

Summary
Human papillomaviruses (HPVs) are non-enveloped DNA viruses that are highly tropic for mucosal and cutaneous epithelia. The HPV life cycle is tightly linked to epithelial cell differentiation, where HPVs only infect the basal proliferating keratinocytes, and progeny virus assembly and release only occurs in differentiated upper-layer keratinocytes. Therefore, human keratinocyte monolayer cultures provide a useful model to study the early stages of HPV infection. However, previous reports have shown some conflicting results of virus-host interactions during HPV entry, which may be partly attributable to the different cell culture models used to examine these steps of HPV infection. Thus, there is a need to have a standardized in vitro model system to study virus-host interactions during HPV entry. Here, we describe the 3 most widely accepted keratinocyte models for studying HPV infection: primary human foreskin keratinocytes (HFK), normal immortalized keratinocytes (NIKS), and transformed HaCaT keratinocytes. We also describe methods to genetically manipulate these cells, enabling the study of candidate host genes that may be important during HPV infection. Lastly, we will outline simple and robust methods to assay HPV infectivity, which can be used to determine whether knockdown or overexpression of a particular gene affects HPV entry.
Keywords: Keratinocyte, HFK, NIKS, HaCaT, Lentivirus, Transduction, Transfection, Puromycin, Papillomavirus, HPV
1. Introduction
Human papillomaviruses (HPVs) are causally associated with multiple human cancers, including 99% of cervical, 50% of other anogenital, and upwards of 25% of head and neck cancers (1-5). These important human pathogens are notably small, roughly 55 nm in diameter with only an 8 kb double-stranded circular DNA genome encoding usually 8-9 genes. They are non-enveloped, with only two capsid proteins, the major capsid protein L1 and the minor capsid protein L2 (6). HPVs have very strict species and tissue tropism; they productively infect only mucosal and cutaneous epithelia, and their life cycle is tightly linked to epithelial cell differentiation (7, 8). Papillomaviruses must first bind to the basement membrane of the epithelium, in order to interact with the basal proliferating keratinocytes they infect (9, 10). These viruses cannot infect differentiated keratinocytes, as cell cycle progression is required for HPV infection (11). This feature of the papillomavirus life cycle long prohibited the production of viruses in a laboratory setting, thus thwarting the study of virus-host interactions during HPV entry. In recent years, methods to produce papillomaviruses independent of epithelial cell differentiation have been developed, allowing progress to be made in the investigation of the early steps of HPV infection (12, 13).
Although our knowledge of HPV entry mechanisms has been greatly advanced since the development of robust HPV production methods, several publications show conflicting data regarding the mechanism of HPV internalization and endocytic trafficking in host cells (14-18). These discrepancies could be at least partly attributable to the different cell culture models used in these studies. For example, experiments conducted in non-keratinocyte 293 and COS-7 cells demonstrated a clathrin-dependent mode of HPV entry (14, 16), which contradicts with data obtained from HeLa and HaCaT cells, showing a clathrin-independent mechanism of HPV entry (18-20). We observed striking differences in HPV16 infectivity among different keratinocytes (Figure 1), and found that host autophagy inhibits HPV16 infection in epithelial cells but not in fibroblasts (data not shown) (21). This highlights the importance of using physiologically relevant cell culture models to study virus-host interactions during HPV entry.
Figure 1. HPV16-LucF infectivity is limited in primary keratinocytes.

The indicated cell lines were inoculated with 10,000 vge/cell of HPV16-LucF and incubated for 48 h. HPV16 infectivity and cell viability were measured by Bright-Glo Luciferase Assay System (Promega) and CellTiter-Glo Luminescent Cell Viability Assay (Promega), respectively. Infectivity data normalized to cell viability are shown as average relative luminescence units (RLU) from quadruplicate samples. The data shown here are from one representative of three independent experiments.
Protocols on how to culture the most widely accepted, physiologically relevant keratinocyte models for HPV infection studies are detailed in sections 2.1 and 3.1. Primary human foreskin keratinocytes (HFKs) are considered the most relevant native host cells for HPV infection. Ironically, however, these cells are extremely difficult to infect with HPV (21-23), indicating that they may have more robust host mechanisms to interfere with HPV entry relative to immortalized and transformed keratinocytes (Figure 1) (21). Indeed, we found that HFKs have higher levels of basal autophagy, recently recognized as a host defense pathway which inhibits HPV16 infection (21). Furthermore, HFKs may have altered heparan-sulfate proteoglycan (HSPG) modifications, which prevent HSPG-facilitated furin cleavage of L2, a prerequisite for infectious entry of papillomaviruses (22). One method reported to overcome the challenges of infecting HFKs is to include exogenous furin in the inoculum, or to pretreat the virus prep with furin, in order to facilitate the required furin cleavage of L2 and enhance infectious entry (22). HFKs usually do not require feeder cells and growth medium is commercially available, however these cells should not be cultured for longer than 4 weeks, as significant morphological changes occur followed by growth arrest by 6 weeks.
Normal immortalized keratinocytes from skin (NIKS) is a spontaneously immortalized near diploid cell line showing normal growth and differentiation. Thus, NIKS are still considered physiologically relevant yet are much more permissive to HPV infection compared to HFKs (Figure 1, ~400-fold increase in infectivity) (21, 23, 24). In addition, as immortalized cells, establishment of genetically modified stable cell lines is feasible with NIKS. NIKS are rather cumbersome to culture, compared to HFK or HaCaT, as they require feeder cells and various growth supplements, many of which should be freshly prepared to support healthy growth without spontaneous differentiation. The HaCaT keratinocytes, currently the most widely used in HPV research, are the most convenient and cost-effective cells to culture. While HaCaTs might still be very useful for many initial experiments, data obtained from these cells should be corroborated with normal keratinocytes, such as NIKS of HFK, in several occasions.
