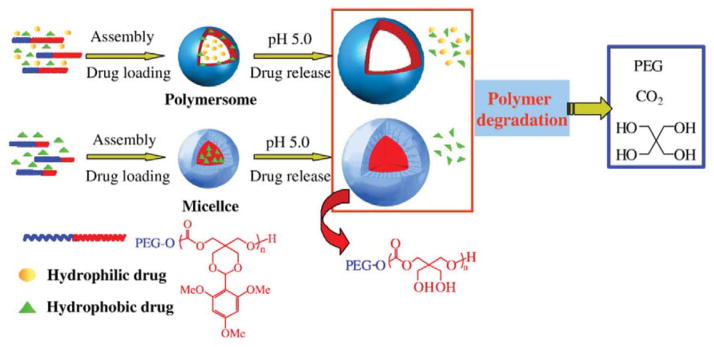
Illustration of pH-sensitive degradable polymersomes based on PEG-PTMBPEC diblock copolymer for triggered release of both hydrophilic and hydrophobic anti-cancer drugs. In comparison, pH-sensitive degradable micelles are typically applied for the encapsulation and release of hydrophobic drugs only. (Reproduced from Ref. [129], with permission from Elsevier)
Another hydrogel system based on reactive APC segments was reported by Zhong et al.147 Acryloly functionalized precursors, oligo(acryloyl carbonate)-b-poly(ethylene glycol)-b-oligo(acryloyl carbonate) (OAC-PEG-OAC, 25 in Fig. 6) triblock copolymers, were prepared by ROP of acryloyl cyclic carbonate (5 in Fig. 6). The hydrogel was formed between thiolated glycol chitosan (GC-SH) and OAC-PEG-OAC via Michael-type addition reaction. Robust hydrogels were formed upon mixing aqueous solutions of GC-SH and OAC-PEG-OAC at relatively low total polymer concentrations of 1.5–4.5 wt% under physiological conditions. Hydrogels with gelation time ranging from 10 s to 17 min and storage moduli varying from 100 to 4300 Pa could be obtained by changing the degree of thiolation of GC-SH, polymer concentrations, thiol/acrylate molar ratios, and the pH. The hydrogels showed good hydrolytic stability in PBS (pH 7.4) at 37°C, whereas much faster degradation occurred in the presence of enzyme. No demonstration of this system for cell encapsulation has been reported yet.
3.2 Drug Delivery Carriers
The use of functional APCs with well-defined chemical compositions and structures for drug delivery applications was pioneered by the Ren-Xi Zhuo,148–158 Xiabing Jing,159–166 James L. Hedrick,33,111,120,121,124,125,128,134,167–171 Yi Yang Yang,33,34,111,120,121,124,125,128,134,155,167–173 Robert M. Waymouth,133,135,167,171,172 Paul A. Wender,133,135 and their colleagues. ROP of functional cyclic carbonates enables an efficient strategy to explore the chemical space to identify APC-based vehicles for optimal drug encapsulation and delivery. Here we review the recent progress in this area based on the encapsulation format of APC-based delivery vehicles.
3.2.1 Micelles for hydrophobic drug delivery
The initial use of APCs for hydrophobic drug delivery relied on the hydrophobic interactions between the drug and the polycarbonate segments. Hydrophobic drugs could be loaded directly on bulk APCs. In the case where hydrophobic segments of the APCs self-assembled to form hydrophobic domains, the drugs could be more stably trapped within these hydrophobic pockets. The release kinetics of the drug is largely governed by the degradation rate or the dissociation of the self-assembled domains of the APCs.
Core/shell nanoparticles or polymeric micelles with a hydrophobic core can be formed through self-assembly of amphiphilic copolymers in an aqueous environment. Diblock and triblock copolymers of PEG and poly(trimethylene carbonate) (PTMC) with relatively long PEG blocks and short PTMC blocks have been shown to form micelles in aqueous solutions.148,174 The critical micellar concentration (CMC) ranged from 35 to 100 mg/L depending on the lengths of the PEG and PTMC blocks, usually decreasing with increasing length of the hydrophobic PTMC segment. Drug loading efficiency as high as 30% has been achieved.148 The degradation rate typically increased with increasing PEG length.
Cyclic carbonate monomers have also been copolymerized with lactones to prepared amphiphilic copolymers to improve the stability of the self-assembled micelles through enhanced hydrophobic interactions. Diblock copolymer polyethylene glycol-b-poly(carbonate-co-lactide) was prepared by copolymerization of lactides and bis-MPA derived monomer 5-methyl-5-benzyloxycarbonyl-1,3-dioxane-2-one (6 in Fig. 4d), with methoxy-poly(ethylene glycol) (mPEG) as an initiator.119 The inclusion of the carbonate moiety facilitated the self-assembly of the copolymers, and the CMC values of these copolymers were up to 10-fold lower than those of PEG-b-PLLA. A non-steroidal anti-androgen for treating early-stage prostate cancer, bicalutamide, was loaded in the copolymer micelles. The bicalutamide loading in the micelles based on the polycarbonate-containing diblock copolymer was about four-fold higher than those achieved with the micelles without the polycarbonate moiety.119
Temperature-sensitive and biodegradable self-assembled micelles were prepared by linking poly(N-isopropylacrylamide) (PNiPAAm) with the hydrophobic APCs.175 PNiPAAm and related copolymers are the most widely investigated temperature-sensitive polymers. Its copolymers with other poly(meth)acrylates were shown to form micelles in response to temperature triggers. However, the non-degradability of these micelles raised the concern of inefficient clearance of the micelles from the body. Lee et al. reported the synthesis of degradable amphiphilic PNiPAAm-b-PTMC by organocatalytic ROP of TMC (1 in Fig. 4d) or 5-methyl-5-benzyloxycarbonyl-1,3-dioxane-2-one (6 in Fig. 4d) using hydroxyl-terminated PNiPAAm as the macroinitiator.175 The PNiPAAm-b-PTMC block copolymers showed temperature-dependent drug release characteristics. At temperatures below the lower critical solution temperature (LCST), slow drug release was observed due to higher stability of the micelles. The drug release became much faster when the temperature was increased to 37 °C or 43 °C (higher than the LCST) to effectively disrupt the micelles. Due to the relatively high hydrophilicity of the PNiPAAm segment, the in vitro degradation of PNiPAAm-b-PTMC was much faster than PTMC. Recently, disc-like micelles were prepared from amphiphilic diblock copolymers containing hydrophilic PEG and hydrophobic cholesterol-functionalized APCs.176 The amphiphilic block copolymers were synthesized through organocatalytic ROP of cholesterol-functionalized cyclic carbonate monomer (18 in Fig. 4d) with MPEG as an initiator. The copolymers exhibited unique self-assembly behaviors as a function of hydrophilic/hydrophobic ratios. The mPEG113-b-P(MTC-Chol)n block copolymers formed disk-like micelles when n = 4 and exhibited stacked-disk-like morphology when n =11. These biodegradable disc-like micelles are expected to exhibit unique biodistribution and cellular uptake patterns as drug delivery carriers.176
