

ABSTRACT
Environmental conditions during perinatal development such as maternal undernutrition, maternal glucocorticoids, placental insufficiency, and maternal sodium overload can program changes in renal Na+ excretion leading to hypertension. Experimental studies indicate that fetal exposure to an adverse maternal environment may reduce glomerular filtration rate by decreasing the surface area of the glomerular capillaries. Moreover, fetal responses to environmental insults during early life that contribute to the development of hypertension may include increased expression of tubular apical or basolateral membrane Na+ transporters and increased production of renal superoxide leading to enhanced Na+ reabsorption. This review will address the role of these potential renal mechanisms in the fetal programming of hypertension in experimental models induced by maternal undernutrition, fetal exposure to glucocorticoids, placental insufficiency, and maternal sodium overload in the rat.
Keywords: developmental origins of health and disease, hypertension, intrauterine growth restriction (IUGR), kidney, oxidative stress
The impact of adverse events during gestational life, on later cardio-renal health of the offspring is highlighted, implicating the importance of fetal life on long-term health.
INTRODUCTION
Renal Na+ excretory function is intrinsically accountable for the long-term control of blood pressure [1, 2]. A renal reduction in Na+ excretion, either by reduced glomerular filtration rate (GFR) or by increased tubular reabsorption of this electrolyte, causes hypertension. Besides genetic determinants, environmental conditions during perinatal development program changes in renal excretory function that lead to hypertension. Irreversible changes that occur during critical periods during early development are designated as the developmental programming of health and disease [3].
Low birth weight in human populations has been hypothesized to correlate to developmental programming of coronary heart disease [4] and hypertension [5] in later life. The hypothesis is based on studies that examined undernourished English populations born from 1911 to 1930 [4] and from 1935 to 1943 [5]. Proof of principle was first established by experimental models of undernutrition in the rat that demonstrated a direct link between adverse influences during fetal life and later increased cardiovascular risk, including the development of hypertension [6–10]. Increased fetal exposure to maternal glucocorticoids that cross the placenta are implicated in the etiology of cardiovascular risk that originate from insults during early life [6] with programmed changes in the fetal hypothalamic-pituitary-adrenal axis indicated to contribute to prenatal undernutrition-induced hypertension [11]. Fetal exposure to synthetic glucocorticoids leads to hypertension in the rat [12–14], requiring caution in their use for the treatment of preterm labor [15, 16]. However, in adequately nourished populations, placental insufficiency due to inadequate vascular adaptation at the uteroplacental interface, as observed in pregnancies complicated by preeclampsia, appears to be the main cause of intrauterine growth restriction (IUGR) and low birth weight [17]. Current models of placental insufficiency that program hypertension in the rat are induced via a reduction in flow introduced at the abdominal aorta below the kidneys and on both ovarian arteries [18] or by ligation of both uterine arteries [19, 20] to reduce uteroplacental flow in late gestation. Furthermore, sodium overload in the pregnant rat also causes placental dysfunction in a manner that resembles preeclampsia in the dam [21] and results in hypertension in the offspring [22–24]. In addition to clarifying the impact of preeclampsia, insight from the effect of maternal sodium overload during gestation per se on fetal development is critical because the human species exists mainly in industrialized areas and consumption of sodium in urbanized regions is approximately ten times greater relative to the amount of sodium consumed by our evolutionary ancestors [25].
Recent investigations confirm the correlation between birth weight and blood pressure in different populations including Chilean children [26], Brazilian children [27–29], and Chinese adults [30]. However, recent studies implicate that the quality of life during the early postnatal years [31–33] also exerts an underlying influence on the development of chronic disease in adult life. Yet, in experimental models of developmental insult, disruptions in the maternal environment may not always induce low birth weight although they may alter the function and physiology of the vital organs of the offspring in later life in a manner that also programs hypertension and increased cardiovascular risk.
Hypertension has been hypothesized to result from a congenital deficiency in nephron number, resulting in a reduced surface area for filtration that programs a subsequent reduction in GFR and an increase in sodium retention and hypertension [34]. Besides a reduction in glomerular surface area, however, hormonal regulation of renal hemodynamics by the renin-angiotensin system (RAS) can also contribute to a reduction in GFR and hypertension. There is evidence that the maternal environment may affect both cornerstones and reduce glomerular surface area and renal hemodynamics. Moreover, fetal exposure to an adverse maternal environment may 1) increase the expression of tubular apical Na+ transporters or of the basolateral membrane Na+ transporters, (Na++K+)ATPase and Na+-ATPase, to increase filtered Na+ reabsorption and/or 2) increase renal superoxide production that may reduce renal medullary blood flow, contributing to the development of hypertension. This review will address the role of these potential renal mechanisms in the fetal programming of hypertension in experimental models of fetal programming induced by maternal undernutrition, maternal glucocorticoids, placental insufficiency, or maternal sodium overload.
NEPHRON NUMBER, GLOMERULAR HYPERTROPHY, AND DEVELOPMENTAL PROGRAMMING OF HYPERTENSION AND RENAL SUSCEPTIBILITY TO DISEASE
Renal volume assessed by ultrasound measurement is decreased in the IUGR fetus [35]. Furthermore, a reduction in the number of nephron is observed in African American and Caucasian individuals from the southeastern United States [36] and in Aborigine populations in Australia [37]. Numerous experimental models of developmental insult induced via maternal low protein [8], fetal exposure to maternal glucocorticoids [14], placental insufficiency [38], and maternal sodium overload [24] also demonstrate a reduction in nephron number that is associated with hypertension. However, findings relating a reduction in nephron number to increased blood pressure are correlative and may not directly indicate a causal role for a decrease in nephron endowment as the main determinant of hypertension. Yet, additional studies implicate that a congenital reduction in nephron number may enhance susceptibility to a secondary insult leading to greater injury or disease.
Nephron number affects the amount of glomerular surface area that is dependent on nephrogenesis when the kidney is not impacted by glomerulosclerosis. Glomerular surface area is one determinant of GFR that can modulate sodium reabsorption and indirectly regulate blood pressure. Whenever GFR is reduced, sodium reabsorption is increased. A reduction in nephron number that initiates during nephrogenesis in the rat is associated with a marked decrease in GFR and hypertension in later life [39], suggesting that a congenital reduction in nephron complement may significantly alter basal blood pressure regulation. Yet, in mice demonstrating a loss of one allele for glial cell line-derived neurotrophic factor that present with approximately 30% fewer nephrons, hypertension is not present when the mice are placed on a normal-salt diet. Yet, hypertension develops in response to a chronic salt load [40], indicating that a congenital reduction in nephron number enhances sensitivity to a secondary insult but may not directly affect the long-term regulation of blood pressure under basal conditions.
Nephrogenesis in the human begins by the 10th week postconception and ends by the 34th week. Although GFR is low at birth, considering the ratio of volume/body mass, normal term babies have a definitive number of nephrons at birth whereas preterm babies may continue nephrogenesis for weeks following birth (for reviews, see [41, 42]). Although renal maturation continues after preterm birth, attaining a continued increase in the generation of glomeruli, a greater percentage of morphologically abnormal glomeruli and a significantly larger cross-sectional area of the renal corpuscle are observed in preterm individuals, suggesting the development of renal hyperfiltration [43]. Thus, maturation of the kidney after birth in the human is associated with impaired nephrogenesis. In addition, due to continuation of nephrogenesis after delivery, preterm babies are more vulnerable than normal term babies to early adverse postnatal environmental influences such as medication [42]. In rats, the main animal species discussed in this review, nephrogenesis begins at the 12th day of fetal life and ends around the 10th day of postnatal life [44, 45]. Rodent models of developmental insult demonstrate sensitivity to an adverse insult that impairs nephrogenesis during pre- [8] and early postnatal life [46], indicating that the window of developmental vulnerability in the rat includes both the prenatal and early postnatal period. However, the normal extension of nephrogenesis into the postnatal period in the rat limits the usefulness of rodent models for understanding how disruptions during nephrogenesis affect later renal health.
Although a reduction in nephron number can be a determinant for decreased GFR, nephron number in rodent offspring of dams submitted to inadequate conditions during the perinatal period can remain unaltered, be reduced, or even increase (Table 1). Fluctuations in the pattern of GFR observed in experimental models of development insult can depend in part on the age of evaluation. Yet, the main contributor to disparate findings may be due to the fact that a reduction in nephron number is normally associated with glomerular hypertrophy (Table 1). The role of hypertrophic compensation on GFR could be verified by micropuncuture measurement of single nephron GFR, which as far as we know has not yet been performed in offspring of mothers adversely affected during gestation.
TABLE 1.
Effects of perinatal maternal environment on GFR and glomerular morphometry in the rat.