To determine whether a particular gene of interest is important during HPV infection, methods to manipulate specific gene expression using RNAi and overexpression are outlined in sections 2.2 and 3.2. Lentiviral vectors are routinely used to deliver plasmids encoding shRNAs or ORFs into keratinocytes including normal keratinocytes HFK and NIKS. In addition to lentiviral transduction, transfection of keratinocytes using recently introduced reagents such as TransIT 2020 and X-tremeGENE HP has proven effective with reasonable transfection efficiencies (~50%) (data not shown). Establishment of specific keratinocyte lines is possible by antibiotic resistance selection and is necessary when a pathway of interest is affected by lentivirus infection or transfection. For example, when studying an innate immune response such as autophagy, we found that lentivirus transduction alone can induce this pathway and confound results (data not shown).
HPV pseudovirions containing luciferase or fluorescent protein genes offer convenient and quantifiable methods to determine relative HPV infectivity. This is instrumental in determining whether a particular gene has a role in the early steps of HPV infection. We routinely use two different types of luciferase reporters, firefly luciferase (FL) and renilla luciferase (RL), and green fluorescence protein (GFP) to produce HPV16 reporter pseudovirions. The pseudovirion hpv16-LucF, developed by Chris Buck (25), expresses both FL and GFP in infected cells at high levels and thus is useful when monitoring infection in HFKs, which exhibit very low levels of infection. We developed hpv16-phRL pseudovirions containing RL only and HPV16RL containing RL inserted into the HPV16 genome. This offers the advantage of comparing luciferase reporter virions containing HPV genomes (HPV16RL) to those containing only the reporter gene (hpv16-phRL). These HPV pseudovirions and chimeric virions are useful when studying viral DNA interactions with host keratinocytes during entry.
2. Materials
2.1 Keratinocyte culture
2.1.1 Primary Human Foreskin Keratinocytes (HFKs)
Human Epidermal Keratinocytes, neonatal (HEKn, aka “HFK”) (Invitrogen Cat. # C-001-5C) (see Note 1)
- EpiLife medium supplemented with Human Keratinocyte Growth Supplement (HKGS):
HKGS (Gibco/Invitrogen Cat. # S-001-5) 5 mL EpiLife with 60 μM Calcium (Gibco/Invitrogen Cat. # MEP1500CA) 500 mL - 0.02% EDTA/PBS:
0.5 M EDTA 526 μL 1X PBS (MediaTech/Fisher Cat. # MT-20-031-CV) 500 mL 0.05% trypsin/EDTA (HyClone/Fisher Cat. # SH30236.01)
- DMEM containing 10% NCS (Newborn Calf Serum):
DMEM w/high glucose (HyClone/Fisher Cat. # SH30022.FS) 500 mL NCS (HyClone/Fisher Cat. # SH30401.01) 50 mL
2.1.2 Normal Immortalized Keratinocytes from Skin (NIKS)
The materials listed below have been adapted from the NIKS culture protocol published by Paul Lambert (26).
DMEM containing 10% NCS (2.1.1, step 5)
1X PBS without calcium and magnesium (MediaTech/Fisher Cat. # MT-20-031-CV)
0.25% trypsin/EDTA (HyClone/Fisher Cat. # SH30042.01)
- 50X Mitomycin C: Wear gloves!
- Use a syringe needle to add 2 mL HBES (2.1.2, step 6) to a 2 mg vial of Mitomycin C (Sigma Cat. # M4287-2MG)
- Transfer to conical tube and bring to 10 mL with HBES
- Filter sterilize and store 1 mL aliquots at −20°C
- E-Complete medium:
DMEM w/high glucose (HyClone/Fisher Cat. # SH30022.FS) 375 mL Ham’s Nutrient Mixture F-12 (HyClone/Fisher Cat. # SH30026.01) 125 mL Fetal Bovine Serum (FBS) (MediaTech/Fisher Cat. # MT-35-011-CV) 12.5 mL 100X Hydrocortisone 5 mL 100X Cholera Toxin 5 mL 100X Adenine 5 mL 100X Epidermal Growth Factor (EGF) 5 mL 100X Insulin (made fresh) 5 mL - HEPES Buffered Earle’s Salts (HBES):
Earle’s Salts 100 mL 1 M HEPES, pH 7.3 (Amresco Cat. # J848) 25 mL Sterile H2O to 1 L total volume - Earle’s Salts:
NaCl 128 g KCl 8 g NaHCO3 74 g NaH2PO4 H2O 2.5 g MgSO4•7H2O 4 g Fe(NO3)3•9H2O 0.002 g Phenol Red 0.1 g Sterile H2O to 2 L total volume *Filter sterilize and store at RT - 100X Hydrocortisone:
- Make 5 mg/mL hydrocortisone stock solution: Add 5 mL cold 100% ethanol to 25 mg vial hydrocortisone (Calbiochem/EMDMillipore Cat. # 3867). Store at −20°C for future use.
- Add 0.8 mL of 5 mg/mL hydrocortisone stock to 100 mL HBES containing 5% FBS (95 mL HBES + 5 mL FBS).
- Filter sterilize and store 10 mL aliquots at −20°C
- 100X Cholera Toxin:
- Make 10 μM cholera toxin stock solution: Use syringe needle to add 1.2 mL sterile water to a 1 mg vial cholera toxin (Sigma Cat # C8052-1MG). Store at 4°C for future use.
- Make 100X cholera toxin from 10 μM stock: Add 100 μL of 10 μM cholera toxin stock to 100 mL HBES containing 0.1% BSA (dissolve 0.1 g BSA into 100 mL HBES by shaking vigorously).