Although the backbones of APCs are hydrophobic in nature, hydrophilicity may be introduced to APCs via side chain functionalization. An amphiphilic graft copolymer comprised of hydrophobic poly(ε-caprolactone) and hydrophilic polycarbonate segments were recently prepared as reduction-sensitive biodegradable micelles by Zhong et al.177 This copolymer was prepared by a two-step process, involving the preparation of functional PCL-co-polycarbonate, PCL-co-P(PDSC), by copolymerization of ε-caprolactone and pyridyl disulfide-functionalized cyclic carbonate monomer (PDSC, 10 in Fig. 4d), followed by the post-polymerization modification with thiolated PEG via thiol–disulfide exchange reaction. The resulting amphiphilic, biodegradable graft copolymer, PCL-g-SS-PEG, formed micelles 110–120 nm in diameter and exhibited particularly low CMC values (< 1mg/L). These biodegradable micelles were prone to rapid shell shedding and aggregation under reductive conditions. Doxorubicin-loaded micelles showed redox-responsive drug releases and pronounced antitumor activity against HeLa cells. Amphiphilic copolymers containing both hydrophobic and hydrophilic APCs were also developed to form micelles for targeted drug delivery applications.120 Sequential copolymerization of hydrophobic (TMC) and hydrophilic diacetonide-protected, carbohydrate-based cyclic carbonate monomers (diacetonide protected glucose, galactose and mannose, or 11, 12, 13 in Fig. 4d) yielded amphiphilic block copolymers with hydrophobic PTMC blocks and hydrophilic carbohydrate-functionalized APC blocks upon deprotection under acidic conditions.120 These glucose and galactose-functionalized block copolymers self-assembled into micelles displaying a high density of sugar moieties on the surface. The delivery of doxorubicin via the galactose-functionalized micelles diaplayed enhanced cytotoxicity towards ASGP-R positive HepG2 cells, to which the micelles selectively target via the surface galactose moieties.
3.2.2 Polymersomes for drug delivery
Polymersomes are similar in microstructures to liposomes which are formed by amphiphilic self-assembling lipids in aqueous media, characterized with a hydrophilic interior and a hydrophilic exterior separated by hydrophobic intermediate components.178 Polymersomes may be exploited to deliver both hydrophilic (drug to be encapsulated within the hydrophilic interior) and hydrophobic (drug to be trapped within the hydrophobic domain) drugs, and exhibit improved stability compared to liposomes. Whether an amphiphilic block copolymer can self-assemble into polymersomes is determined mainly by the hydropholic/hydrophilic balance, molecular weight, and the effective interaction parameter of its hydrophobic block with H2O (χ).178 Biodegradable polymersomes prepared from block copolymers based on poly(ethylene glycol)-b-poly(trimethylnene carbonates) and poly(trimethylene carbonate)-b-poly(l-glutamic acid)130–132 have been reported.
Recently, Zhong et al. reported a stimuli-sensitive degradable polymersome containing pH-responsive polycarbonate segments (Fig. 7).129 These polymersomes were based on diblock copolymer of PEG and the APC containing acid-labile, trimethoxybenzylidene acetal-functionalized side chains (PTMBPEC). The copolymer with appropriate molecular compositions, PEG(1.9k)-PTMBPEC(6k), was shown to spontaneously form polymersomes 100–200 nm in diameter in aqueous solutions. The copolymer with longer PEG segment, PEG(5k)-PTMBPEC(5.8k), on the other hand, formed micelles under the same condition. The acetal protection groups on the APC side chains were stable at pH of 7.4, but were rapidly deprotected at pH 4.0 and 5.0, exhibiting a half-life of 0.5 and 3 days, respectively. Both paclitaxel (PTX, hydrophobic) and doxorubicin hydrochloride (DOX · HCl, hydrophilic) could be loaded to the PEG(1.9k)-PTMBPEC(6k)-based polymersomes, while the PEG(5k)-PTMBPEC(5.8k)-based micelles could only be loaded with the hydrophobic PTX. Both carriers exhibited pH-dependent drug release profiles and the release rate increased significantly with lower pH. The PTX release from the polymersome were much faster than from the micelle, likely due to the more significant dimensional changes in polymersomes upon the cleavage of the acetal groups.
Temperature-induced fusion and fission of the polymersome prepared from poly(trimethylene carbonate)-b-Poly(l-glutamic acid) were also reported.179 Polymersome budding and fission occurred when the temperature was increased above the melting temperature of the PTMC component while the fusion events were observed when the temperature was decreased. This phenomenon provides another potential strategy for controlled release of therapeutics via polymersomes.
3.2.3 Degradable polycationic polycarbonates for DNA and siRNA delivery
Gene therapy has emerged as a promising strategy for the treatment of genetic diseases. Cationic polymeric non-viral vectors have received a lot attentions for potentially safer delivery of negatively charged DNA or siRNA cargos. Many earlier non-viral gene-delivery studies used commercially available, non-degradable polycations such as poly(L-lysine), polyethylenimine (PEI), and polyamidoamine dendrimers (PAMAM), which exhibit fairly good transfection efficiency but significant cytotoxicity. Biodegradable, polycationic APCs have recently been explored as improved delivery vehicles for DNA (or siRNA).
Zhuo and colleagues prepared a series of amine-functionalized APCs by a three-step process, including lipase-catalyzed ROP of ally functionalized cyclic carbonate monomer, conversion of ally groups to epoxy groups, and finally the grafting of polyethylenimine (PEI) to the polymer via nucleophile opening of the epoxy by the primary amine residues of the PEI(Fig. 8a).158 Poly(5-methyl-5-allyloxycarbonyl-trimethylene carbonate) (PMAC) was first synthesized in bulk, catalyzed by immobilized porcine pancreas lipase (IPPL). Upon epoxidation of the allyl group by 3-chloroperoxybenzoic acid, the polymer was reacted with low molecular weight PEIx in ~100% efficiency to give PEI-grafted polycarbonate (PAMC-g-PEIx) with controlled molecular weight and a slightly broad PDI. Due to the shielding effect of the PMAC backbone on the positive charge density, PMAC-g-PEIx polyplexes exhibited much lower cytotoxicity compared to its PEI counterparts. PMAC-g-PEIx could form positively charged nano-sized particles (30–90 nm) with pDNA. In vitro transfection experiments in 293T cells showed that the PMAC-g-PEIx/DNA complexes exhibiteds enhanced transfection efficiency compared with PEI25K.