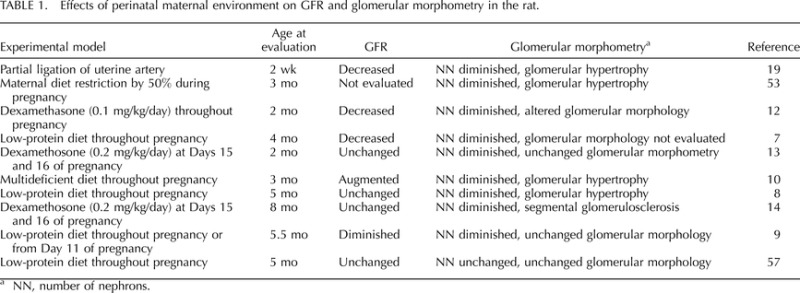
NN, number of nephrons.
The correlation between glomerular hypertrophy and a lower number of nephrons is observed in hypertensive white and African American individuals in the southeastern United States [47] and also in Senegalese men [48]. Chronic renal failure and microalbuminuria are more prevalent in low birth weight inhabitants of the southeastern United States [49] and Aborigines in the Australia's northern territories [50], respectively. Thus, these findings indicate that a reduction in nephron number may increase susceptibility to renal injury and disease. A comparison between two strain of rats, Lewis and Fisher 344, demonstrate that the latter, which exhibits a reduction in number of nephron, is more vulnerable to progressive chronic disease due to 5/6 nephrectomy than the former [51]. Similarly, rats submitted to IUGR by placental ischemia demonstrate an increase in renal vascular resistance, a reduction in GFR and renal injury in response to mild renal ischemia (15 min) followed by reperfusion that does not impair renal function and or result in renal injury in control counterparts [52]. Thus, these observations indicate that a congenital deficiency in nephron number may not be a unique determinant of hypertension in adult life; however, it does appear to contribute to a worsened prognosis of renal dysfunction in response to a secondary insult. Thus, additional molecular events in oligonephronic individuals contribute to the development of hypertension and renal disease.
Maternal Undernutrition and Nephron Number
The importance of timing of a developmental insult is indicated in experimental studies that vary exposure of the fetus to nutrient restriction during gestational life. Restriction of protein in the maternal diet from Days 1 to 11 of gestation does not alter the nephron number in male and female offspring. Yet, glomerular number, which is indicative of nephron number, is reduced in male offspring exposed to a maternal low-protein diet from Day 12 of gestation until birth [9] and male offspring exposed to global undernutrition throughout gestation [8]. Thus, the last 2 wk of gestation appears to serve as a crucial period whereby undernutrition can compromise nephrogenesis in the rat [7, 53]. Furthermore, exposure to a maternal low-protein diet during lactation in rat also compromises nephrogenesis, demonstrating that the crucial period of vulnerability extends into early postnatal life in the rat [54]. A reduction in nephron number is observed even when the maternal undernutrition is induced by restriction of only one nutrient in the rat such as protein [7], vitamin A [55], or iron [56], by global food restriction [53], or by a multideficient diet that is rich in carbohydrates that is not deficient in calories [10]. Compensatory glomerular hypertrophy is reported in the neonate rat that exhibits a nephron deficiency of 25% to 30% [53]. Glomerular hypertrophy is also observed in adult rats exposed to undernutrition during prenatal life [8, 10]. However, GFR is not reduced [8], indicating that glomerular hypertrophy that occurs following a reduction in nephron number sustains renal function and thus implicating other causative factors in the etiology of hypertension programmed in response to undernutrition during early life. Moderate maternal protein restriction programs hypertension associated with a reduction in nephron number in male, but not female, rat offspring [8, 57], suggesting a sex-specific impact of prenatal undernutrition on nephrogenesis. Kidney development and nephrogenesis depends in part on angiotensin II (Ang II) and is associated with glial cell-derived neurotrophic factor production [58–60]. Woods and coworkers [8] demonstrate that renin and Ang II expression is reduced in the kidney of male, but not female, rat offspring at birth following exposure to maternal undernutrition during fetal life [8, 57]. Thus, the sex difference in intrarenal RAS expression during development suggests that sex-specific adult outcomes related to nephron number and blood pressure may originate during fetal life. Although not completely understood, sex differences in gene expression in human and murine kidneys under baseline and disease conditions [61], if they could be further elucidated, could be utilized as preventative targets for sex-specific treatment of cardiovascular and renal disease. Besides RAS compromise, oligonephronia in undernourished rats is also attributed to apoptosis [62, 63]. Undernutrition in the sheep also compromises nephron number [64].
Fetal Exposure to Maternal Glucocorticoids and Nephron Number
Other models of developmental insult also demonstrate a reduction in nephron number. Prenatal exposure to dexamethasone, a synthetic glucocorticoid that is not appropriately metabolized by 11β-hydroxysteroid dehydrogenase 2 (11βHSD2) in the placenta, throughout pregnancy [12, 65] or from Days 15 to 18 of gestation leads to a reduction in nephron number in the offspring [13, 14]. However, only male offspring are hypertensive [14]. Maternal exposure to natural glucocorticoids induced by inhibition of 11βHSD2, the enzyme that serves as the barrier for fetal exposure to maternal glucocorticoids, also programs a reduction in nephron number [66]. As observed in models of maternal undernutrition, oligonephronia induced by inappropriate fetal exposure to maternal dexamethasone is also correlated with increased apoptosis [67].
Maternal Sodium Overload During Gestation and Nephron Number
Consumption of a high-salt diet by pregnant ewes during the final phase of nephrogenesis reduces the number of nephrons in the offspring [68]. Similarly in rats, exposure to maternal high dietary salt intake during pregnancy and lactation periods compromises nephrogenesis in the offspring [24]. Sodium overload may influence fetal development through its effects on decreasing maternal plasma renin [69] and Ang II [70] levels, correlating with reduced renal Ang II formation in the offspring during nephrogenesis [71]. The RAS is fundamental to nephrogenesis [58–60, 46]; therefore suppression of the RAS during fetal and early postnatal life may have long-term repercussions on renal function [23, 46]. Likewise, a 0.9% NaCl solution administered throughout pregnancy does not alter the number of nephrons, though it can arrest renal development [71]. Moreover, when the sodium overload progresses from gestation to weaning, it can be associated with an increase in the glomerulosclerosis index [72]. In addition, perinatal exposure to a maternal low-sodium diet during fetal life not only programs a reduction in nephron number, it is also induces high blood pressure in the offspring [24].
Placental Insufficiency and Nephron Number
When IUGR is induced by ligation of the two uterine arteries on Day 19 of gestation, it is associated with a reduction in the nephron number that is accompanied by an increase in markers of renal apoptosis [20]. Male, but not female, offspring are hypertensive in young adulthood [18, 73, 74]. Yet, in adult life, both male and female offspring demonstrate glomerular hypertrophy, whereas renal function is impaired to a greater degree in male compared to female counterparts [75]. Newborn piglets born with low birth weight compared to littermates also demonstrate a reduction in nephron number associated with a reduction in GFR and compromised urinary Na+ excretion [76]. Thus, IUGR affects nephron number regardless of the species studied and in a manner that is sex-specific despite the method of developmental insult.
To summarize, insults during fetal life often program a reduction in nephron number that corresponds to the development of hypertension in later life. Programming of reduced nephron number and hypertension is timing-specific and linked to the period of nephrogenesis. In addition, programming of reduced nephron number and hypertension may be sex-specific with the origins of sex differences in the fetal response to a developmental insult originating during fetal life due to sex-specific effects on gene expression that affect renal development. Although nephron number correlates to hypertension in these models of developmental insult, other causative factors may be important in the long-term regulation of impaired blood pressure.
THE RAS AND DEVELOPMENTAL PROGRAMMING OF HYPERTENSION
The RAS plays a key role in the long-term control of blood pressure through its actions on the kidney and the vasculature [77]. The RAS is a regulator of efferent arterial resistance contributing to the control of GFR by altering glomerular hydrostatic pressure, the main determinant of GFR [78]. Thus, a decrease in GFR mediated by programmed alterations in regulatory systems such as the RAS are implicated in the development and maintenance of hypertension programmed in response to a developmental insult.