- Filter sterilize and store 50 mL aliquots at 4°C
- 100X Adenine:
- Dissolve 242 mg adenine (Sigma Cat. # A9795) in 100 mL 0.05 M HCl by stirring for 1 hr
- Filter sterilize and store 10 mL aliquots at −20°C
- 100X Epidermal Growth Factor (EGF):
- Add 20 mL sterile water to a 200 μg vial human recombinant EGF (R&D Systems, Cat. # 236-EG-200)
- Transfer to conical tube and add 180 mL HBES containing 0.1% BSA (dissolve 0.1g BSA into 100 mL HBES by shaking vigorously)
- Filter sterilize and store 10 mL aliquots at −20°C
- 100X Insulin: Prepare immediately before use and never freeze!!
- Dissolve 12.5 mg insulin (Calbiochem/EMD Millipore Cat. # 407709-50MG) in 25 mL 0.005 M HCl
- Add 2-3 mL FBS to a filter and vacuum it through the filter, discarding the flow-through
- Filter sterilize insulin using the above FBS-blocked filter
2.1.3 HaCaT Keratinocytes
- E-medium containing 5% FBS:
DMEM w/high glucose (HyClone/Fisher Cat. # SH30022.FS) 375 mL Ham’s Nutrient Mixture F-12 (HyClone/Fisher Cat. # SH30026.01) 125 mL Fetal Bovine Serum (FBS) (MediaTech/Fisher Cat. # MT-35-011-CV) 25 mL 1X PBS without calcium and magnesium (MediaTech/Fisher Cat. # MT-20-031-CV)
0.02% EDTA/PBS (section 2.1.1, step 3)
0.25% trypsin/EDTA (HyClone/Fisher Cat. # SH30042.01)
2.2 Gene Knockdown and Overexpression in Keratinocytes
2.2.1 Production of Lentiviruses for Transducing Keratinocytes
- DMEM containing 10% FBS:
DMEM w/high glucose (HyClone/Fisher Cat. # SH30022.FS) 500 mL Fetal Bovine Serum (FBS, MediaTech/Fisher Cat. # MT-35-011-CV) 50 mL Opti-MEM Reduced Serum Medium (Gibco/Invitrogen Cat. # 31985070)
- Lentivirus packaging plasmids:
- pMDG.2 (VSV-G) (Addgene Plasmid # 12259)
- pCMV-deltaR8.2 (Addgene Plasmid # 12263)
- Proviral plasmid of choice
- Polyethylenimine (PEI)
- Dissolve Linear PEI (Polysciences Cat. # 23966-2) in H2O heated to 80°C to yield 1 mg/mL solution.
- Allow solution to cool to RT.
- Adjust pH to 7.0 with 5 M HCl.
- Filter sterilize and store at −80°C. Can also be stored at 4°C for up to 4 months.
0.45 μm PVDF syringe filters, 13 mm diameter (Fisherbrand Cat. # 09-720-4)
Hank’s Buffered Saline Solution (HBSS, MediaTech/Fisher Cat. # MT-21-023-CV)
Bovine Serum Albumin (BSA, Amresco Cat. # 0332)
Ultra-Clear Thinwall ultracentrifuge tubes for SW41Ti rotor, 13.2 mL, 14×89 mm (Beckman Coulter Cat. # 344059)
20% sucrose/HBSS: Dissolve 20 g sucrose into 100 mL 1X HBSS and filter sterilize.
2.2.2 Transduction of Keratinocytes
Lentiviruses: as prepared in section 2.2.1
Keratinocytes and keratinocyte culture medium (section 2.1)
2.2.3 Transfection of Keratinocytes
Keratinocytes and keratinocyte growth medium (section 2.1)
Opti-MEM Reduced Serum Medium (Gibco/Invitrogen Cat. # 31985070)
X-tremeGENE HP DNA Transfection Reagent (Roche Cat. # 06366244001): for transfection of HFK and NIKS
TransIT®-2020 Transfection Reagent (Mirus Bio Cat. # MIR 5404): for transfection of HaCaT
DNA to be transfected: prepared as desired
2.2.4 Selection of puromycin-resistant cells
Keratinocytes transduced or transfected with puromycin-resistance gene
10 mg/mL puromycin: Add 10 mL sterile water to 100 mg vial puromycin dihydrochloride (MP Biomedicals Cat. # 210055280). Store aliquots at −20°C. Vortex well upon thawing.
2.3 Human Papillomavirus Infectivity Assay
2.3.1 Luciferase Reporter Assay: hpv16-LucF, hpv16-phRL, HPV16RL, or other luciferase-containing HPV
hpv16-LucF, hpv16-phRL, HPV16RL, or other luciferase-containing HPV: Prepared as described (12, 27, 28) (see Note 2)
Bright-Glo™ Luciferase Assay System (Promega Cat. # E2620)
Renilla-Glo® Luciferase Assay System (Promega Cat. # E2720)
CellTiter-Glo® Luminescent Cell Viability Assay (Promega Cat. # G7572)
DMEM (HyClone/Fisher Cat. # SH30022.FS)
Opaque 96-well plates (Life Science Products/VWR Cat. # 89093-598)
GloMax®-Multi+ Detection System with Instinct Software (Promega), or other comparable luminometer
2.3.2 Fluorescence Microscopy: hpv16-LucF, hpv16-EGFP, HPV16GFP, or other fluorescent protein gene-containing HPV
hpv16-LucF, hpv16-EGFP and HPV16GFP
Fluorescence microscope
Flow cytometer
Optional: SYTOX® Dead Cell Stain (Invitrogen)
2.3.3 Reverse Transcription-Quantitative PCR (RT-qPCR): HPV containing full-length genome
HPV containing full-length genome, prepared as described (12, 13, 27).