FIGURE 8.

Representative preparations of degradable cationic APCs for DNA and siRNA deliveries. (a) a 3-step method for preparing PEI-grafted polycarbonate, reproduced from Ref. [158], with permission from Elsevier; (b) a 3-step method for preparing amine-functionalized polycarbonates, reproduced from Ref. [34], with permission from Elsevier; (c) a 2-step method for preparing cationic APCs, adapted from Ref. [33], with permission from Elsevier; (d) synthesis of a guanidinium-rich amphipathic carbonate co-oligomers, adapted from Ref. [135], Copyright (2012) National Academy of Sciences, USA.
Yang and colleagues also reported amine-functionalized APC for gene delivery using a similar strategy (Fig. 8b),34 which involved organocatalytic ROP of protected carboxyl-functionalized cyclic carbonate monomer, deprotection to expose the carboxyl groups, and the conjugation of aliphatic amines to the carboxyls by amidation. Specifically, a series of benzyl-protected polycarbonates with well defined molecular weight and narrow polydispersity (Mn = 4,500 to 8,400 g/mol, PDI <1.20) were first prepared by organocatalytic ROP of 5-methyl-5-benzyloxycarbonyl-1,3-dioxan-2-one. Carboxylic acid-functionalized polycarbonate was then obtained after the removal of benzyl groups via Pd/C catalyzed hydrogenation. The amine-functionalized polycarbonate was prepared by further reaction with a variety of aliphatic amines (triethylenetetramine, tetraethylenepentamine and pentaethylenehexamine). The degree of amine conjugation was estimated to be ~60%. These functional APCs readily formed nanoparticles upon direct dissolution in water. The CMC values ranged from 22 to 45 mg/L depending on the molecular weight of the copolymer and the type of aliphatic amine conjugated. These amine-functionalized APCs readily attracted DNA to form polycarbonate/DNA complexes 200 to 1000 nm in size. Transfection using these polymeric vectors mediated luciferase expression in HEK293, HepG2 and 4T1 cell lines at efficiencies comparable or superior to that enabled by the PEI control. Moreover, the cytotoxicity of these polycarbonates much much less compared to PEI.
The same group further optimized this DNA delivery platform by employing a 2-step instead of the 3-step reaction (Fig. 8c).33 A series of cationic APCs with well-defined molecular weights and narrow polydispersities were developed using organocatalytic ROP of haloalkyl-functionalized cyclic carbonates derived from bis-MPA, followed by quaternization with bis-tertiary amine. The resulting cationic APCs were able to bind to and condense DNA to form polycarbonate/DNA nanocomplexes (83 to 124 nm). The nanocomplexes induced high luciferase expression efficiency in all four cell lines examined at relatively low N/P ratios in the presence of serum.
Cationic APCs have also been designed for the delivery of siRNA to induce RNA interference. RNA interference (RNAi) has been recognized as a general endogenous mechanism adopted by many organisms to silence the expression of genes that control various cellular events and to protect the cell from viral replication.180 synthetic small interfering RNA (siRNA) are polyanionic, polar, and large double-stranded RNA molecules, typically consisting of a 19–23 base-paired region with two 3’ overhanging nucleotides. The introduction of siRNAs into cultured cells can trigger highly efficient gene silencing through the degradation of the endogenous mRNA whose sequence is complementary to the siRNA, making siRNAs a promising therapeutic modality for the treatment of cancer, viral infections, ocular disorders, and genetic diseases. The delivery of siRNA across the cell membrane and nucleus without degradation is the key to the success of RNAi therapeutics.
Wendera’s group successfully delivered siRNA into cells to achieve 90% knockdown of a selected target protein by using amphiphilic carbonate co-oligomers, which composed of guanidinium-rich side chains for binding siRNA through electrostatic and hydrogen-bonding interactions and hydrophobic side chains for facilitating cellular entry.135 The co-oligomers were prepared by sequential or one-pot copolymerization of a series bis-MPA derived cyclic carbonate monomers with biocompatible lipid side chains (ethyl, hexyl, or dodecyl) or cholesterol and Boc-protected guanidine monomers using benzyl alcohol or monomethyl poly(ethylene glycol) (MPEG) as an initiator (Fig. 8d). Block or random co-oligomers with controlled composition and length were obtained. Removal of the Boc groups with trifluoroacetic acid yielded the desired amphiphilic carbonate co-oligomers containing both hydrophobic alkyl side chains and hydrophilic/charged guanidine groups. The size of the siRNA:co-oligomer complexes ranged from ~200 nm to 1.5 μm in diameter depending on the co-oligomer type and the siRNA/co-oligomer ratio. A preliminary screening experiment on the delivery efficiency by siRNA/co-oligomer complexes showed that dodecylated co-oligomers achieved an average of 86% knockdown of the target protein with high specificity under a serum-free condition. Interestingly, the shorter co-oligomers were found to outperform their longer counterparts within each hydrophobic side chain series. Random co-oligomers did not perform as consistently as their block co-oligomer counterparts. By mixing different co-oligomers with defined block compositions, even greater diversity in the siRNA complexation system, and thus siRNA delivery performances, could be accomplished.
CONCLUSIONS
In summary, APCs, a type of long-known but under-utilized degradable polymers, have been rejuvenated with new functionalities and properties. A wide range of APCs and APC-based copolymers have been prepared using a combination of improved polymerization techniques and novel functional monomers. Practical industrial applications of APCs, however, are still rare. The successful translation of APCs for industrial uses will require further improvements in many aspects, including the development of more universal/versatile catalyst systems, deeper understanding of the polymerization mechanisms and kinetics as a function of monomer structures, and ultimately the development of a predictive model to guide the rational/iterative design of functional polymers for the various targeted applications. With the flexibility provided by APCs in adjusting polymer/copolymer degradation rate, hydrophlicity/hydrophocity, and thermal-mechanical properties, many fundamental questions, such as polymeric structure-properties relationship and cell-biomaterials interactions, can be more systematically interrogated, ultimately benefiting their biomedical applications and beyond.
Acknowledgments
This work was supported by the National Institutes of Health (R01AR055615, R01GM088678 and R21AR056866).
