

ABSTRACT
As the central component of canonical TGFbeta superfamily signaling, SMAD4 is a critical regulator of organ development, patterning, tumorigenesis, and many other biological processes. Because numerous TGFbeta superfamily ligands are expressed in developing testes, there may exist specific requirements for SMAD4 in individual testicular cell types. Previously, we reported that expansion of the fetal testis cords requires expression of SMAD4 by the Sertoli cell lineage. To further uncover the role of Smad4 in murine testes, we produced conditional knockout mice lacking Smad4 in either Leydig cells or in both Sertoli and Leydig cells simultaneously. Loss of Smad4 concomitantly in Sertoli and Leydig cells led to underdevelopment of the testis cords during fetal life and mild testicular dysgenesis in young adulthood (decreased testis size, partially dysgenic seminiferous tubules, and low sperm production). When the Sertoli/Leydig cell Smad4 conditional knockout mice aged (56- to 62-wk old), the testis phenotypes became exacerbated with the appearance of hemorrhagic tumors, Leydig cell adenomas, and a complete loss of spermatogenesis. In contrast, loss of Smad4 in Leydig cells alone did not appreciably alter fetal and adult testis development. Our findings support a cell type-specific requirement of Smad4 in testis development and suppression of testicular tumors.
Keywords: azoospermia, hemorrhage, Leydig cell hyperplasia, mouse, Smad, teratoma, testicular dysgenesis, testis
Concurrent deletion of Smad4 in mouse Sertoli and Leydig cells results in altered testis development during embryogenesis and defects in testis function with age with the eventual onset of hemorrhagic testicular tumors.
INTRODUCTION
The transforming growth factor β (TGFβ) superfamily of growth factors regulate a host of cellular processes ranging from axis formation and tissue patterning during fetal development to the modulation of diseases such as hereditary hemorrhagic telangiectasia and cancer [1–3]. Thus far, two different canonical SMAD pathways have been identified that can be activated by TGFβ superfamily ligands. The identity of the ligand determines which SMAD pathway is utilized by the target cell; specifically, activins and TGFβs activate SMAD2 and SMAD3 receptor SMADs, whereas bone morphogenetic proteins (BMPs) activate receptor SMAD1, SMAD5, and SMAD8 [4]. In canonical TGFβ superfamily signaling, SMAD4 serves as the central regulator, binding to either set of receptor SMADs and escorting them into the nucleus to modulate gene transcription [5, 6].
The TGFβ superfamily has been implicated in the development of many embryonic tissues, including the testes. Numerous TGFβ superfamily ligands, including anti-Müllerian hormone (AMH), TGFβ1, TGFβ2, TGFβ3, activin A, and activin B have been studied in fetal testes through the use of various knockout mouse technologies [7–12]. In mice, Smad4 mRNA transcripts are present in Sertoli cells, gonocytes, and interstitial cells at the time of birth [13]. Because SMAD4 is the central component of canonical TGFβ superfamily signaling, we hypothesized disruption of Smad4 expression in testicular somatic cells would alter fetal development and thus adult function of the testes. Indeed, loss of Smad4 in Sertoli cells led to dysgenesis of testis cords as a result of decreased Sertoli cell proliferation [12]. In this study, we first investigated the importance of Smad4 in the interstitial Leydig cells, which express high levels of SMAD4 and are established targets of AMH [7, 14]. Furthermore, we postulated that given the potential for cross-talk between the two cell populations, concurrent loss of Smad4 expression in Sertoli and Leydig cells might uncover new roles for Smad4 in these testicular somatic cells not revealed by removal of Smad4 in either cell population alone. Therefore, we also created and analyzed a mouse model lacking Smad4 in both the Sertoli and Leydig cell populations.