RNeasy mini kit (Qiagen Cat. # 74106)
RNase-free DNase set (Qiagen Cat. # 79254)
Oligo d(T)12-18 Primer (Invitrogen Cat. # 18418012)
SuperScript II Reverse Transcriptase (Life Sciences Cat. # 18064-014)
Primers for amplification of HPV genes: for HPV16 W12 genome, primers to amplify early genes have been published (11)
Hard-Shell® Thin-Wall 96-Well Skirted Plates (Bio-Rad Cat. # HSP-9901) or comparable 96-well plates for qPCR
Bio-rad CFX Connect™ Real-Time PCR Detection System (Bio-Rad Cat. # 185-5200) or comparable qPCR machine
FastStart Universal SYBR Green Master Mix (Rox) (Roche Applied Science Cat. # 04913850001)
3. Methods
3.1 Keratinocyte culture
3.1.1 Primary Human Foreskin Keratinocytes (HFKs)
Maintain HFKs in EpiLife medium supplemented with HKGS at 37°C / 5% CO2. Change medium every other day when cells are sparse, and every day as cell approach 50% confluency. Passage cells when they reach 80% confluency. Follow the protocol below to passage HFKs. (see Note 3)
Briefly wash cells once with 0.02% EDTA/PBS. Incubate in 0.05% trypsin/EDTA for 6 min at 37°C / 5% CO2.
Rock and tap the dish to dislodge the cells, then add DMEM containing 10% NCS to neutralize the trypsin (see Note 4). Pipet up and down to obtain a single cell suspension.
Pellet the cells by centrifugation in swinging bucket rotor at 200 g for 5 min.
Resuspend cell pellet in EpiLife medium containing HKGS, and plate as desired. If HFK cells are seeded at a density of 1×104cells/cm2 they reach confluency in 3-4 days (see Note 5).
3.1.2 Normal Immortalized Keratinocytes from Skin (NIKS)
The NIKS culture protocol described below is modified from the previously described protocol for epithelial cell culture (26). Maintain NIKS in E-complete medium and co-culture with growth-arrested 3T3 mouse fibroblasts (aka “feeder” cells) (see Note 6) at 37°C / 5% CO2. Note that preparation of 3T3s for NIKS co-culture requires at least 2-4 hrs. Change the medium on NIKS every 24-48 hrs (see Note 7). Confluency is a critical issue for NIKS. NIKS should be passaged prior to reaching 80% confluency (as soon as colonies begin touching one another), or when cells in the middle of the colonies appear unhealthy (large, flat, “fried egg” phenotype). Furthermore, NIKS do not proliferate well when too sparse, so never plate below 10% confluency. The following protocol details maintenance and preparation of 3T3 cells for use as feeders in NIKS cultures, as well as NIKS passaging.
- Maintain 3T3 cultures for future use in NIKS co-cultures. 3T3s are maintained in DMEM containing 10% NCS at 37°C / 5% CO2. Frequent medium changes are not necessary for these cells. Passage cells prior to reaching 100% confluency. To passage 3T3 cells:
- Wash once with 1X PBS. Incubate in 0.25% trypsin for 2 min at 37°C / 5% CO2.
- Add DMEM containing 10% NCS to neutralize trypsin and dilute as desired. If cells are split 1:8 from a confluent dish, they typically reach 90% confluency in 2 days. (see Note 8)
- Prepare 3T3 feeder cells for NIKS co-culture. The desired confluency of 3T3 feeder cells in NIKS co-cultures is ~25%, thus 1 confluent dish of 3T3 cells will yield 4 dishes of feeder cells for NIKS co-cultures. The following is written for (1) 10 cm dish of 3T3s.
- Dilute 100 μL of 50X mitomycin C into 5 mL DMEM containing 10% NCS. Aspirate medium from (1) 10 cm dish of 3T3s and add the diluted mitomycin C.
- Incubate for 2-4 hrs at 37°C / 5% CO2.
- Wash cells twice with 10 mL 1X PBS. Incubate in 0.25% trypsin for 2 min at 37°C / 5% CO2 (see Note 9).
- Add either DMEM containing 10% NCS or E-complete to neutralize trypsin. If 3T3 feeders will be added to NIKS culture immediately, then suspend cells in E-complete. 3T3 feeders can be maintained up to 24 hrs after mitomycin C treatment and before adding to NIKS culture if kept in DMEM containing 10% NCS.
- Remove 3T3 feeder cells from NIKS cultures to be passaged. As NIKS are far more adherent to tissue culture plastic than mitomycin C-treated 3T3s, the feeders can easily be removed without dislodging NIKS, as follows.
- Wash NIKS/feeder co-culture once with 0.02% EDTA/PBS. Incubate in 0.05% trypsin/EDTA for 60 sec in 37°C / 5% CO2 incubator (see Note 10).
- Rock and tap the dish thoroughly to dislodge the feeders. Check under the microscope to observe that all feeders are lifted and NIKS are still adhered. NIKS should appear rounded but stay adhered to the dish.
- Remove dislodged feeders by washing twice with 1X PBS.
Incubate NIKS in 0.25% trypsin for 8 min at 37°C / 5% CO2.
Rock and tap the dish to dislodge NIKS.
Add E-complete to neutralize the trypsin and pipet up and down thoroughly to obtain a single cell suspension.