REFERENCES AND NOTES
- 1.Thomas S, Visakh PM. Handbook of Engineering and Speciality Thermoplastics. John Wiley & Sons, Inc; 2011. pp. 1–14. [Google Scholar]
- 2.Brunelle DJ, Korn MR. Advances in polycarbonates. An American Chemical Society Publication; 2005. [Google Scholar]
- 3.LeGrand DG, Bendler JT. Handbook of polycarbonate science and technology. CRC Press; 2000. [Google Scholar]
- 4.Sugimoto H, Inoue S. Pure Appl Chem. 2006;78:1823–1834. [Google Scholar]
- 5.Luinstra GA. Polym Rev. 2008;48:192–219. [Google Scholar]
- 6.D’Alessandro DM, Smit B, Long JR. Angewandte Chemie International Edition. 2010;49:6058–6082. doi: 10.1002/anie.201000431. [DOI] [PubMed] [Google Scholar]
- 7.Fukuoka S, Kawamura M, Komiya K, Tojo M, Hachiya H, Hasegawa K, Aminaka M, Okamoto H, Fukawa I, Konno S. Green Chem. 2003;5:497–507. [Google Scholar]
- 8.Meier MAR, Weckhuysen BM, Bruijnincx PCA, Buchard A, Bakewell C, Weiner J, Williams C. Organometallics and Renewables. Vol. 39. Springer; Berlin Heidelberg: 2012. pp. 175–224. [Google Scholar]
- 9.Feng J, Zhuo RX, Zhang XZ. Prog Polym Sci. 2012;37:211–236. [Google Scholar]
- 10.Tempelaar S, Mespouille L, Coulembier O, Dubois P, Dove AP. Chem Soc Rev. 2013;42:1312–1336. doi: 10.1039/c2cs35268k. [DOI] [PubMed] [Google Scholar]
- 11.Inoue S, Koinuma H, Tsuruta T. J Polym Sci, Part B: Polym Letters. 1969;7:287. [Google Scholar]
- 12.Bisht KS, Svirkin YY, Henderson LA, Gross RA, Kaplan DL, Swift G. Macromolecules. 1997;30:7735–7742. [Google Scholar]
- 13.Darensbourg DJ, Ganguly P, Choi W. Inorg Chem. 2006;45:3831–3833. doi: 10.1021/ic052109j. [DOI] [PubMed] [Google Scholar]
- 14.Nederberg F, Lohmeijer BG, Leibfarth F, Pratt RC, Choi J, Dove AP, Waymouth RM, Hedrick JL. Biomacromolecules. 2007;8:153–160. doi: 10.1021/bm060795n. [DOI] [PubMed] [Google Scholar]
- 15.Yamamoto Y, Kaihara S, Toshima K, Matsumura S. Macromol Biosci. 2009;9:968–978. doi: 10.1002/mabi.200900039. [DOI] [PubMed] [Google Scholar]
- 16.Park JH, Jeon JY, Lee JJ, Jang Y, Varghese JK, Lee BY. Macromolecules. 2013;46:3301–3308. [Google Scholar]
- 17.Venkataraman S, Veronica N, Voo ZX, Hedrick JL, Yang YY. Polym Chem. 2013;4:2945–2948. [Google Scholar]
- 18.Engler AC, Chan JMW, Coady DJ, O’Brien JM, Sardon H, Nelson A, Sanders DP, Yang YY, Hedrick JL. Macromolecules. 2013;46:1283–1290. [Google Scholar]
- 19.Mindemark J, Bowden T. Polym Chem. 2012;3:1399–1401. [Google Scholar]
- 20.Wang LS, Cheng SX, Zhuo RX. Macromol Rapid Commun. 2004;25:959–963. [Google Scholar]
- 21.Tempelaar S, Mespouille L, Dubois P, Dove AP. Macromolecules. 2011;44:2084–2091. [Google Scholar]
- 22.Tempelaar S, Barker IA, Truong VX, Hall DJ, Mespouille L, Dubois P, Dove AP. Polym Chem. 2013;4:174–183. [Google Scholar]
- 23.Sanders DP, Fukushima K, Coady DJ, Nelson A, Fujiwara M, Yasumoto M, Hedrick JL. J Am Chem Soc. 2010;132:14724–14726. doi: 10.1021/ja105332k. [DOI] [PubMed] [Google Scholar]
- 24.Wang XL, Zhuo RX, Liu LJ, He F, Liu G. J Polym Sci, Part A: Polym Chem. 2002;40:70–75. [Google Scholar]
- 25.Mei HJ, Zhong ZL, Long FF, Zhuo RX. Macromol Rapid Commun. 2006;27:1894–1899. [Google Scholar]
- 26.Pratt RC, Nederberg F, Waymouth RM, Hedrick JL. Chem Commun. 2008;0:114–116. doi: 10.1039/b713925j. [DOI] [PubMed] [Google Scholar]
- 27.Wang R, Chen W, Meng F, Cheng R, Deng C, Feijen J, Zhong Z. Macromolecules. 2011;44:6009–6016. [Google Scholar]
- 28.Xu JW, Prifti F, Song J. Macromolecules. 2011;44:2660–2667. doi: 10.1021/ma200021m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen W, Yang H, Wang R, Cheng R, Meng F, Wei W, Zhong Z. Macromolecules. 2009;43:201–207. [Google Scholar]
- 30.Zhang XJ, Cai MM, Zhong ZL, Zhuo RX. Macromol Rapid Commun. 2012;33:693–697. doi: 10.1002/marc.201100765. [DOI] [PubMed] [Google Scholar]
- 31.Xu JW, Filion TM, Prifti F, Song J. Chem Asian J. 2011;6:2730–2737. doi: 10.1002/asia.201100411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartolini C, Mespouille L, Verbruggen I, Willem R, Dubois P. Soft Matter. 2011;7:9628–9637. [Google Scholar]
- 33.Ong ZY, Fukushima K, Coady DJ, Yang YY, Ee PLR, Hedrick JL. J Controlled Release. 2011;152:120–126. doi: 10.1016/j.jconrel.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 34.Seow WY, Yang YY. J Controlled Release. 2009;139:40–47. doi: 10.1016/j.jconrel.2009.05.028. [DOI] [PubMed] [Google Scholar]
- 35.Chiu FC, Lai CS, Lee RS. J Appl Polym Sci. 2007;106:283–292. [Google Scholar]
- 36.Zhou Y, Zhuo RX, Liu ZL. Polymer. 2004;45:5459–5463. [Google Scholar]
- 37.Ono Y. Catal Today. 1997;35:15–25. [Google Scholar]
- 38.Zhu WX, Huang X, Li CC, Xiao YN, Zhang D, Guan GH. Polym Int. 2011;60:1060–1067. [Google Scholar]
- 39.Park JH, Jeon JY, Lee JJ, Jang Y, Varghese JK, Lee BY. Macromolecules. 2013 [Google Scholar]
- 40.Jiang Z, Liu C, Xie W, Gross RA. Macromolecules. 2007;40:7934–7943. [Google Scholar]
- 41.Zhu W, Huang X, Li C, Xiao Y, Zhang D, Guan G. Polym Int. 2011;60:1060–1067. [Google Scholar]
- 42.Foy E, Farrell JB, Higginbotham CL. J Appl Polym Sci. 2009;111:217–227. [Google Scholar]
- 43.Darensbourg DJ, Mackiewicz RM, Phelps AL, Billodeaux DR. Acc Chem Res. 2004;37:836–844. doi: 10.1021/ar030240u. [DOI] [PubMed] [Google Scholar]