Developmental Insults and the RAS
Langley-Evans and Jackson [79] report that plasma angiotensin converting enzyme-1 (ACE-1) activity is significantly increased in rats in offspring exposed to a casein-deficient diet during fetal life. The importance of the RAS in the developmental programming of hypertension in this model is demonstrated by the ability of the ACE inhibitor, captopril, to normalize blood pressure in undernourished offspring relative to their control counterparts [79]. Vehaskari et al. [80] report a reduction in plasma renin activity associated with an increase in plasma aldosterone levels in young rats exposed to maternal low protein during fetal life. Renal expression of the Ang II receptor (AT1R) is also increased in this model [81] with the relative importance of the RAS demonstrated by normalization of the blood pressure by chronic RAS blockade [82]. Alterations in expression of the RAS are also common to other models of developmental insult [83], and blockade of the RAS also abolishes hypertension in offspring of dams submitted to placental insufficiency [84]. Renin and ACE mRNA expression are increased in the kidneys and adipose tissue of adult offspring exposed to maternal dexamethasone, a synthetic steroid, during fetal life [85]. Changes in RAS components are also observed in the offspring exposed to maternal sodium overload during fetal or early postnatal life [24, 72]. However, Mesquita et al. [86] report that exposure to a low-protein diet throughout pregnancy in the rat reduces expression of the Ang II receptors, AT1R and AT2R. The reduction in AT1R is paralleled by a reduction in the expressions of janus kinase 2, which is a non-receptor-type tyrosine kinase that regulates signal transducers and activators of transcription. In the fetal brain of undernourished mice, components such as mRNA of angiotensinogen and ACE-1 are increased, while mRNA levels of AT2R are decreased. Hypomethylation of the CpG islands in the promoter regions of ACE-1 gene and upregulation of the microRNAs, mmu-mir-27a and 27b, which regulate ACE-1 mRNA translation, are also seen in the fetal brain of prenatal undernourished mice [87]. These findings give support that changes in DNA methylation and microRNA are key mediators of hypertension at adult life. Furthermore, intracerebroventricular blockade of the central actions of the RAS ameliorates hypertension in low-protein offspring, implicating a central role for the RAS in the programming of hypertension [88].
To summarize, classical hormones that regulate GFR such as Ang II contribute to hypertension programmed by prenatal undernutrition [79, 80, 86], maternal sodium overload during pregnancy and lactation [24, 72], placental insufficiency [84], and fetal exposure to maternal glucocorticoids during Day 13 of gestation to term [85]. Although insight from these studies implicate an important role for the RAS in the developmental programming of hypertension, the exact role of Ang II in the etiology of programmed hypertension and impaired renal hemodynamics is still not clearly demonstrated from reports published thus far. However, the impact of the RAS on programming of hypertension in response to a developmental insult may result from its influence on sodium reabsorption in the proximal tubule.
TUBULAR SODIUM TRANSPORTERS AND HYPERTENSION
The bulk of sodium reabsorption in the apical membrane of proximal tubule epithelium is largely carried out by the Na+-H+-transporter, while parallel transport of sodium in the basolateral membrane is mainly carried out by the (Na++K+)ATPase and Na+-ATPase transporters. In the thick ascending limb and distal tubule, the bulk of sodium transport in the apical membrane involves the Na+K+2Cl− (NKCC2) and Na+Cl− cotransporters whereas parallel transport in the basolateral membrane is mainly conducted via the (Na++K+)ATPase transporter. The (Na++K+)ATPase transporter is an epithelial polarized enzyme that maintains overall intracellular Na+ equilibration and creates the electrochemical gradient for electrolyte reabsorption in the basolateral membrane of the tubular kidney. Less widespread, the α-subunit of Na+-ATPase was recently purified and cloned from the basolateral membrane of epithelial bowel of guinea pig [89]. This enzyme is responsible for approximately 10% of the reabsorption of filtered sodium [90] in the proximal tubule, and it is also designated as the ouabain-insensitive Na+-ATPase transporter or a second sodium pump. Although recently cloned, the role of Na+-ATPase in sodium reabsorption in the proximal tubule has been extensively investigated [91–93] and implicated in the development of hypertension in the spontaneously hypertensive rat (SHR) [89, 94]. Specifically, increased activity of the NKCC2 transporter along the thick ascending limb of the Milan hypertensive rat [95] and increased activity of Na+-H+-transporter along the proximal tubule in juvenile SHRs [96–98] highlights the importance of these apical membrane Na+ transporters in the maintenance of hypertension. Sodium ion reabsorption along the proximal tubule plays a remarkable role in the development of hypertension in the SHR [96–98] with Na+ reabsorption increased during the juvenile age [96] and with activity reduced in the adult SHR [96, 98], implicating that temporal changes in sodium transporter expression may contribute to the etiology and maintenance of hypertension.
Maternal Undernutrition, Prenatal Glucocorticoid Exposure, and Tubular Sodium Transporters
Alterations in tubular Na+ reabsorption may be one mechanism by which exposure to a developmental insult programs an increase in blood pressure. Renal mRNA expression of the α1 and β1 subunits of the (Na++K+)ATPase are increased in adult rat offspring exposed to undernutrition during fetal life [99]. This alteration in sodium transport expression is associated with the development of hypertension, implicating a role for impaired sodium transport in the etiology of hypertension programmed in response to fetal undernutrition [99]. Maternal undernutrition also programs a reduction in renal expression of 11βHSD2 mRNA, the enzyme that catalyzes the inactivation of cortisol to corticosterone in the rat (cortisol in humans) that coincides with an increase in renal expression of the glucocorticoid receptor mRNA [99]. Glucocorticoids increases (Na++K+)ATPase activity in the thick ascending limb [100]. Thus, these hormones could contribute to the developmental programming of hypertension in prenatal undernourished rats.
Bumetanide-sensitive cotransporter NKCC2 mRNA and protein expression are increased in the thick ascending limb [101, 102] of rats exposed to prenatal undernutrition. In addition, the thiazide-sensitive Na+Cl− cotransporter located in the distal convoluted tubule is also increased in animals exposed to undernutrition during fetal life [101, 102]. However, increases in (Na++K+)ATPase activity in later life are preceded by a reduction in whole kidney (Na++K+)ATPase activity at 1 mo of age in rats submitted to a maternal low-protein diet during prenatal life, indicating that developmental insults may program temporal changes in expression of tubular sodium transporter activity [103]. The importance of glucocorticoids in the programming of altered sodium transporter expression is demonstrated in experimental studies whereby changes in expression of the basolateral or apical membrane sodium transporters are programmed by direct prenatal exposure to inappropriate levels of maternal dexamethasone (Table 2). Dexamethasone administered in the second half of pregnancy [85] or during four critical days of nephrogenesis in the rat (Days 15 to 18 of pregnancy) [104, 105] programs a marked increase in sodium transporter expression. These programmed changes in expression include an increase in mRNA expression of the α1 subunit of (Na++K+)ATPase [85], expression of NHE3 on the brush border membrane vesicles of the proximal tubule [102], and expression of NKCC2 and Na+Cl− cotransporter in the renal cortex [104]. These changes in transporter expression are correlated with increased blood pressure in these models of developmental insult [104, 105]. Thus, these studies indicate a strong role for programmed alterations in sodium transport expression in the developmental programming of hypertension.
TABLE 2.
Effect of perinatal maternal environment on renal Na+ transporters in the rat.
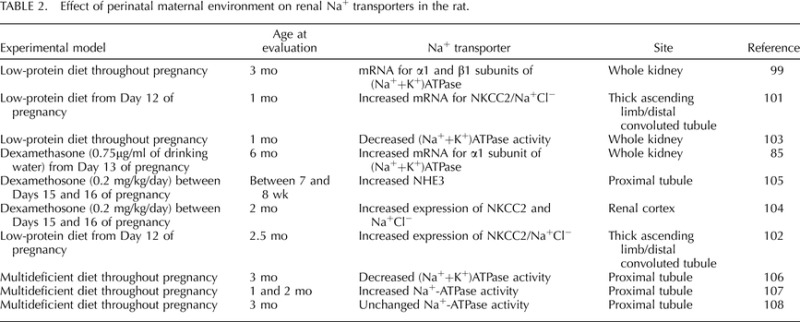
Based on evidence that changes in Na+-ATPase activity may be involved in hypertension, Na+-ATPase activity has been investigated in offspring exposed to maternal undernutrition during perinatal life [54, 106–108]. In rats exposed to a multideficient diet during prenatal life, Na+-ATPase activity is increased at 30 and 60 days of age or during the establishment of programmed hypertension [108]. However, Na+-ATPase activity is unchanged at an age of 90 days [108]. The activity of this enzyme in the proximal tubule is regulated in part by Ang II with protein kinase C and A contributing as intracellular downstream mediators [109]. Rats exposed to a multideficient diet during prenatal life demonstrate a decrease in expression and activity of protein kinase C and an increase in expression and activity of protein kinase A in the proximal tubule membranes associated with a loss of responsiveness to physiological concentrations of Ang II in adult life [108]. Besides hyporesponsiveness to Ang II, these animals present a reduction in activity of the (Na++K+)ATPase transporter [106], suggesting a reduction in proximal tubule Na+ reabsorption in adult animals. Taken together, these findings demonstrate the following: first, that exposure to a multideficient diet during prenatal life programs temporal changes in Na+ transporters similar to what is observed in the SHRs [96–98], and second, that increases in Na+ reabsorption may precede the development of hypertension in prenatal undernourished rats.
Increases in Na+ transporters, either in prenatal undernourished rats [99, 101, 102] or in prenatal rats exposed to maternal dexamethasone is correlated to increased levels of blood pressure [102, 104]. Bilateral renal denervation during adult life abolishes the increase in NKCC2 and Na+Cl− expression in the renal cortex in conjunction with hypertension programmed by prenatal exposure to dexamethasone [104]. Thus, this study suggests that the renal nerves may contribute to the etiology of hypertension programmed by perinatal exposure to glucocorticoids via alterations in Na+ tubular transport.