Dilute NIKS as desired, and plate onto new dishes containing feeders, or add feeders to NIKS in suspension and plate together. (see Note 11)
3.1.3 HaCaT Keratinocytes
Maintain HaCaTs in E-medium containing 5% FBS at 37°C / 5% CO2. Change medium every 2 days, and split cells when they reach 80% confluency. Passage HaCaTs as follows:
Wash HaCaTs once with 1X PBS. Incubate in 0.02% EDTA/PBS for 15 min at 37°C / 5% CO2.
Aspirate 0.02% EDTA/PBS and incubate in 0.25% trypsin/EDTA for 6 min at 37 °C.
Rock and tap the dish to dislodge HaCaTs.
Add E-medium containing 5% FBS to neutralize trypsin. Pipet up and down to collect cells in single-cell suspension.
Dilute cells as desired. If cells are split 1:8 they will reach confluency in 2-3 days.
3.2 Gene Knockdown and Overexpression in Keratinocytes
3.2.1 Production of Lentiviruses for Transducing Keratinocytes
As keratinocytes have historically been difficult to transfect without cytotoxicity, lentiviral transduction is commonly used to deliver DNA constructs for shRNA knockdown or ORF overexpression. This protocol utilizes PEI as a transfection reagent to deliver lentivirus-packaging plasmids into 293FT cells. This protocol is written for transfection of one 10 cm dish. Scale as necessary.
- Maintain 293FT cells for future use. 293FT cells are maintained in DMEM containing 10% FBS at 37°C / 5% CO2 (see Note 12), and should be passaged prior to reaching 90% confluency (see Note 13). Frequent medium changes are not recommended for these cells. Care should be taken not to disturb the cell monolayer, as 293FTs are only weakly adherent and will become dislodged easily by physical force (i.e. pipetting). To passage 293FTs:
- Wash cells once with 1X PBS. Incubate in 0.25% trypsin/EDTA for 5 min at 37°C / 5% CO2.
- Rock and tap the dish to dislodge cells.
- Add DMEM containing 10% FBS to neutralize trypsin, and pipet up and down to obtain a single-cell suspension.
- Dilute cells as desired. If a confluent dish split 1:12 they typically reach 90% confluency in 2 days.
One day prior to transfection, plate 4.5 × 106 293FT cells per 10 cm dish (to be 60-80% confluent next day) in DMEM containing 10% FBS.
- Transfect 293FT cells:
- Dilute DNA into 1 mL Opti-MEM and mix well:
- 5.3 μg pMDG.2 (VSV-G)
- 8 μg pCMV-deltaR8.2
- 10.7 μg proviral plasmid (see Note 14)
- Dilute 72 μL of 1 μg/μL PEI into 1 mL Opti-MEM and mix well.
- Combine diluted DNA and diluted PEI, and immediately vortex for 5 sec.
- Incubate at RT for 15 min.
- Add transfection mixture to 10 mL DMEM containing 10% FBS and mix by pipetting.
- Aspirate medium from 293FT cells and add above transfection mixture.
- Incubate at 37°C / 5% CO2 overnight.
Replace transfection medium with fresh Opti-MEM (cells should not be incubated in transfection mixture for longer than 18 hrs). (see Note 15)
- Harvest lentivirus at 48 - 72 hrs post transfection:
- Harvest cells and lentivirus-containing medium by pipetting up and down (no need to trypsinize). (see Note 16)
- Pellet cell debris by centrifugation at 3200 g for 5 min.
- Collect supernatant and pass through 0.45 μm PVDF syringe filter to remove remaining cell debris.
- Store 1 mL aliquots at −80°C or (optional) concentrate/purify lentiviruses (see Note 17). Lentiviruses should not be frozen and thawed more than 3 times.
- If desired, lentiviruses can be concentrated as follows (see Note 18):
- Add 5 mL of filtered lentivirus supernatant to SW41Ti ultracentrifuge tube.
- Underlay 2 mL of 20% sucrose/HBSS using 2 mL pipet, being careful not to mix layers.
- Add remaining lentivirus supernatant to top, being careful not to disturb the interface.
- Balance tubes with HBSS.
- Ultracentrifuge at 65,000 g for 2 hrs at 4°C.
- Pour off supernatant into waste beaker containing 10 % bleach and let tubes sit upside down over a kimwipe for 5 min. Remove excess liquid from side of tube with kimwipe or by aspiration.
- Resuspend pellet with desired volume of HBSS containing 1 % BSA (usually 50X concentration – 200 μL) by pipetting up and down vigorously (avoid frothing!). Vortex low speed for 30 min at 4°C to fully resuspend the virus pellet.
- Transfer to 1.5 mL tube, vortex and centrifuge max speed for 30 sec.
- Transfer supernatant to new tube and store aliquots at −80°C.
3.2.2 Transduction of Keratinocytes
HFK and NIKS cells transduce with relatively high efficiency, however HaCaT cells exhibit low transduction efficiency. This protocol is written for HFK and NIKS only.
One day prior to transduction, plate keratinocytes at a density of 1×104 cells/cm2.
Thaw lentivirus on ice (see Note 19), and dilute as desired into appropriate culture medium (see Note 20).
Add diluted lentivirus to keratinocytes and incubate overnight at 37°C / 5% CO2.
Change medium next day. (see Note 21)
Assay cells at 48-72 hrs post transduction.
3.2.3 Transfection of Keratinocytes
We found that keratinocytes can be transfected with reasonable efficiencies with the following reagents (~50%, data not shown). Follow the manufacturer’s instructions for the following reagents. The optimal ratios of transfection reagent to DNA are listed below, however further optimization may be necessary.
| HFK and NIKS: | X-tremeGENE HP DNA Transfection Reagent (Roche) |
| Optimal transfection reagent to DNA ratio: 2:1 | |
| HaCaT: | TransIT®-2020 Transfection Reagent (Mirus Bio) |
| Optimal transfection reagent to DNA ratio: 3:1 |
3.2.4 Selection of puromycin-resistant cells
We routinely use puromycin resistance selection to generate stable keratinocyte lines. The following outlines our procedure for puromycin resistance selection.