- 44.Sugimoto H, Inoue S. J Polym Sci, Part A: Polym Chem. 2004;42:5561–5573. [Google Scholar]
- 45.Omae I. Catal Today. 2006;115:33–52. [Google Scholar]
- 46.Buchard A, Bakewell CM, Weiner J, Williams CK. Organometallics and Renewables. 2012;39:175–224. [Google Scholar]
- 47.Lu XB, Ren WM, Wu GP. Acc Chem Res. 2012;45:1721–1735. doi: 10.1021/ar300035z. [DOI] [PubMed] [Google Scholar]
- 48.Aida T, Inoue S. J Am Chem Soc. 1983;105:1304–1309. [Google Scholar]
- 49.Darensbourg DJ, Niezgoda SA, Draper JD, Reibenspies JH. J Am Chem Soc. 1998;120:4690–4698. [Google Scholar]
- 50.Cheng M, Moore DR, Reczek JJ, Chamberlain BM, Lobkovsky EB, Coates GW. J Am Chem Soc. 2001;123:8738–8749. doi: 10.1021/ja003850n. [DOI] [PubMed] [Google Scholar]
- 51.Moore DR, Cheng M, Lobkovsky EB, Coates GW. J Am Chem Soc. 2003;125:11911–11924. doi: 10.1021/ja030085e. [DOI] [PubMed] [Google Scholar]
- 52.Cheng M, Moore DR, Reczek JJ, Chamberlain BM, Lobkovsky EB, Coates GW. J Am Chem Soc. 2001;123:8738–8749. doi: 10.1021/ja003850n. [DOI] [PubMed] [Google Scholar]
- 53.Luinstra GA, Haas GR, Molnar F, Bernhart V, Eberhardt R, Rieger B. Chem-Eur J. 2005;11:6298–6314. doi: 10.1002/chem.200500356. [DOI] [PubMed] [Google Scholar]
- 54.Cohen CT, Chu T, Coates GW. J Am Chem Soc. 2005;127:10869–10878. doi: 10.1021/ja051744l. [DOI] [PubMed] [Google Scholar]
- 55.Ren WM, Zhang X, Liu Y, Li JF, Wang H, Lu XB. Macromolecules. 2010;43:1396–1402. [Google Scholar]
- 56.Ren WM, Liu ZW, Wen YQ, Zhang R, Lu XB. J Am Chem Soc. 2009;131:11509–11518. doi: 10.1021/ja9033999. [DOI] [PubMed] [Google Scholar]
- 57.Coates GW, Moore DR. Angew Chem, Int Ed. 2004;43:6618–6639. doi: 10.1002/anie.200460442. [DOI] [PubMed] [Google Scholar]
- 58.Lu XB, Wang Y. Angew Chem, Int Ed. 2004;43:3574–3577. doi: 10.1002/anie.200453998. [DOI] [PubMed] [Google Scholar]
- 59.Koning C, Wildeson J, Parton R, Plum B, Steeman P, Darensbourg DJ. Polymer. 2001;42:3995–4004. [Google Scholar]
- 60.Luinstra GA, Borchardt E. Synthetic Biodegradable Polymers. 2012;245:29–48. [Google Scholar]
- 61.Lukaszczyk J, Jaszcz K, Kuran W, Listos T. Macromol Biosci. 2001;1:282–289. [Google Scholar]
- 62.Tao YH, Wang XH, Zhao XJ, Li J, Wang FS. J Polym Sci, Part A: Polym Chem. 2006;44:5329–5336. [Google Scholar]
- 63.Geschwind J, Frey H. Macromolecules. 2013;46:3280–3287. [Google Scholar]
- 64.Zhang H, Grinstaff MW. J Am Chem Soc. 2013;135:6806–6809. doi: 10.1021/ja402558m. [DOI] [PubMed] [Google Scholar]
- 65.Geschwind J, Frey H. Macromol Rapid Commun. 2013;34:150–155. doi: 10.1002/marc.201200682. [DOI] [PubMed] [Google Scholar]
- 66.Geschwind J, Wurm F, Frey H. Macromol Chem Phys. 2013;214:892–901. [Google Scholar]
- 67.Darensbourg DJ, Moncada AI, Choi W, Reibenspies JH. J Am Chem Soc. 2008;130:6523–6533. doi: 10.1021/ja800302c. [DOI] [PubMed] [Google Scholar]
- 68.Darensbourg DJ, Moncada AI. Macromolecules. 2009;42:4063–4070. [Google Scholar]
- 69.Darensbourg DJ, Moncada AI. Macromolecules. 2010;43:5996–6003. [Google Scholar]
- 70.Carothers WH, Dorough GL, Natta FJv. J Am Chem Soc. 1932;54:761–772. [Google Scholar]
- 71.Nederberg F, Connor EF, Möller M, Glauser T, Hedrick JL. Angew Chem, Int Ed. 2001;40:2712–2715. [PubMed] [Google Scholar]
- 72.Helou M, Miserque O, Brusson JM, Carpentier JF, Guillaume SM. Chem-Eur J. 2010;16:13805–13813. doi: 10.1002/chem.201001111. [DOI] [PubMed] [Google Scholar]
- 73.Makiguchi K, Ogasawara Y, Kikuchi S, Satoh T, Kakuchi T. Macromolecules. 2013;46:1772–1782. [Google Scholar]
- 74.Campos JM, Ribeiro MR, Ribeiro MF, Deffieux A, Peruch F. Macromol Chem Phys. 2013;214:85–93. [Google Scholar]
- 75.Nemoto N, Sanda F, Endo T. Journal of Polymer Science Part A: Polymer Chemistry. 2001;39:1305–1317. [Google Scholar]
- 76.Delcroix D, Martín-Vaca B, Bourissou D, Navarro C. Macromolecules. 2010;43:8828–8835. [Google Scholar]
- 77.Al-Azemi TF, Bisht KS. Macromolecules. 1999;32:6536–6540. [Google Scholar]
- 78.Al-Azemi TF, Harmon JP, Bisht KS. Biomacromolecules. 2000;1:493–500. doi: 10.1021/bm005552o. [DOI] [PubMed] [Google Scholar]
- 79.Gross RA, Kalra B, Kumar A. Appl Microbiol Biotechnol. 2001;55:655–660. doi: 10.1007/s002530100617. [DOI] [PubMed] [Google Scholar]
- 80.Feng J, Zhuo RX, He F, Wang XL. Macromol Symp. 2003;195:237–240. [Google Scholar]
- 81.He F, Wang YP, Liu G, Jia HL, Feng J, Zhuo RX. Polymer. 2008;49:1185–1190. [Google Scholar]
- 82.Wu R, Al-Azemi TF, Bisht KS. Biomacromolecules. 2008;9:2921–2928. doi: 10.1021/bm800696q. [DOI] [PubMed] [Google Scholar]
- 83.Liu G, He F, Wang YP, Feng J, Zhuo RX. Chin Chem Lett. 2006;17:137–139. [Google Scholar]
- 84.Su W, Luo XH, Wang HF, Li L, Feng J, Zhang XZ, Zhuo RX. Macromol Rapid Commun. 2011;32:390–396. doi: 10.1002/marc.201000600. [DOI] [PubMed] [Google Scholar]
- 85.Simon J, Olsson JV, Kim H, Tenney IF, Waymouth RM. Macromolecules. 2012;45:9275–9281. [Google Scholar]
- 86.Tryznowski M, Tomczyk K, Fras Z, Gregorowicz J, Rokicki G, Wawrzynska E, Parzuchowski PG. Macromolecules. 2012;45:6819–6829. [Google Scholar]
- 87.Vandenberg EJ, Tian D. Macromolecules. 1999;32:3613–3619. [Google Scholar]
- 88.Chen W, Meng FH, Li F, Ji SJ, Zhong ZY. Biomacromolecules. 2009;10:1727–1735. doi: 10.1021/bm900074d. [DOI] [PubMed] [Google Scholar]
- 89.Allen SD. 2009/065,528:2009. WO Patent App PCT/US.