OXIDATIVE STRESS AND THE DEVELOPMENTAL PROGRAMMING OF HYPERTENSION
Oxidative stress involves an imbalance in reactive oxygen species and may include the production of increased levels of hydrogen peroxide, superoxide anion radicals, and hydroxyl anions. One consequence of superoxide elevation in the renal medulla is a reduction in medullary blood flow and an increase in Na+ reabsorption leading to sustained hypertension [110, 111]. A component of superoxide's action in renal tissue is mediated by a reduction in local nitric oxide levels [112] that can increase Na+ reabsorption due to augmentation of NKCC2 activity in the ascending limb of Henle [113]. Furthermore, it is known that the oxidative stress alters the interplay between the mineralocorticoid receptor and one of its intracellular signaling molecules, the small guanosine triphosphatase Rac1, to increase Na+ reabsorption and induce hypertension [114]. Because Rac1 is also a NADPH-oxidase subunit, the action of aldosterone, the main mineralocorticoid, results in an increase in oxidative stress resulting from NADPH-oxidase activation and increased Na+ reabsorption throughout upregulation of epithelial sodium channels, the Na+Cl− cotransporter and (Na++K+)ATPase transporter located in the distal portion of the tubule [115].
Maternal Undernutrition, Placental Insufficiency, and Maternal Sodium Overload and Renal Oxidative Stress
Oxidative stress is increased in the kidneys of prenatal undernourished rats [106, 108, 116–118] as well as in the offspring of mothers submitted to placental insufficiency [119] and sodium overloads [120, 121]. Hypertension in male offspring of mothers submitted to placental insufficiency is abolished by treatment with tempol, a mimetic of superoxide dismutase [118], indicating a key role for oxidative stress in the etiology of programmed hypertension. Suppression of the RAS during nephrogenesis may contribute to the increase in oxidative stress in adult life. Blockade of the RAS during the postnatal period of the rat that corresponds to nephrogenesis programs hypertension in the adult rat that is reduced by treatment with tempol [122]. An increase in renal markers of oxidative stress are also observed in different animal models of developmental insult [106, 108, 116–118], including adult offspring exposed to prenatal undernutrition [79, 80], placental insufficiency [84], or models that involve suppression of the RAS during early life [8, 71, 83, 123]. Importantly, increased oxidative stress is linked to an elevation in blood pressure in the male rat relative to the female rat [119, 122, 124], suggesting that oxidative stress may contribute to the sexual dimorphic control of blood pressure and cardiovascular risk induced by a prenatal insult.
Oxidative stress is appropriately considered a cause, a consequence, or a potentiating factor in the development of hypertension (for a review, see [125]). Pretreatment of undernourished mothers during gestation with antioxidants prevents the development of hypertension in the offspring [107]. In addition, maternal undernutrition is associated with an increase in placental oxidative stress, indicating that exposure to oxidative stress has its origins in early life [106]. Moreover, epigenetic processes may contribute to the increase in oxidative stress that is responsible for endothelial dysfunction [126] in prenatal undernourished rats. The efficacy of antioxidant tools to prevent renal dysfunction reinforces the role of oxidative stress as a cause of perinatal programmed hypertension.
INTERVENTIONS TO MITIGATE PROGRAMMED OUTCOMES
Several studies indicate that early interventions may be preventative against the fetal programming of later renal dysfunction or hypertension. Maternal treatment with the antioxidant α-tocopherol during lactation in prenatally undernourished rats [123] or cross-fostering pups at birth of placental insufficient dams [73] prevents oligonephronia. Oligonephronia induced by maternal undernutrition is also prevented by administration of ouabain in parallel with maternal undernutrition during pregnancy. Ouabain, an inhibitor of (Na++K+)ATPase transporters, increases intracellular calcium and prevents apoptosis to ameliorate reductions in nephron endowment [62, 63]. Treatment with tempol during pregnancy prevents prenatal glucocorticoid-induced aorta dysfunction and hypertension [127] in addition to endothelial dysfunction programmed by undernutrition in the prenatal rat [126]. Moreover, rats prenatally treated with dexamethasone and cross-fostered to mothers on a diet rich in n-3 long-chain polyunsaturated fatty acids (n-3 PUFA) do not demonstrate hyperleptinemia or develop hypertension [128]. Supplementation with n-3 PUFA attenuates oxidative stress, inflammation, and tubulointerstitial fibrosis in the remnant kidney of 5/6 nephrectomized rats [129], suggesting that in addition to its actions to reduce leptin, n-3 PUFA may also contribute to a reduction in renal oxidative stress. Thus, the benefit of n-3 PUFA administered during lactation to rats prenatally exposed to dexamethasone [128] may also involve attenuation of oxidative stress and provides further evidence implicating a role for oxidative stress in the early origins of hypertension induced in response to adverse influences that originate from the maternal environment.
The critical period for development of tubular Na+ transporters includes not only the prenatal period, but also early postnatal life in the rat (for a review, see [130]). Several studies strongly suggest that the period for developmental programming of Na+ transporters extends beyond lactation in the rat: treatment with enalapril, an ACE inhibitor [131] or exposure to a low-sodium diet [132] for a period of 3 wk after weaning prevents adult hypertension in rats expose to undernutrition during prenatal life. Treatment with an ACE inhibitor from 2 to 6 wk of life changes the course of hypertension in SHRs in later life [133], and enalapril administered for a period of 3 wk after weaning reduces activity of (Na++K+)ATPase and Na+-ATPase in the proximal tubule [120]. Despite these promising effects, it is important to emphasize that ACE inhibitors and AT1R antagonists are potential teratogenic agents to humans [134, 135] and thus, their use is contraindicated during pregnancy. Caveats include the numerous studies demonstrating that the use of RAS inhibitors program adverse effects when administered prenatally in the rat [122, 136–138] as compared to their action when administered after weaning [120, 131, 133]. Taken together, these studies indicate early lifestyle changes including antioxidant supplementation, a balanced diet, salt restriction, or even therapeutic maneuvers can serve as potential targets and may prove effective in counteracting the adverse programming of chronic health in the event that adverse environmental influences during development cannot be avoided.
CONCLUSIONS
Although it is clear that nephrogenesis can be compromised by perinatal responses to an adverse maternal environment and that renal hemodynamics may be affected by altered regulation of classic hormones such as Ang II, GFR is not always reduced in experimental models of programmed insult (Table 1), in part due to the development of glomerular hypertrophy. Thus, a congenital reduction in the number of nephrons presents as a status that worsens renal function in the face of later insults [36, 48, 49, 51, 52, 139]. On the other hand, molecular programming events such as changes in tubular sodium transporters (Table 2) and increased renal oxidative stress seem to be crucial in the development of programmed hypertension (Fig. 1). Therefore, hypertension induced in response to adverse maternal influences during fetal life may involve a reduction in GFR and/or an increase in sodium reabsorption (Fig. 1). Early interventions to ensure proper nephrogenesis and reduce inappropriate increases in renal oxidative stress may be potential targets in the development of therapeutic agents in the prevention of later programmed renal dysfunction.
FIG. 1.
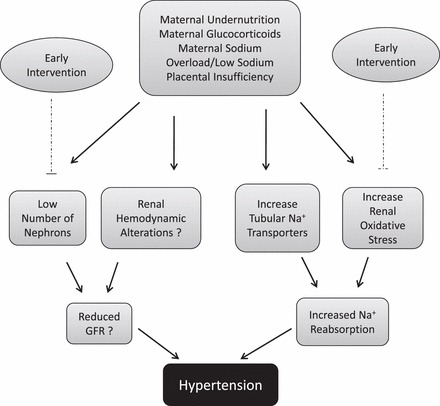
Renal role in the development of hypertension in the offspring of mothers showing a perturbed maternal environment. Black line, programmed alteration; dashed line, inhibition (indicative of early intervention that could prevent renal dysfunction). GFR is not always predictable due to glomerular hypertrophy (see Table 1).
Supplementary Material
Footnotes
B.T.A. is supported by NIH grants HL074927 and HL51971. A.D.P. is supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil, 9135/11-1).