- Add puromycin-containing growth medium to cells at 48 hrs post transduction or transfection (above). Optimal puromycin concentrations:
- HFK: 1.0 μg/mL
- NIKS: 1.5 μg/mL
- HaCaT: 2.5 μg/mL
Culture cells according to standard protocol (section 3.1), replenishing puromycin-containing medium every other day. Puromycin will kill non-transduced/transfected cells in 2-3 days.
To generate stable cell lines, continue selection process for 1-2 weeks post transduction, or 2-3 weeks post transfection.
3.3 Human Papillomavirus Infectivity Assays
3.3.1 Luciferase Reporter Assay: hpv16-LucF, hpv16-phRL, HPV16RL, or other luciferase-containing HPV
One day before infection, plate 4×103 keratinocytes/well of a 96-well plate (see Note 22). Plate in quadruplicate for each sample for statistical analysis.
Next day, inoculate keratinocytes with 1,000-10,000 vge/cell (MOI 2-20) hpv16-LucF, hpv16-phRL, HPV16RL, or other luciferase-containing HPV.
- At 48 hrs post infection, perform luciferase assay and cell viability assay to determine relative infectivity and viability, respectively (see Note 23):
- Thaw and equilibrate Bright-Glo™ (FL assay) or Renilla-Glo™ (RL assay), and CellTiter-Glo® (viability assay) reagents at RT.
- Make master mixes: Combine Bright-Glo™ or Renilla-Glo™, and CellTiter-Glo® reagents with an equal volume of DMEM at RT.
- Aspirate culture medium from wells and add 50-60 μL of desired master mix per well.
- Incubate at RT for 2-3 min for Bright-Glo™ or 10 min for Renilla-Glo™ and CellTiter-Glo®.
- Transfer to opaque 96-well plate, being cautious to avoid introducing bubbles.
- Read luminescence with GloMax®-Multi+ Detection System or comparable luminometer.
- Normalize luciferase activity to cell viability.
3.3.2 Fluorescence Microscopy: hpv16-LucF, hpv16-EGFP, HPV16GFP, or other fluorescent protein gene-containing HPV
One day before infection, plate 1×104 keratinocytes/cm2 onto 24-well or 12-well plate.
Next day, inoculate keratinocytes with 1,000-10,000 vge/cell (MOI 2-20) hpv16-LucF, hpv16-EGFP, HPV16GFP, or other fluorescent protein gene-containing HPV.
Check expression of fluorescence protein in infected cells using fluorescence microscope.
At 48 hrs post infection, determine the percentage of cells infected by flow cytometric quantification of fluorescent protein expression. Dead cells can be excluded by staining with SYTOX® Dead Cell Stain, according to the manufacturer’s instructions.
3.3.3 Reverse Transcription-Quantitative PCR (RT-qPCR): HPV containing full-length genome
One day before infection, plate 1×105 keratinocytes/well of a 6-well plate.
Next day, inoculate keratinocytes with 1,000-10,000 vge/cell (MOI 2-20) HPV containing full-length genome.
At 48 hrs post infection, extract total RNA from keratinocytes using RNeasy mini kit and RNase-free DNase set, according to the manufacturer’s instructions for on-column DNase treatment.
- Reverse transcribe total RNA to generate first strand complementary DNA (cDNA) using oligo d(T)12-18 primers:
- Set up oligo d(T) priming reaction:
RNA sample (<25 μg) 10 μL 100 μM oligo d(T) primer 1 μL - Incubate at 70°C for 10 min, then cool to 4°C
- Set up cDNA synthesis reaction:
Priming reaction mix 11 μL 5X First-strand reaction buffer 4 μL 0.1 M DTT 2 μL 10 mM dNTP mix 1 μL Superscript II Reverse Transcriptase 1 μL Nuclease-free H2O 1 μL - Incubate at 42°C for 1 hr, then cool to 4°C
- Store at −20°C for future use
- Set up RT-qPCR reaction mixture. Primer sequences for HPV16 early genes are published (11).
- 0.5 μM forward primer
- 0.5 μM reverse primer
- 50 ng cDNA template
- 10 μL FastStart Universal SYBR Green Master (Rox)
- Nuclease-free H2O to 20 μL total volume
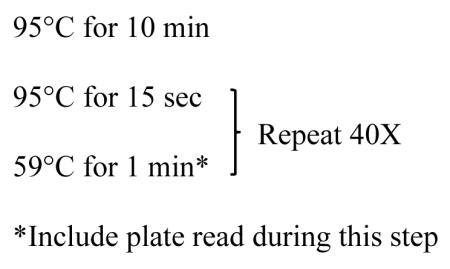
Perform qPCR using the following temperature protocol:
Acknowledgements
We thank Paul Lambert and Denis Lee for technical assistance regarding NIKS cell culture, and for providing NIKS and HaCaT keratinocytes, as well as the 293FT packaging cells. We acknowledge John Schiller for providing pLucF and p16Shell plasmids, and Jerry Schaack for providing pMDG.2 and useful suggestions for lentivirus production and purification. We also thank Paul Lambert, Zhaohui Qian, and members of the Pyeon laboratory for useful support and suggestions.