- 90.Allen SD, Coates GW, Cherian AE, Simoneau CA, Gridnev AA, Farmer JJ. 20,130,066,044:2013. US Patent.
- 91.Darensbourg DJ, Andreatta JR, Moncada AI. Carbon Dioxide as Chemical Feedstock. 2010:213–248. [Google Scholar]
- 92.Fritz N, Dao H, Allen SAB, Kohl PA. Int J Adhes Adhes. 2012;38:45–49. [Google Scholar]
- 93.Füßl A, Sandler JKW, Nalawade S, Steinke TH, Warzelhan V, Künkel A, Hahn K, Lohmann J, Brym AK. 20,130,059,141:2013. US Patent.
- 94.Ku SY, Liman CD, Cochran JE, Toney MF, Chabinyc ML, Hawker CJ. Advanced Materials. 2011;23:2289–2293. doi: 10.1002/adma.201100028. [DOI] [PubMed] [Google Scholar]
- 95.Tominaga Y, Shimomura T, Nakamura M. Polymer. 2010;51:4295–4298. [Google Scholar]
- 96.Welle A, Kroger M, Doring M, Niederer K, Pindel E, Chronakis IS. Biomaterials. 2007;28:2211–2219. doi: 10.1016/j.biomaterials.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 97.Wu RZ, Zhang JF, Fan YW, Stoute D, Lallier T, Xu XM. Biomedical Materials. 2011:6. doi: 10.1088/1748-6041/6/3/035004. [DOI] [PubMed] [Google Scholar]
- 98.Hou Q, Grijpma DW, Feijen J. Acta Biomater. 2009;5:1543–1551. doi: 10.1016/j.actbio.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 99.Bat E, Feijen J, Grijpma DW. Biomacromolecules. 2010;11:2692–2699. doi: 10.1021/bm1007234. [DOI] [PubMed] [Google Scholar]
- 100.Bat E, Kothman BHM, Higuera GA, van Blitterswijk CA, Feijen J, Grijpma DW. Biomaterials. 2010;31:8696–8705. doi: 10.1016/j.biomaterials.2010.07.102. [DOI] [PubMed] [Google Scholar]
- 101.Bat E, van Kooten TG, Feijen J, Grijpma DW. Acta Biomater. 2011;7:1939–1948. doi: 10.1016/j.actbio.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 102.Dargaville BL, Vaquette C, Peng H, Rasoul F, Chau YQ, Cooper-White JJ, Campbell JH, Whittaker AK. Biomacromolecules. 2011;12:3856–3869. doi: 10.1021/bm201291e. [DOI] [PubMed] [Google Scholar]
- 103.Schülller-Ravoo S, Feijen J, Grijpma DW. Acta Biomater. 2012;8:3576–3585. doi: 10.1016/j.actbio.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 104.Kim SY, Kim HJ, Lee KE, Han SS, Sohn YS, Jeong B. Macromolecules. 2007;40:5519–5525. [Google Scholar]
- 105.Bat E, Grijpma DW, Feijen J. J Controlled Release. 2008;132:e37–e39. [Google Scholar]
- 106.Park SH, Choi BG, Joo MK, Han DK, Sohn YS, Jeong B. Macromolecules. 2008;41:6486–6492. [Google Scholar]
- 107.Zawaneh PN, Singh SP, Padera RF, Henderson PW, Spector JA, Putnam D. Proc Natl Acad Sci U S A. 2010;107:11014–11019. doi: 10.1073/pnas.0811529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang C, Aung A, Liao L, Varghese S. Soft Matter. 2009;5:3831–3834. [Google Scholar]
- 109.Nederberg F, Trang V, Pratt RC, Kim SH, Colson J, Nelson A, Frank CW, Hedrick JL, Dubois P, Mespouille L. Soft Matter. 2010;6:2006–2012. [Google Scholar]
- 110.Zhang C, Sangaj N, Hwang Y, Phadke A, Chang CW, Varghese S. Acta Biomater. 2011;7:3362–3369. doi: 10.1016/j.actbio.2011.05.024. [DOI] [PubMed] [Google Scholar]
- 111.Li Y, Yang C, Khan M, Liu SQ, Hedrick JL, Yang YY, Ee PLR. Biomaterials. 2012;33:6533–6541. doi: 10.1016/j.biomaterials.2012.05.043. [DOI] [PubMed] [Google Scholar]
- 112.Nitta K, Miyake J, Watanabe J, Ikeda Y. Biomacromolecules. 2012;13:1002–1009. doi: 10.1021/bm201703y. [DOI] [PubMed] [Google Scholar]
- 113.Sharifi S, Blanquer SBG, van Kooten TG, Grijpma DW. Acta Biomater. 2012;8:4233–4243. doi: 10.1016/j.actbio.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 114.Fan C, Zhang C, Jing Y, Liao L, Liu L. RSC Advances. 2013;3:157–165. [Google Scholar]
- 115.Kawalec M, Dove AP, Mespouille L, Dubois P. Polym Chem. 2013;4:1260–1270. [Google Scholar]
- 116.Truong VX, Barker IA, Tan M, Mespouille L, Dubois P, Dove AP. Journal of Materials Chemistry B. 2013;1:221–229. doi: 10.1039/c2tb00148a. [DOI] [PubMed] [Google Scholar]
- 117.Peng T, Su J, Cheng SX, Zhuo RX. J Biomater Sci, Polym Ed. 2006;17:941–951. doi: 10.1163/156856206777996899. [DOI] [PubMed] [Google Scholar]
- 118.Hu XL, Jing XB. Expert Opin Drug Deliv. 2009;6:1079–1090. doi: 10.1517/17425240903158917. [DOI] [PubMed] [Google Scholar]
- 119.Danquah M, Fujiwara T, Mahato RI. Biomaterials. 2010;31:2358–2370. doi: 10.1016/j.biomaterials.2009.11.081. [DOI] [PubMed] [Google Scholar]