REFERENCES
- Crawford MP, Richardson TQ, Guyton AC. Renal servocontrol of arterial blood pressure. J Appl Physiol 1967; 22 (1): 139–142. [DOI] [PubMed] [Google Scholar]
- Guyton AC. The surprising kidney-fluid mechanism for pressure control—its infinite gain! Hypertension 1990; 16 (6): 725–730. [DOI] [PubMed] [Google Scholar]
- Barker DJ. The origins of the developmental origins theory. J Intern Med 2007; 261 (5): 412–417. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet 1989; 2 (8663): 577–580. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. BMJ 1990; 301: 259–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner DS, Jackson AA, Langley-Evans SC. The effect of prenatal diet and glucocorticoids on growth and systolic blood pressure in the rat. Proc Nutr Soc 1998; 57 (2): 235–240. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Welham SJ, Jackson AA. Fetal exposure to a maternal low protein diet impairs nephrogenesis and promotes hypertension in the rat. Life Sci 1999; 64: 965–974. [DOI] [PubMed] [Google Scholar]
- Woods LL, Ingelfinger JR, Nyengaard JR, Rasch R. Maternal protein restriction suppresses the newborn renin-angiotensin system and programs adult hypertension in rats. Pediatr Res 2001; 49: 460–467. [DOI] [PubMed] [Google Scholar]
- Woods LL, Weeks DA, Rasch R. Programming of adult blood pressure by maternal protein restriction: role of nephrogenesis. Kidney Int 2004; 65: 1339–1348. [DOI] [PubMed] [Google Scholar]
- Paixão AD, Maciel CR, Teles MB, Figueiredo-Silva J. Regional Brazilian diet-induced low birth weight is correlated with changes in renal hemodynamics and glomerular morphometry in adult age. Biol Neonate 2001; 80: 239–246. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Nwagwu M. Impaired growth and increased glucocorticoid-sensitive enzyme activities in tissues of rat fetuses exposed to maternal low protein diets. Life Sci 1998; 63 (7): 605–615. [DOI] [PubMed] [Google Scholar]
- Celsi G, Kistner A, Aizman R, Eklöf AC, Ceccatelli S, de Santiago A, Jacobson SH. Prenatal dexamethasone causes oligonephronia, sodium retention, and higher blood pressure in the offspring. Pediatr Res 1998; 44 (3): 317–322. [DOI] [PubMed] [Google Scholar]
- Ortiz LA, Quan A, Weinberg A, Baum M. Effect of prenatal dexamethasone on rat renal development. Kidney Int 2001; 59 (5): 1663–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz LA, Quan A, Zarzar F, Weinberg A, Baum M. Prenatal dexamethasone programs hypertension and renal injury in the rat. Hypertension 2003; 41 (2): 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Horm Behav 2011; 59 (3): 279–289. [DOI] [PubMed] [Google Scholar]
- Alexander N, Rosenlöcher F, Stalder T, Linke J, Distler W, Morgner J, Kirschbaum C. Impact of antenatal synthetic glucocorticoid exposure on endocrine stress reactivity in term-born children. J Clin Endocrinol Metab 2012; 97 (10): 3538–3544. [DOI] [PubMed] [Google Scholar]
- Henriksen T, Clausen T. The fetal origins hypothesis: placental insufficiency and inheritance versus maternal malnutrition in well-nourished populations. Acta Obstet Gynecol Scand 2002; 81 (2): 112–114. [DOI] [PubMed] [Google Scholar]
- Alexander BT. Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension 2003; 41 (3): 457–462. [DOI] [PubMed] [Google Scholar]
- Merlet-Bénichou C, Gilbert T, Muffat-Joly M, Lelièvre-Pégorier M, Leroy B. Intrauterine growth retardation leads to a permanent nephron deficit in the rat. Pediatr Nephrol 1994; 8 (2): 175–180. [DOI] [PubMed] [Google Scholar]
- Pham TD, MacLennan NK, Chiu CT, Laksana GS, Hsu JL, Lane RH. Uteroplacental insufficiency increases apoptosis and alters p53 gene methylation in the full-term IUGR rat kidney. Am J Physiol Regul Integr Comp Physiol 2003; 285 (5): R962–R970. [DOI] [PubMed] [Google Scholar]
- Beauséjour A, Bibeau K, Lavoie JC, St-Louis J, Brochu M. Placental oxidative stress in a rat model of preeclampsia. Placenta 2007; 28 (1): 52–58. [DOI] [PubMed] [Google Scholar]
- Contreras RJ, Wong DL, Henderson R, Curtis KS, Smith JC. High dietary NaCl early in development enhances mean arterial pressure of adult rats. Physiol Behav 2000; 71 (1–2): 173–181. [DOI] [PubMed] [Google Scholar]
- da Silva AA, de Noronha IL, de Oliveira IB, Malheiros DM, Heimann JC. Renin-angiotensin system function and blood pressure in adult rats after perinatal salt overload. Nutr Metab Cardiovasc Dis 2003; 13 (3): 133–139. [DOI] [PubMed] [Google Scholar]
- Koleganova N, Piecha G, Ritz E, Becker LE, Müller A, Weckbach M, Nyengaard JR, Schirmacher P, Gross-Weissmann ML. Both high and low maternal salt intake in pregnancy alter kidney development in the offspring. Am J Physiol Renal Physiol 2011; 301 (2): F344–F354. [DOI] [PubMed] [Google Scholar]
- Meneton P, Jeunemaitre X, de Wardener HE, MacGregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev 2005; 85 (2): 679–715. [DOI] [PubMed] [Google Scholar]
- Martinez-Aguayo A, Aglony M, Bancalari R, Avalos C, Bolte L, Garcia H, Loureiro C, Carvajal C, Campino C, Inostroza A, Fardella C. Birth weight is inversely associated with blood pressure and serum aldosterone and cortisol levels in children. Clin Endocrinol (Oxf) 2012; 76 (5): 713–718. [DOI] [PubMed] [Google Scholar]
- Strufaldi MW, Silva EM, Franco MC, Puccini RF. Blood pressure levels in childhood: probing the relative importance of birth weight and current size. Eur J Pediatr 2009; 168 (5): 619–624. [DOI] [PubMed] [Google Scholar]
- Salgado CM, Jardim PC, Teles FB, Nunes MC. Influence of low birth weight on microalbuminuria and blood pressure of school children. Clin Nephrol 2009; 71 (4): 367–374. [DOI] [PubMed] [Google Scholar]
- Pereira JA, Rondó PH, Lemos JO, Pacheco de Souza JM, Dias RS. The influence of birthweight on arterial blood pressure of children. Clin Nutr 2010; 29 (3): 337–340. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang ZX, George LK, Wang ZS, Fan ZJ, Xu T, Zhou XL, Han SM, Wen HB, Zeng Y. Birth measurements, family history, and environmental factors associated with later-life hypertensive status. Am J Hypertens 2012; 25 (4): 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG, Forsén T, Tuomilehto J, Osmond C, Barker DJ. Early growth and coronary heart disease in later life: longitudinal study. BMJ 2001; 322 (7292): 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkner B, Hulman S, Kushner H. Effect of birth weight on blood pressure and body size in early adolescence. Hypertension 2004; 43 (2): 203–207. [DOI] [PubMed] [Google Scholar]
- Eriksson JG. Early growth and coronary heart disease and type 2 diabetes: findings from the Helsinki Birth Cohort Study (HBCS). Am J Clin Nutr 2011; 94 (6 Suppl): 1799S–1802S. [DOI] [PubMed] [Google Scholar]
- Mackenzie HS, Brenner BM. Fewer nephrons at birth: a missing link in the etiology of essential hypertension? Am J Kidney Dis 1995; 26: 91–98. [DOI] [PubMed] [Google Scholar]
- Silver LE, Decamps PJ, Korst LM, Platt LD, Castro L. Intrauterine growth restriction is accompanied by decreased renal volume in the human fetus. Am J Obstet Gynecol 2003; 188 (5): 1320–1325. [DOI] [PubMed] [Google Scholar]
- Hughson M, Farris AB, III, , Douglas-Denton R, Hoy WE, Bertram JF. Glomerular number and size in autopsy kidneys: the relationship to birth weight. Kidney Int 2003; 63 (6): 2113–2122. [DOI] [PubMed] [Google Scholar]
- Hoy WE, Hughson MD, Singh GR, Douglas-Denton R, Bertram JF. Reduced nephron number and glomerulomegaly in Australian Aborigines: a group at high risk for renal disease and hypertension. Kidney Int 2006; 70 (1): 104–110. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Westcott K, Siebel AL, Owens JA, Moritz KM. Growth restriction before or after birth reduces nephron number and increases blood pressure in male rats. Kidney Int 2008; 74 (2): 187–195. [DOI] [PubMed] [Google Scholar]
- Woods LL. Neonatal uninephrectomy causes hypertension in adult rats. Am J Physiol 1999; 276 (4 Pt 2): R974–R978. [DOI] [PubMed] [Google Scholar]