4. Notes
Invitrogen also sells HEKp, which are HEKn pooled from multiple donors. This protocol will work with either HEKn or HEKp. It is also works well with HFK cells purchased from Lonza (NHEK-Neo, Lonza Cat. # 192906). Among the keratinocyte medium we tested, HFK cultures appear most healthy when cultured in EpiLife medium containing HKGS (data not shown).
hpv16-phRL and HPV16RL virions are packaged in 293FT cells as previous described (13). hpv16-phRL pseudovirions contain the phRL-SV40 plasmid (Promega), and HPV16RL virions contain a chimeric HPV DNA, generated by insertion of the hRL reporter gene from phRL-SV40 (Promega) into the L2 and 5′ L1 genes of the HPV16 W12 genome (28). For more information regarding these constructs, contact Dohun Pyeon at dohun.pyeon@ucdenver.edu.
Alternatively, HFKs can be cultured following the NIKS culture protocol (see section 3.1.2). This method of culturing HFKs has been reported to extend their lifetime to more than twice the number of passages compared with culturing in EpiLife medium supplemented with HKGS (29). Importantly, we found that HPV16-LucF infectivity of HFKs is dramatically enhanced when they are cultured according to the NIKS protocol (data not shown). We do not currently know why HPV16 infectivity is enhanced, but we speculate that the serum in the E-complete medium may have growth factors and HSPGs which aid in the entry of HPV16 into keratinocytes (Ozbun, MA, personal communication).
Any serum-containing medium can be used here to neutralize trypsin, as the cells will be pelleted and resuspended in EpiLife. EpiLife medium supplemented with HKGS will not effectively neutralize trypsin.
Each lot of HFKs varies slightly in doubling time, as each lot is obtained from a different donor, or set of multiple donors. Therefore, seeding density for each batch may need optimization.
If desired, NIKS cells can be cultured without feeders in reduced-calcium E-complete medium (Paul Lambert, personal communication). We found that even under reduced-calcium environment, a large percentage of NIKS appeared to differentiate within 24 hrs, as their morphology changed from cuboidal to elongated, and these elongated cells did not appear to divide. For this reason, if an experiment requires the absence of feeders, we limit the feeder-free culture period to 2-3 hrs.
3T3 feeders in the NIKS co-culture will additionally utilize a significant amount of nutrients from the medium, so frequent medium changes are necessary regardless of NIKS confluency.
Because feeders are needed so often, we maintain at least (2) or (3) 10 cm dishes of 3T3s of varying confluency. From a confluent dish, we recommend routinely splitting at a ratio of 1:8, 1:4, and 1:2.
If NIKS will be added directly to this dish of feeders, trypsinization is not necessary.
Alternatively, feeders can be removed by pipetting up and down after incubation with 0.02% EDTA/PBS for 1-2 min at RT. Harsh pipetting necessary to thoroughly dislodge feeders sometimes results in physical damage to adhered NIKS, thus we prefer to remove feeders with trypsin to avoid harsh pipetting.
Upon addition to NIKS culture, 3T3 feeders should be replenished every 2 days to maintain NIKS in a healthy, proliferating state. If NIKS cells appear stressed, flattened and larger morphology, and a clear nuclear membrane and stress granules (“fried egg”), replacing the feeders in fresh E-complete often recovers these cells.
293FT cells can be cultured in DMEM containing 10% NCS rather than FBS. Though the cells appear to proliferate very well in NCS-containing medium, we find that culturing them in FBS-containing medium increases transfection efficiency.
293FT cells easily transform further in over-confluent conditions. We find that when 293FT cells are kept in an over-confluent state, they become less adherent.
Optional: add 1 μg lentiviral GFP expression plasmid (such as TurboGFP) to visualize transduced cells and calculate transduction efficiency.
We obtain significantly higher titers of lentivirus when packaging is carried out in Opti-MEM reduced-serum medium instead of DMEM containing 10% FBS. We speculate that even though FBS in the medium increases transfection efficiency (see Note 12), it later has an inhibitory effect on virus packaging. 293FT cells cultured in Opti-MEM will display significant cytotoxicity, however production of infectious lentivirus is enhanced.
Biohazard warning. Lentiviruses are HIV-derivative viruses and they must be handled in a BSL2+ environment. You must contact your institution’s Bio-Safety office to receive permission and specific instructions on how to handle these viruses. We use 10% bleach to sterilize work areas, followed by water and 70% ethanol to avoid corrosion of stainless steel surfaces.
Lentiviruses can be stored at 4°C for up to 3 days without significant loss of titer.
Further purification/concentration of lentiviruses by ultracentrifugation is recommended if they will be used to transduce HFKs, as HFKs do not tolerate Opti-MEM very well. Note that ultracentrifugation of lentiviruses results in a significant loss of overall titer.
Rapid thawing and multiple freeze-thaw cycles will result in significant loss of virus titer.
Typically, lentiviruses are diluted 1:10 into culture medium. However, if it is suspected that the titer is lower than normal, lentiviruses can be diluted 1:1 with culture medium. If lentivirus was concentrated, adjust volume according to concentration factor.
If desired, lentiviruses can be removed 4 hrs after inoculation, as most lentiviruses will have adsorbed to the cells within a few hours. We have not observed any significant loss in transduction efficiency by replacing infection medium after 4 hrs.
Keratinocytes may be cultured in sterile, opaque 96-well plates if desired, however it may be preferable to culture cells in clear 96-well plates in order to visualize the cell condition throughout the experiment. In this case, the lysate can simply be transferred to an opaque 96-well plate immediately prior to measuring luminescence.
The CellTiter-Glo® cell viability assay usually shows very high signal (>107 RLU for 96-well plate), which can leak into neighboring wells of the Bright-Glo™ infectivity assay, thereby skewing the infectivity data. This can be avoided by performing the Bright-Glo™ assay prior to the CellTiter-Glo® assay, or by separating the two assays by several wells or on separate plates.