- 120.Suriano F, Pratt R, Tan JPK, Wiradharma N, Nelson A, Yang YY, Dubois P, Hedrick JL. Biomaterials. 2010;31:2637–2645. doi: 10.1016/j.biomaterials.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 121.Lee ALZ, Venkataraman S, Sirat SBM, Gao SJ, Hedrick JL, Yang YY. Biomaterials. 2012;33:1921–1928. doi: 10.1016/j.biomaterials.2011.11.032. [DOI] [PubMed] [Google Scholar]
- 122.Qiao Y, Yang C, Coady DJ, Ong ZY, Hedrick JL, Yang YY. Biomaterials. 2012;33:1146–1153. doi: 10.1016/j.biomaterials.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 123.Wang HF, Luo XH, Liu CW, Feng J, Zhang XZ, Zhuo RX. Acta Biomater. 2012;8:589–598. doi: 10.1016/j.actbio.2011.08.030. [DOI] [PubMed] [Google Scholar]
- 124.Yang C, Attia ABE, Tan JPK, Ke XY, Gao SJ, Hedrick JL, Yang YY. Biomaterials. 2012;33:2971–2979. doi: 10.1016/j.biomaterials.2011.11.035. [DOI] [PubMed] [Google Scholar]
- 125.Attia ABE, Yang C, Tan JPK, Gao SJ, Williams DF, Hedrick JL, Yang YY. Biomaterials. 2013;34:3132–3140. doi: 10.1016/j.biomaterials.2013.01.042. [DOI] [PubMed] [Google Scholar]
- 126.Cajot S, Lecomte P, Jerome C, Riva R. Polym Chem. 2013;4:1025–1037. [Google Scholar]
- 127.Danquah M, Fujiwara T, Mahato RI. J Polym Sci, Part A: Polym Chem. 2013;51:347–362. [Google Scholar]
- 128.Engler AC, Chan JMW, Fukushima K, Coady DJ, Yang YY, Hedrick JL. Acs Macro Letters. 2013;2:332–336. doi: 10.1021/mz400069u. [DOI] [PubMed] [Google Scholar]
- 129.Chen W, Meng FH, Cheng R, Zhong ZY. J Controlled Release. 2010;142:40–46. doi: 10.1016/j.jconrel.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 130.Sanson C, Schatz C, Le Meins JF, Brulet A, Soum A, Lecommandoux S. Langmuir. 2010;26:2751–2760. doi: 10.1021/la902786t. [DOI] [PubMed] [Google Scholar]
- 131.Sanson C, Schatz C, Le Meins JF, Soum A, Thevenot J, Garanger E, Lecommandoux S. J Controlled Release. 2010;147:428–435. doi: 10.1016/j.jconrel.2010.07.123. [DOI] [PubMed] [Google Scholar]
- 132.Sanson C, Diou O, Thevenot J, Ibarboure E, Soum A, Brulet A, Miraux S, Thiaudiere E, Tan S, Brisson A, Dupuis V, Sandre O, Lecommandoux S. ACS Nano. 2011;5:1122–1140. doi: 10.1021/nn102762f. [DOI] [PubMed] [Google Scholar]
- 133.Cooley CB, Trantow BM, Nederberg F, Kiesewetter MK, Hedrick JL, Waymouth RM, Wender PA. J Am Chem Soc. 2009;131:16401–16403. doi: 10.1021/ja907363k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang C, Ong ZY, Yang YY, Ee PLR, Hedrick JL. Macromol Rapid Commun. 2011;32:1826–1833. doi: 10.1002/marc.201100350. [DOI] [PubMed] [Google Scholar]
- 135.Geihe EI, Cooley CB, Simon JR, Kiesewetter MK, Edward JA, Hickerson RP, Kaspar RL, Hedrick JL, Waymouth RM, Wender PA. Proc Natl Acad Sci U S A. 2012;109:13171–13176. doi: 10.1073/pnas.1211361109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hoffman AS. Adv Drug Deliv Rev. 2012;64:18–23. [Google Scholar]
- 137.Omidian H, Rocca JG, Park K. J Controlled Release. 2005;102:3–12. doi: 10.1016/j.jconrel.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 138.Tran NQ, Joung YK, Lih E, Park KD. Biomacromolecules. 2011;12:2872–2880. doi: 10.1021/bm200326g. [DOI] [PubMed] [Google Scholar]
- 139.Kickhofen B, Wokalek H, Scheel D, Ruh H. Biomaterials. 1986;7:67–72. doi: 10.1016/0142-9612(86)90092-x. [DOI] [PubMed] [Google Scholar]
- 140.Bertone P, Snyder M. FEBS J. 2005;272:5400–5411. doi: 10.1111/j.1742-4658.2005.04970.x. [DOI] [PubMed] [Google Scholar]
- 141.Hoare TR, Kohane DS. Polymer. 2008;49:1993–2007. [Google Scholar]
- 142.Alvarez-Lorenzo C, Yanez F, Concheiro A. J Drug Deliv Sci Tec. 2010;20:237–248. [Google Scholar]
- 143.Lee KY, Mooney DJ. Chem Rev. 2001;101:1869–1879. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 144.Drury JL, Mooney DJ. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 145.Annabi N, Nichol JW, Zhong X, Ji CD, Koshy S, Khademhosseini A, Dehghani F. Tissue Eng PT B-Rev. 2010;16:371–383. doi: 10.1089/ten.teb.2009.0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Matsuda T, Kwon IK, Kidoaki S. Biomacromolecules. 2004;5:295–305. doi: 10.1021/bm034231k. [DOI] [PubMed] [Google Scholar]
- 147.Yu Y, Deng C, Meng F, Shi Q, Feijen J, Zhong Z. J Biomed Mater Res, Part A. 2011;99A:316–326. doi: 10.1002/jbm.a.33199. [DOI] [PubMed] [Google Scholar]
- 148.Zhang Y, Zhuo RX. Biomaterials. 2005;26:2089–2094. doi: 10.1016/j.biomaterials.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 149.Zhou Y, Zhuo RX, Liu ZL, Xu D. Polymer. 2005;46:5752–5757. [Google Scholar]