MATERIALS AND METHODS
Generation of Conditional Knockout Mice
Leydig cell Smad4 conditional knockout (cKO) mice (Amhr2cre/+;Smad4fl/−) were generated by mating Smad4+/− mice to AMH type 2 receptor (Amhr2)-Cre or Amhr2cre/+ transgenic mice; the resulting Amhr2cre/+;Smad4+/− mice were then crossed to Smad4fl/fl animals [15]. To produce Sertoli and Leydig cell Smad4 cKO mice (Sf1cre/+;Smad4fl/−), Smad4+/− animals were mated to Steroidogenic factor 1 or Sf1cre/+ transgenic mice [16]. The resulting Sf1cre/+;Smad4+/− mice were then crossed to Smad4fl/fl animals [17]. All the mouse strains were maintained on a mixed C57BL/6J/129 genetic background. For fetal analysis, timed matings were produced by housing female mice with males overnight and checking for vaginal plugs the next morning (E0.5 = noon of the day when a vaginal plug was found). Fetal tissue was collected from E12.5 to E19.5. For adult analysis, Sertoli and Leydig cell Smad4 cKO, Sertoli and Leydig cell Smad4 control (Sf1cre/+;Smad4+/fl), Leydig cell Smad4 cKO, and Leydig cell Smad4 control (Amhr2cre/+;Smad4+/fl) males were collected at 12- to 16- or at 56- to 62-wk of age. All the procedures described were reviewed and approved by the Institutional Animal Care and Use Committees of University of Illinois and National Institute of Environmental Health Sciences and were performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD).
Immunohistochemistry and Histology
For immunohistochemistry of fetal samples, testes were fixed in 4% paraformaldehyde, dehydrated through a sucrose gradient, and cryosectioned. The sections were incubated with primary antibody against laminin (1:200, L9393; Sigma) and fluorescein isothiocyanate-conjugated secondary antibody (1:500, Jackson Immuno, Inc.) as previously described [12]. For histological analysis, E19.5 and adult testis samples were fixed in Bouin solution, and paraffin sections were stained with hematoxylin and eosin (H&E).
Daily Sperm Production
Analysis and calculation of daily sperm production (DSP) followed the procedure of Joyce et al. [18] with slight modifications. Testes were homogenized for 30 sec using a Polytron homogenizer, and spermatids were then counted on a hemocytometer.
Computer-Assisted Sperm Analysis
To assess the quantity of epididymal sperm, the left cauda epididymis of young adult (12- to 16-wk old) or aged adult (56- to 62-wk-old) mice was removed within 2 min of carbon dioxide asphyxiation. Cauda epididymides were minced in 1 ml of dmKRBT (120 mM NaCl, 2 mM KCl, 2 mM CaCl2, 10 mM NaHCO3, 1.2 mM MgSO4·7H20, 5.6 mM glucose, 1.1 mM sodium pyruvate, 25 mM TAPSO [2-{N-tris<hydroxymethyl>methylamino}-2 hydroxy propane sulfonic acid], 18.5 mM sucrose, 6 mg/ml BSA, pH 7.3), and the sperm were allowed to swim out for 5 min at 37°C. The medium was then diluted and placed onto a glass cannula for computer-assisted sperm analysis using the integrated visual optical system motility analyzer (Hamilton-Throne Research). The operational settings of the integrated visual optical system were the standard mouse parameters recommended by the manufacturer. For each sample, six scans were analyzed.
Hormone Analysis
Plasma FSH levels were measured in duplicate by radioimmunoassay according to instructions with kits from the National Hormone and Pituitary Distribution Program. The sensitivity of the FSH assay was 1.0 ng/ml, and the intraassay coefficient of variability was 1.22%. Radioimmunoassay results were calculated by four-parameter logistic analysis using AssayZap (BioSoft).
Statistical Analysis
Statistical differences were determined via two-tailed t-test comparisons.