References
- 1.zur Hausen H. Papillomavirus infections--a major cause of human cancers. Biochimica et biophysica acta. 1996;1288:F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 2.Gillison ML, Shah KV. Human papillomavirus-associated head and neck squamous cell carcinoma: mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. Current opinion in oncology. 2001;13:183–188. doi: 10.1097/00001622-200105000-00009. [DOI] [PubMed] [Google Scholar]
- 3.zur Hausen H. Viruses in human cancers. European journal of cancer (Oxford, England : 1990) 1999;35:1174–1181. doi: 10.1016/s0959-8049(99)00113-6. [DOI] [PubMed] [Google Scholar]
- 4.Burd EM. Human papillomavirus and cervical cancer. Clinical Microbiology Reviews. 2003;16:1–17. doi: 10.1128/CMR.16.1.1-17.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillison ML, Lowy DR. A causal role for human papillomavirus in head and neck cancer. Lancet. 2004;363:1488–1489. doi: 10.1016/S0140-6736(04)16194-1. [DOI] [PubMed] [Google Scholar]
- 6.Modis Y, Trus BL, Harrison SC. Atomic model of the papillomavirus capsid. The EMBO Journal. 2002;21:4754–4762. doi: 10.1093/emboj/cdf494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nature reviews. Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 8.Stubenrauch F, Laimins LA. Human papillomavirus life cycle: active and latent phases. Seminars in cancer biology. 1999;9:379–386. doi: 10.1006/scbi.1999.0141. [DOI] [PubMed] [Google Scholar]
- 9.Joyce JG, Tung JS, Przysiecki CT, et al. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. The Journal of biological chemistry. 1999;274:5810–5822. doi: 10.1074/jbc.274.9.5810. [DOI] [PubMed] [Google Scholar]
- 10.Giroglou T, Florin L, Schafer F, et al. Human papillomavirus infection requires cell surface heparan sulfate. Journal of Virology. 2001;75:1565–1570. doi: 10.1128/JVI.75.3.1565-1570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pyeon D, Pearce SM, Lank SM, et al. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathogens. 2009;5:e1000318. doi: 10.1371/journal.ppat.1000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buck CB, Pastrana DV, Lowy DR, et al. Efficient intracellular assembly of papillomaviral vectors. Journal of Virology. 2004;78:751–757. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pyeon D, Lambert PF, Ahlquist P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:9311–9316. doi: 10.1073/pnas.0504020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bousarghin L, Touzé A, Sizaret P-Y, et al. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. Journal of Virology. 2003;77:3846–3850. doi: 10.1128/JVI.77.6.3846-3850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Day PM, Lowy DR, Schiller JT. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology. 2003;307:1–11. doi: 10.1016/s0042-6822(02)00143-5. [DOI] [PubMed] [Google Scholar]
- 16.Abban CY, Bradbury NA, Meneses PI. HPV16 and BPV1 infection can be blocked by the dynamin inhibitor dynasore. American journal of therapeutics. 2008;15:304–311. doi: 10.1097/MJT.0b013e3181754134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laniosz V, Dabydeen SA, Havens MA, et al. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is brefeldin a sensitive. Journal of Virology. 2009;83:8221–8232. doi: 10.1128/JVI.00576-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schelhaas M, Shah B, Holzer M, et al. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathogens. 2012;8:e1002657. doi: 10.1371/journal.ppat.1002657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spoden G, Freitag K, Husmann M, et al. Clathrin- and caveolin-independent entry of human papillomavirus type 16--involvement of tetraspanin-enriched microdomains (TEMs) PLoS ONE. 2008;3:e3313. doi: 10.1371/journal.pone.0003313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spoden G, Kühling L, Cordes N, et al. Human papillomavirus types 16, 18, and 31 share similar endocytic requirements for entry. Journal of Virology. 2013;87:7765–7773. doi: 10.1128/JVI.00370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffin LM, Cicchini L, Pyeon D. Human papillomavirus infection is inhibited by host autophagy in primary human keratinocytes. Virology. 2013;437:12–19. doi: 10.1016/j.virol.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day PM, Lowy DR, Schiller JT. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. Journal of Virology. 2008;82:12565–12568. doi: 10.1128/JVI.01631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozbun MA. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. Journal of Virology. 2002;76:11291–11300. doi: 10.1128/JVI.76.22.11291-11300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen-Hoffmann BL, Schlosser SJ, Ivarie CA, et al. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. The Journal of investigative dermatology. 2000;114:444–455. doi: 10.1046/j.1523-1747.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- 25.Johnson KM, Kines RC, Roberts JN, et al. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. Journal of Virology. 2009;83:2067–2074. doi: 10.1128/JVI.02190-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambert PF, Ozbun MA, Collins A, et al. Using an immortalized cell line to study the HPV life cycle in organotypic “raft” cultures. Methods in molecular medicine. 2005;119:141–155. doi: 10.1385/1-59259-982-6:141. [DOI] [PubMed] [Google Scholar]
- 27.Buck CB, Thompson CD, Pang Y-YS, et al. Maturation of papillomavirus capsids. Journal of Virology. 2005;79:2839–2846. doi: 10.1128/JVI.79.5.2839-2846.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu T, Griffin LM, Guo K, et al. APOBEC3A functions as a restriction factor of Human Papillomavirus. (Manuscript submitted) [DOI] [PMC free article] [PubMed]
- 29.Fu B, Quintero J, Baker CC. Keratinocyte growth conditions modulate telomerase expression, senescence, and immortalization by human papillomavirus type 16 E6 and E7 oncogenes. Cancer Research. 2003;63:7815–7824. [PubMed] [Google Scholar]