- 150.Zhang Y, Zhuo RX. J Biomed Mater Res, Part A. 2006;76A:674–680. doi: 10.1002/jbm.a.30395. [DOI] [PubMed] [Google Scholar]
- 151.Peng T, Su J, Cheng X, Zhuo RX. J Mater Sci - Mater Med. 2007;18:1765–1769. doi: 10.1007/s10856-007-3022-9. [DOI] [PubMed] [Google Scholar]
- 152.Zou T, Li SL, Hu ZY, Cheng SX, Zhuo RX. J Biomater Sci, Polym Ed. 2007;18:519–530. doi: 10.1163/156856207780852497. [DOI] [PubMed] [Google Scholar]
- 153.Zhang XJ, Mei HJ, Hu C, Zhong ZL, Zhuo RX. Macromolecules. 2009;42:1010–1016. [Google Scholar]
- 154.Zhang XJ, Chen FJ, Zhong ZL, Zhuo RX. Macromol Rapid Commun. 2010;31:2155–2159. doi: 10.1002/marc.201000392. [DOI] [PubMed] [Google Scholar]
- 155.Jia HZ, Wang HF, Liu CW, Li C, Yang J, Xu XD, Feng J, Zhang XZ, Zhuo RX. Soft Matter. 2012;8:6906–6912. [Google Scholar]
- 156.Wang HF, Jia HZ, Cheng SX, Feng J, Zhang XZ, Zhuo RX. Pharm Res. 2012;29:1582–1594. doi: 10.1007/s11095-012-0669-9. [DOI] [PubMed] [Google Scholar]
- 157.Zhang XJ, Zhang ZG, Zhong ZL, Zhuo RX. J Polym Sci, Part A: Polym Chem. 2012;50:2687–2696. [Google Scholar]
- 158.Wang CF, Lin YX, Jiang T, He F, Zhuo RX. Biomaterials. 2009;30:4824–4832. doi: 10.1016/j.biomaterials.2009.05.053. [DOI] [PubMed] [Google Scholar]
- 159.Hu XL, Liu S, Chen XS, Mo GJ, Xie ZG, Jing XB. Biomacromolecules. 2008;9:553–560. doi: 10.1021/bm701092j. [DOI] [PubMed] [Google Scholar]
- 160.Hu XL, Liu S, Huang YB, Chen XS, Jing XB. Biomacromolecules. 2010;11:2094–2102. doi: 10.1021/bm100458n. [DOI] [PubMed] [Google Scholar]
- 161.Kuang HH, Wu SH, Meng FB, Xie ZG, Jing XB, Huang YB. J Mater Chem. 2012;22:24832–24840. [Google Scholar]
- 162.Ma PA, Liu S, Huang YB, Chen XS, Zhang LP, Jing XB. Biomaterials. 2010;31:2646–2654. doi: 10.1016/j.biomaterials.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 163.Wan YH, Zheng YH, Song XF, Hu XL, Liu S, Tong T, Jing XB. J Biomater Sci, Polym Ed. 2011;22:1131–1146. doi: 10.1163/092050610X500570. [DOI] [PubMed] [Google Scholar]
- 164.Wu SH, Kuang HH, Meng FB, Wu YJ, Li XY, Jing XB, Huang YB. J Mater Chem. 2012;22:15348–15356. [Google Scholar]
- 165.Xie ZG, Guan HL, Chen L, Tian HY, Chen XS, Jing XB. Polymer. 2005;46:10523–10530. [Google Scholar]
- 166.Yan LS, Wu WB, Zhao W, Qi RG, Cui DM, Xie ZG, Huang YB, Tong T, Jing XB. Polym Chem. 2012;3:2403–2412. [Google Scholar]
- 167.Nederberg F, Appel E, Tan JPK, Kim SH, Fukushima K, Sly J, Miller RD, Waymouth RM, Yang YY, Hedrick JL. Biomacromolecules. 2009;10:1460–1468. doi: 10.1021/bm900056g. [DOI] [PubMed] [Google Scholar]
- 168.Kim SH, Tan JPK, Nederberg F, Fukushima K, Colson J, Yang CA, Nelson A, Yang YY, Hedrick JL. Biomaterials. 2010;31:8063–8071. doi: 10.1016/j.biomaterials.2010.07.018. [DOI] [PubMed] [Google Scholar]
- 169.Tan JPK, Kim SH, Nederberg F, Fukushima K, Coady DJ, Nelson A, Yang YY, Hedrick JL. Macromol Rapid Commun. 2010;31:1187–1192. doi: 10.1002/marc.201000105. [DOI] [PubMed] [Google Scholar]
- 170.Yang CA, Tan JPK, Cheng W, Attia ABE, Ting CTY, Nelson A, Hedrick JL, Yang YY. Nano Today. 2010;5:515–523. [Google Scholar]
- 171.Kim SH, Tan JPK, Fukushima K, Nederberg F, Yang YY, Waymouth RM, Hedrick JL. Biomaterials. 2011;32:5505–5514. doi: 10.1016/j.biomaterials.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 172.Fukushima K, Pratt RC, Nederberg F, Tan JPK, Yang YY, Waymouth RM, Hedrick JL. Biomacromolecules. 2008;9:3051–3056. doi: 10.1021/bm800526k. [DOI] [PubMed] [Google Scholar]
- 173.Chen W, Yang HC, Wang R, Cheng R, Meng FH, Wei WX, Zhong ZY. Macromolecules. 2010;43:201–207. [Google Scholar]
- 174.Zhang Z, Grijpma DW, Feijen J. J Controlled Release. 2006;111:263–270. doi: 10.1016/j.jconrel.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 175.Lee RS, Chen WH. React Funct Polym. 2011;71:455–462. [Google Scholar]
- 176.Venkataraman S, Lee AL, Maune HT, Hedrick JL, Prabhu VM, Yang YY. Macromolecules. 2013;46:4839–4846. [Google Scholar]
- 177.Chen W, Zou Y, Jia J, Meng F, Cheng R, Deng C, Feijen J, Zhong Z. Macromolecules. 2013;46:699–707. [Google Scholar]
- 178.Lee JS, Feijen J. J Controlled Release. 2012;161:473–483. doi: 10.1016/j.jconrel.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 179.Sanson C, Le Meins JF, Schatz C, Soum A, Lecommandoux S. Soft Matter. 2010;6:1722–1730. [Google Scholar]
- 180.Whitehead KA, Langer R, Anderson DG. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]