- Ruta LA, Dickinson H, Thomas MC, Denton KM, Anderson WP, Kett MM. High-salt diet reveals the hypertensive and renal effects of reduced nephron endowment. Am J Physiol Renal Physiol 2010; 298 (6): F1384–F1392. [DOI] [PubMed] [Google Scholar]
- Quigley R. Developmental changes in renal function. Curr Opin Pediatr 2012; 24 (2): 184–190. [DOI] [PubMed] [Google Scholar]
- Fanni D, Gerosa C, Nemolato S, Mocci C, Pichiri G, Coni P, Congiu T, Piludu M, Piras M, Fraschini M, Zaffanello M, Iacovidou N, et al. “Physiological” renal regenerating medicine in VLBW preterm infants: could a dream come true? J Matern Fetal Neonatal Med 2012; 25 (Suppl 3): 41–48. [DOI] [PubMed] [Google Scholar]
- Sutherland MR, Gubhaju L, Moore L, Kent AL, Dahlstrom JE, Horne RS, Hoy WE, Bertram JF, Black MJ. Accelerated maturation and abnormal morphology in the preterm neonatal kidney. J Am Soc Nephrol 2011; 22 (7): 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon S. Developmental changes in nephron number, proximal tubular length and superficial nephron glomerular filtration rate of rats. J Physiol 1977; 272: 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufro-McReddie A, Romano LM, Harris JM, Ferder L, Gomez RA. Angiotensin II regulates nephrogenesis and renal vascular development. Am J Physiol 1995; 269: F110–F115. [DOI] [PubMed] [Google Scholar]
- Saez F, Castells MT, Zuasti A, Salazar F, Reverte V, Loria A, Salazar FJ. Sex differences in the renal changes elicited by angiotensin II blockade during the nephrogenic period. Hypertension 2007; 49 (6): 1429–1435. [DOI] [PubMed] [Google Scholar]
- Hughson MD, Douglas-Denton R, Bertram JF, Hoy WE. Hypertension, glomerular number, and birth weight in African Americans and white subjects in the southeastern United States. Kidney Int 2006; 69 (4): 671–678. [DOI] [PubMed] [Google Scholar]
- McNamara BJ, Diouf B, Hughson MD, Hoy WE, Bertram JF. Associations between age, body size and nephron number with individual glomerular volumes in urban West African males. Nephrol Dial Transplant 2009; 24: 1500–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackland DT, Bendall HE, Osmond C, Egan BM, Barker DJ. Low birth weights contribute to the high rates of early-onset chronic renal failure in the southeastern United States. Arch Intern Med 2000; 160: 1472–1476. [DOI] [PubMed] [Google Scholar]
- Hoy WE, Rees M, Kile E, Mathews JD, Wanng Z. A new dimension to the Barker hypothesis: low birthweight and susceptibility to renal disease. Kidney Int 1999; 56: 1072–1077. [DOI] [PubMed] [Google Scholar]
- Szabo AJ, Muller V, Chen GF, Samsell LJ, Erdely A, Baylis C. Nephron number determines susceptibility to renal mass reduction-induced CKD in Lewis and Fisher 344 rats: implications for development of experimentally induced chronic allograft nephropathy. Nephrol Dial Transplant 2008; 23: 2492–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda NB. Low birth weight increases susceptibility to renal injury in a rat model of mild ischemia-reperfusion. Am J Physiol Renal Physiol 2011; 301 (2): F420–F426. [DOI] [PubMed] [Google Scholar]
- Lucas SR. Costa Silva VL, Miraglia SM, Zaladek-Gil F. Functional and morphometric evaluation of offspring kidney after intrauterine undernutrition. Pediatr Nephrol 1997; 11: 719–723. [DOI] [PubMed] [Google Scholar]
- Luzardo R, Silva PA, Einicker-Lamas M, Ortiz-Costa S, do Carmo Mda G, Vieira-Filho LD, Paixão AD, Lara LS, Vieyra A. Metabolic programming during lactation stimulates renal Na+ transport in the adult offspring due to an early impact on local angiotensin II pathways. PLoS One 2011; 6 (7): e21232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelievre-Pegorier M, Vilar J, Ferrier ML, Moreau E, Freund N, Gilbert T, Merlet-Benichou C. Mild vitamin A deficiency leads to inborn nephron deficit in the rat. Kidney Int 1998; 54: 1455–1462. [DOI] [PubMed] [Google Scholar]
- Drake KA, Sauerbry MJ, Blohowiak SE, Repyak KS, Kling PJ. Iron deficiency and renal development in the newborn rat. Pediatr Res 2009; 66: 619–624. [DOI] [PubMed] [Google Scholar]
- Woods LL, Ingelfinger JR, Rasch R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am J Physiol Regul Integr Comp Physiol 2005; 289 (4): R1131–R1136. [DOI] [PubMed] [Google Scholar]
- Iosipiv IV, Schroeder M. A role for angiotensin II AT1 receptors in ureteric bud cell branching. Am J Physiol Renal Physiol 2003; 285 (2): F199–F207. [DOI] [PubMed] [Google Scholar]
- Yosypiv IV, Schroeder M, El-Dahr SS. Angiotensin II type 1 receptor-EGF receptor cross-talk regulates ureteric bud branching morphogenesis. J Am Soc Nephrol 2006; 17 (4): 1005–1014. [DOI] [PubMed] [Google Scholar]
- Yosypiv IV, Boh MK, Spera MA, El-Dahr SS. Downregulation of Spry-1, an inhibitor of GDNF/Ret, causes angiotensin II-induced ureteric bud branching. Kidney Int 2008; 74 (10): 1287–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si H, Banga RS, Kapitsinou P, Ramaiah M, Lawrence J, Kambhampati G, Gruenwald A, Bottinger E, Glicklich D, Tellis V, Greenstein S, Thomas DB, et al. Human and murine kidneys show gender- and species-specific gene expression differences in response to injury. PLoS One 2009; 4 (3): e4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Khodus GR, Kruusmägi M, Kamali-Zare P, Liu XL, Eklöf AC, Zelenin S, Brismar H, Aperia A. Ouabain protects against adverse developmental programming of the kidney. Nat Commun 2010; 1: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodus GR, Kruusmägi M, Li J, Liu XL, Aperia A. Calcium signaling triggered by ouabain protects the embryonic kidney from adverse developmental programming. Pediatr Nephrol 2011; 26 (9): 1479–1482. [DOI] [PubMed] [Google Scholar]
- Gilbert JS, Lang AL, Grant AR, Nijland MJ. Maternal nutrient restriction in sheep: hypertension and decreased nephron number in offspring at 9 months of age. J Physiol 2005; 565 (Pt 1): 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins JP, Monteiro JC, Paixão AD. Renal function in adult rats subjected to prenatal dexamethasone. Clin Exp Pharmacol Physiol 2003; 30 (1–2): 32–37. [DOI] [PubMed] [Google Scholar]
- McMullen S, Langley-Evans SC. Sex-specific effects of prenatal low-protein and carbenoxolone exposure on renal angiotensin receptor expression in rats. Hypertension 2005; 46 (6): 1374–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson H, Walker DW, Wintour EM, Moritz K. Maternal dexamethasone treatment at midgestation reduces nephron number and alters renal gene expression in the fetal spiny mouse. Am J Physiol Regul Integr Comp Physiol 2007; 292 (1): R453–R461. [DOI] [PubMed] [Google Scholar]
- Tay S, Blache D, Gregg K, Revell D. Consumption of a high-salt diet by ewes during pregnancy alters nephrogenesis in 5-month-old offspring. Animal 2012; 6 (11): 1803–1810. [DOI] [PubMed] [Google Scholar]
- Beauséjour A, Auger K, St-Louis J, Brochu M. High-sodium intake prevents pregnancy-induced decrease of blood pressure in the rat. Am J Physiol Heart Circ Physiol 2003; 285 (1): H375–H383. [DOI] [PubMed] [Google Scholar]
- Ding Y, Lv J, Mao C, Zhang H, Wang A, Zhu L, Zhu H, Xu Z. High-salt diet during pregnancy and angiotensin-related cardiac changes. J Hypertens 2010; 28 (6): 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbi AP, Costa RS, Coimbra TM. Postnatal renal development of rats from mothers that received increased sodium intake. Pediatr Nephrol 2004; 19 (11): 1212–1218. [DOI] [PubMed] [Google Scholar]
- Marin EC, Balbi AP, Francescato HD. Alves da Silva CG, Costa RS, Coimbra TM. Renal structure and function evaluation of rats from dams that received increased sodium intake during pregnancy and lactation submitted or not to 5/6 nephrectomy. Ren Fail 2008; 30 (5): 547–555. [DOI] [PubMed] [Google Scholar]
- Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA, Moritz KM. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J Am Soc Nephrol 2007; 18 (6): 1688–1696. [DOI] [PubMed] [Google Scholar]
- Moritz KM, Mazzuca MQ, Siebel AL, Mibus A, Arena D, Tare M, Owens JA, Wlodek ME. Uteroplacental insufficiency causes a nephron deficit, modest renal insufficiency but no hypertension with ageing in female rats. J Physiol 2009; 587 (Pt 11): 2635–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baserga M, Bares AL, Hale MA, Callaway CW, McKnight RA, Lane PH, Lane RH. Uteroplacental insufficiency affects kidney VEGF expression in a model of IUGR with compensatory glomerular hypertrophy and hypertension. Early Hum Dev 2009; 85 (6): 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer R, Walter B, Bauer K, Klupsch R, Patt S, Zwiener U. Intrauterine growth restriction reduces nephron number and renal excretory function in newborn piglets. Acta Physiol Scand 2002; 176 (2): 83–90. [DOI] [PubMed] [Google Scholar]
- Crowley SD, Coffman TM. Recent advances involving the renin-angiotensin system. Exp Cell Res 2012; 318 (9): 1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner PR, Hall JE, Guyton AC. Control of glomerular filtration rate: role of intrarenally formed angiotensin II. Am J Physiol 1984; 246 (6 Pt 2): F897–F906. [DOI] [PubMed] [Google Scholar]
- Langley-Evans SC, Jackson AA. Captopril normalises systolic blood pressure in rats with hypertension induced by fetal exposure to maternal low protein diets. Comp Biochem Physiol A Physiol 1995; 110: 223–228. [DOI] [PubMed] [Google Scholar]
- Vehaskari VM, Aviles DH, Manning J. Prenatal programming of adult hypertension in the rat. Kidney Int 2001; 59: 238–245. [DOI] [PubMed] [Google Scholar]
- Vehaskari VM, Stewart T, Lafont D, Soyez C, Seth D, Manning J. Kidney angiotensin and angiotensin receptor expression in prenatally programmed hypertension. Am J Physiol Renal Physiol 2004; 287 (2): F262–F267. [DOI] [PubMed] [Google Scholar]
- Manning J, Vehaskari VM. Low birth weight-associated adult hypertension in the rat. Pediatr Nephrol 2001; 16 (5): 417–422. [DOI] [PubMed] [Google Scholar]
- Grigore D, Ojeda NB, Robertson EB, Dawson AS, Huffman C, Bourassa E, Speth RC, Brosnihan KB, Alexander BT. Placental insufficiency results in temporal alterations in the renin angiotensin system in male hypertensive growth restricted offspring. Am J Physiol Regul Integr Comp Physiol 2007; 293 (2): R804–R811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojeda NB, Grigore D, Yanes LL, Iliescu R, Robertson EB, Zhang H, Alexander BT. Testosterone contributes to marked elevations in mean arterial pressure in adult male intrauterine growth restricted offspring. Am J Physiol Regul Integr Comp Physiol 2007; 292 (2): R758–R763. [DOI] [PubMed] [Google Scholar]
- Wyrwoll CS, Mark PJ, Waddell BJ. Developmental programming of renal glucocorticoid sensitivity and the renin-angiotensin system. Hypertension 2007; 50: 579–584. [DOI] [PubMed] [Google Scholar]
- Mesquita FF, Gontijo JA, Boer PA. Expression of renin-angiotensin system signaling compounds in maternal protein-restricted rats: effect on renal sodium excretion and blood pressure. Nephrol Dial Transplant 2010; 25: 380–388. [DOI] [PubMed] [Google Scholar]
- Goyal R, Goyal D, Leitzke A, Gheorghe CP, Longo LD. Brain renin-angiotensin system: fetal epigenetic programming by maternal protein restriction during pregnancy. Reprod Sci 2010; 17 (3): 227–238. [DOI] [PubMed] [Google Scholar]
- Pladys P, Lahaie I, Cambonie G, Thibault G, Lê NL, Abran D, Nuyt AM. Role of brain and peripheral angiotensin II in hypertension and altered arterial baroreflex programmed during fetal life in rat. Pediatr Res 2004; 55 (6): 1042–1049. [DOI] [PubMed] [Google Scholar]
- Rocafull MA, Thomas LE, del Castillo JR. The second sodium pump: from the function to the gene. Pflugers Arch 2012; 463 (6): 755–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruso-Neves C, Rangel LB, Lara LS, Lopes AG. Regulation of the renal proximal tubule second sodium pump by angiotensins. Braz J Med Biol Res 2001; 34 (8): 1079–1084. [DOI] [PubMed] [Google Scholar]
- Proverbio F, Proverbio T, Marín R. Ouabain-insensitive Na+-stimulated ATPase activity of basolateral plasma membranes from guinea-pig kidney cortex cells. II. Effect of Ca2+. Biochim Biophys Acta 1982; 688 (3): 757–763. [DOI] [PubMed] [Google Scholar]
- Proverbio F, Proverbio T, Marín R. Na+-ATPase is a different entity from the (Na+ + K+)-ATPase in rat kidney basolateral plasma membranes. Biochim Biophys Acta 1986; 858 (1): 202–205. [DOI] [PubMed] [Google Scholar]
- Caruso-Neves C, Rangel LB, Vives D, Vieyra A, Coka-Guevara S, Lopes AG. Ouabain-insensitive Na+-ATPase activity is an effector protein for cAMP regulation in basolateral membranes of the proximal tubule. Biochim Biophys Acta 2000; 1468 (1–2): 107–114. [DOI] [PubMed] [Google Scholar]
- Queiroz-Madeira EP, Lara LS, Wengert M, Landgraf SS, Líbano-Soares JD, Zapata-Sudo G, Sudo RT, Takiya CM, Gomes-Quintana E, Lopes AG, Caruso-Neves C. Na+-ATPase in spontaneous hypertensive rats: possible AT1 receptor target in the development of hypertension. Biochim Biophys Acta 2010; 1798 (3): 360–366. [DOI] [PubMed] [Google Scholar]
- Carmosino M, Rizzo F, Ferrari P, Torielli L, Ferrandi M, Bianchi G, Svelto M, Valenti G. NKCC2 is activated in Milan hypertensive rats contributing to the maintenance of salt-sensitive hypertension. Pflugers Arch 2011; 462 (2): 281–291. [DOI] [PubMed] [Google Scholar]
- Aldred KL, Harris PJ, Eitle E. Increased proximal tubule NHE-3 and H+-ATPase activities in spontaneously hypertensive rats. J Hypertens 2000; 18 (5): 623–628. [DOI] [PubMed] [Google Scholar]
- Panico C, Luo Z, Damiano S, Artigiano F, Gill P, Welch WJ. Renal proximal tubular reabsorption is reduced in adult spontaneously hypertensive rats: roles of superoxide and Na+/H+ exchanger 3. Hypertension 2009; 54 (6): 1291–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crajoinas RO, Lessa LM, Carraro-Lacroix LR, Davel AP, Pacheco BP, Rossoni LV, Malnic G, Girardi AC. Posttranslational mechanisms associated with reduced NHE3 activity in adult vs. young prehypertensive SHR. Am J Physiol Renal Physiol 2010; 299 (4): F872–F881. [DOI] [PubMed] [Google Scholar]
- Bertram C, Trowern AR, Copin N, Jackson AA, Whorwood CB. The maternal diet during pregnancy programs altered expression of the glucocorticoid receptor and type 2 11β-hydroxysteroid dehydrogenase: potential molecular mechanisms underlying the programming of hypertension in utero. Endocrinology 2001; 142: 2841–2853. [DOI] [PubMed] [Google Scholar]
- Féraille E, Doucet A. Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiol Rev 2001; 81: 345–418. [DOI] [PubMed] [Google Scholar]
- Manning J, Beutler K, Knepper MA, Vehaskari VM. Upregulation of renal BSC1 and TSC in prenatally programmed hypertension. Am J Physiol Renal Physiol 2002; 283 (1): F202–F206. [DOI] [PubMed] [Google Scholar]
- Dagan A, Habib S, Gattineni J, Dwarakanath V, Baum M. Prenatal programming of rat thick ascending limb chloride transport by low-protein diet and dexamethasone. Am J Physiol Regul Integr Comp Physiol 2009; 297 (1): R93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton N, Al-Wasil SH, Bond H, Berry JL, Denton J, Freemont AJ. The effect of a low-protein diet in pregnancy on offspring renal calcium handling. Am J Physiol Regul Integr Comp Physiol 2007; 293 (2): R759–R765. [DOI] [PubMed] [Google Scholar]
- Dagan A, Kwon HM, Dwarakanath V, Baum M. Effect of renal denervation on prenatal programming of hypertension and renal tubular transporter abundance. Am J Physiol Renal Physiol 2008; 295 (1): F29–F34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagan A, Gattineni J, Cook V, Baum M. Prenatal programming of rat proximal tubule Na+/H+ exchanger by dexamethasone. Am J Physiol Regul Integr Comp Physiol 2007; 292 (3): R1230–R1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira-Filho LD, Lara LS, Silva PA, Luzardo R, Einicker-Lamas M, Cardoso HD, Paixão AD, Vieyra A. Placental oxidative stress in malnourished rats and changes in kidney proximal tubule sodium ATPases in offspring. Clin Exp Pharmacol Physiol 2009; 36 (12): 1157–1163. [DOI] [PubMed] [Google Scholar]
- Vieira-Filho LD, Farias JS, Cabral EV, Silva PA, Paixao AD, Vieyra A. Early changes on proximal tubule Na+-ATPase activity precede blood pressure elevation and renal dysfunction induced by intrauterine undernutrition: reprogramming by alpha-tocopherol. FASEB J 2013; 27: 907.3. 23180826 [Google Scholar]
- Vieira-Filho LD, Lara LS, Silva PA, Santos FT, Luzardo R, Oliveira FS, Paixão AD, Vieyra A. Placental malnutrition changes the regulatory network of renal Na-ATPase in adult rat progeny: reprogramming by maternal α-tocopherol during lactation. Arch Biochem Biophys 2011; 505 (1): 91–97. [DOI] [PubMed] [Google Scholar]
- Rangel LB, Lopes AG, Lara LS, Carvalho TL, Silva IV, Oliveira MM, Einicker-Lamas M, Vieyra A, Nogaroli L, Caruso-Neves C. PI-PLCbeta is involved in the modulation of the proximal tubule Na+-ATPase by angiotensin II. Regul Pept 2005; 127 (1–3): 177–182. [DOI] [PubMed] [Google Scholar]
- Feng D, Yang C, Geurts AM, Kurth T, Liang M, Lazar J, Mattson DL, O'Connor PM, Cowley AW., Jr. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab 2012; 15 (2): 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Cowley AW, Jr, , Ito S. Molecular mechanisms and therapeutic strategies of chronic renal injury: physiological role of angiotensin II-induced oxidative stress in renal medulla. J Pharmacol Sci 2006; 100: 2–8. [DOI] [PubMed] [Google Scholar]
- Evans RG, Fitzgerald SM. Nitric oxide and superoxide in the renal medulla: a delicate balancing act. Curr Opin Nephrol Hypertens 2005; 14 (1): 9–15. [DOI] [PubMed] [Google Scholar]
- Evans RG, Majid DS, Eppel GA. Mechanisms mediating pressure natriuresis: what we know and what we need to find out. Clin Exp Pharmacol Physiol 2005; 32 (5–6): 400–409. [DOI] [PubMed] [Google Scholar]
- Kawarazaki H, Ando K, Shibata S, Muraoka K, Fujita M, Kawarasaki C, Fujita T. Mineralocorticoid receptor-Rac1 activation and oxidative stress play major roles in salt-induced hypertension and kidney injury in prepubertal rats. J Hypertens 2012; 30 (10): 1977–1985. [DOI] [PubMed] [Google Scholar]
- Rozansky DJ. The role of aldosterone in renal sodium transport. Semin Nephrol 2006; 26 (2): 173–181. [DOI] [PubMed] [Google Scholar]
- Stewart T, Jung FF, Manning J, Vehaskari VM. Kidney immune cell infiltration and oxidative stress contribute to prenatally programmed hypertension. Kidney Int 2005; 68 (5): 2180–2188. [DOI] [PubMed] [Google Scholar]
- Magalhães JC, da Silveira AB, Mota DL, Paixão AD. Renal function in juvenile rats subjected to prenatal malnutrition and chronic salt overload. Exp Physiol 2006; 91: 611–619. [DOI] [PubMed] [Google Scholar]
- Silva LA, Vieira-Filho LD, Barreto IS, Cabral EV, Vieyra A, Paixão AD. Prenatal undernutrition changes renovascular responses of nimesulide in rat kidneys. Basic Clin Pharmacol Toxicol 2011; 108: 115–121. [DOI] [PubMed] [Google Scholar]
- Ojeda NB, Hennington BS, Williamson DT, Hill ML, Betson NE, Sartori-Valinotti JC, Reckelhoff JF, Royals TP, Alexander BT. Oxidative stress contributes to sex differences in blood pressure in adult growth-restricted offspring. Hypertension 2012; 60 (1): 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral EV, Vieira-Filho LD, Silva PA, Nascimento WS, Aires RS, Oliveira FS, Luzardo R, Vieyra A, Paixão AD. Perinatal Na+ overload programs raised renal proximal Na+ transport and enalapril-sensitive alterations of Ang II signaling pathways during adulthood. PLoS One 2012; 7 (8): e43791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso HD, Cabral EV, Vieira-Filho LD, Vieyra A, Paixão AD. Fetal development and renal function in adult rats prenatally subjected to sodium overload. Pediatr Nephrol 2009; 24 (10): 1959–1965. [DOI] [PubMed] [Google Scholar]
- Reverte V, Tapia A, Baile G, Gambini J, Gíménez I, Llinas MT, Salazar FJ. Role of angiotensin II in arterial pressure and renal hemodynamics in rats with altered renal development: age- and sex-dependent differences. Am J Physiol Renal Physiol 2013; 304 (1): F33–F40. [DOI] [PubMed] [Google Scholar]
- Vieira-Filho LD, Cabral EV, Santos FT, Coimbra TM, Paixão AD. Alpha-tocopherol prevents intrauterine undernutrition-induced oligonephronia in rats. Pediatr Nephrol 2011; 26 (11): 2019–2029. [DOI] [PubMed] [Google Scholar]
- Bhatia K, Elmarakby AA, El-Remessy AB, Sullivan JC. Oxidative stress contributes to sex differences in angiotensin II-mediated hypertension in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol 2012; 302 (2): R274–R282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araujo M, Wilcox CS. Oxidative stress in hypertension: role of the kidney. Antioxid Redox Signal 2013; (in press). Published online ahead of print 30 April 2013; DOI 10.1089/ars.2013.5259. [DOI] [PMC free article] [PubMed]
- Rexhaj E, Bloch J, Jayet PY, Rimoldi SF, Dessen P, Mathieu C, Tolsa JF, Nicod P, Scherrer U, Sartori C. Fetal programming of pulmonary vascular dysfunction in mice: role of epigenetic mechanisms. Am J Physiol Heart Circ Physiol 2011; 301 (1): H247–H252. [DOI] [PubMed] [Google Scholar]
- Roghair RD, Wemmie JA, Volk KA, Scholz TD, Lamb FS, Segar JL. Maternal antioxidant blocks programmed cardiovascular and behavioural stress responses in adult mice. Clin Sci (Lond) 2011; 121 (10): 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrwoll CS, Mark PJ, Mori TA, Puddey IB, Waddell BJ. Prevention of programmed hyperleptinemia and hypertension by postnatal dietary omega-3 fatty acids. Endocrinology 2006; 147: 599–606. [DOI] [PubMed] [Google Scholar]
- An WS, Kim HJ, Cho KH, Vaziri ND. Omega-3 fatty acid supplementation attenuates oxidative stress, inflammation, and tubulointerstitial fibrosis in the remnant kidney. Am J Physiol Renal Physiol 2009; 297 (4): F895–F903. [DOI] [PubMed] [Google Scholar]
- Baum M. Developmental changes in proximal tubule NaCl transport. Pediatr Nephrol 2008; 23 (2): 185–194. [DOI] [PubMed] [Google Scholar]
- Manning J, Vehaskari VM. Postnatal modulation of prenatally programmed hypertension by dietary Na and ACE inhibition. Am J Physiol Regul Integr Comp Physiol 2005; 288: R80–R84. [DOI] [PubMed] [Google Scholar]
- Stewart T, Ascani J, Craver RD, Vehaskari VM. Role of postnatal dietary sodium in prenatally programmed hypertension. Pediatr Nephrol 2009; 24: 1727–1733. [DOI] [PubMed] [Google Scholar]
- Harrap SB, Van der Merwe WM, Griffin SA, Macpherson F, Lever AF. Brief angiotensin converting enzyme inhibitor treatment in young spontaneously hypertensive rats reduces blood pressure long-term. Hypertension 1990; 16 (6): 603–614. [DOI] [PubMed] [Google Scholar]
- Quan A. Fetopathy associated with exposure to angiotensin converting enzyme inhibitors and angiotensin receptor antagonists. Early Hum Dev 2006; 82 (1): 23–28. [DOI] [PubMed] [Google Scholar]
- Laube GF, Kemper MJ, Schubiger G, Neuhaus TJ. Angiotensin-converting enzyme inhibitor fetopathy: long-term outcome. Arch Dis Child Fetal Neonatal Ed 2007; 92 (5): F402–F403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson AB, Nitescu N, Chen Y, Guron GS, Marcussen N, Matejka GL, Friberg P. IGF-I treatment attenuates renal abnormalities induced by neonatal ACE inhibition. Am J Physiol Regul Integr Comp Physiol 2000; 279 (3): R1050–R1060. [DOI] [PubMed] [Google Scholar]
- Lasaitiene D, Chen Y, Guron G, Marcussen N, Tarkowski A, Telemo E, Friberg P. Perturbed medullary tubulogenesis in neonatal rat exposed to renin-angiotensin system inhibition. Nephrol Dial Transplant 2003; 18 (12): 2534–2541. [DOI] [PubMed] [Google Scholar]
- Coleman CM, Minor JJ, Burt LE, Thornhill BA, Forbes MS, Chevalier RL. Angiotensin AT1-receptor inhibition exacerbates renal injury resulting from partial unilateral ureteral obstruction in the neonatal rat. Am J Physiol Renal Physiol 2007; 293 (1): F262–F268. [DOI] [PubMed] [Google Scholar]
- Vickers MH, Breier BH, Cutfield WS, Hofman PL, Gluckman PD. Fetal origins of hyperphagia, obesity, and hypertension and postnatal amplification by hypercaloric nutrition. Am J Physiol Endocrinol Metab 2000; 279 (1): E83–E87. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.