RESULTS
Loss of Smad4 in Both Sertoli and Leydig Cells, but Not Leydig Cells Alone, Results in Testis Cord Dysgenesis
To investigate the requirement for Smad4 in somatic cells of murine fetal testes, we inactivated Smad4 either in the fetal Leydig cells via Amhr2-Cre (hereafter referred to as Leydig Smad4 cKO) or in the precursors of both Sertoli and Leydig cells via Sf1-Cre (hereafter referred to as Sertoli/Leydig Smad4 cKO). Previously, we reported that loss of Smad4 in Sertoli cells via Amh-Cre in fetal testes resulted in underdevelopment of the testis cords after E15.5 and that this developmental defect led to oligozoospermia and testicular dysgenesis in adulthood [12]. To determine whether similar abnormalities arose in Leydig Smad4 cKO and Sertoli/Leydig Smad4 cKO embryos, we analyzed testis morphogenesis at E15.5 (before the onset of testis cord expansion) and at E19.5 (the time of birth). Immunofluorescence for laminin was used to demarcate the basal lamina at the boundary of the testis cords in transverse sections (Fig. 1). In the control (Fig. 1A), Leydig Smad4 cKO (Fig. 1B), and Sertoli/Leydig Smad4 cKO (Fig. 1C) testes, testis cords displayed the anticipated transverse circular loop structure at E15.5. This indicated establishment of the testis cords was not altered despite the early expression of gonadal Cre recombinase in the mouse strains we chose (by E10.5 in Sf1-Cre and E12.5 in Amhr2-Cre). Around the time of birth (E19.5), transverse sections of testes from control (Fig. 1D) and Leydig Smad4 cKO (Fig. 1E) contained numerous small testis cord cross-sections, hallmarks of proper testis cord coiling and expansion. In contrast, testis cords in transverse sections from E19.5 Sertoli/Leydig Smad4 cKO mice (Fig. 1F) resembled more closely those of E15.5 testes than of E19.5 control testes (Fig. 1D). Despite differences in testis cord morphology, both Leydig Smad4 cKO and Sertoli/Leydig Smad4 cKO mice were normally masculinized and did not differ from controls with regard to anogenital distance, testicular descent, or development of secondary sexual organs (data not shown). Fetal analyses revealed that loss of Smad4 in both Sertoli and Leydig cells alters testis cord development during late embryogenesis, similar to the effect of loss of Smad4 in the fetal Sertoli cells [12]. In contrast, loss of Smad4 in the Leydig cells alone does not grossly affect testis cord structure during fetal life.
FIG. 1.

Testis morphogenesis in control and Smad4 conditional knockout mouse embryos at E15.5 and E19.5. A–F) Developmental time course of testes from control (A, D), Leydig Smad4 cKO (B, E), and Sertoli/Leydig Smad4 cKO (C, F) embryos. Bars = 100 μm. White arrows indicate underdeveloped testis cords. G–I) Whole mount images of testes in representative E19.5 control (G), Leydig Smad4 cKO (H), and Sertoli/Leydig Smad4 cKO mice (I). The epididymis was removed to allow for easier visualization of testicular hemorrhages in the Sertoli/Leydig Smad4 cKO testis (I). Yellow arrowhead indicates hemorrhagic regions. E, epididymis.
In addition to the testis cord and seminiferous tubule defects in Sertoli/Leydig Smad4 cKO newborns, we also observed abnormalities in testis vasculature. Hemorrhages (arrowheads in Fig. 1I) as well as aberrations in testicular shape were found in ∼80% of Sertoli/Leydig Smad4 cKO testes at E19.5. These hemorrhages extended from the testis surface toward the central regions. We did not observe hemorrhages or alterations of testicular shape in Leydig Smad4 cKO or control newborn mice (Fig. 1, G and H).
Leydig Smad4 cKO Male Mice Exhibit Normal Testicular Functions in Adulthood
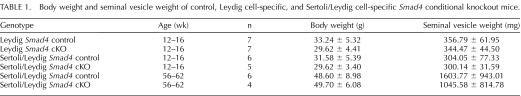
Although we did not detect gross alteration of fetal testis development in Leydig Smad4 cKO mice, we speculated the loss of Smad4 in Leydig cells could affect testicular functions in adulthood. We analyzed reproductive parameters (testis weight, DSP, sperm concentration, and plasma FSH levels) and testis histology in 12- to 16-wk-old Leydig Smad4 cKO mice as well as strain-specific controls (Fig. 2). No statistically significant differences were observed in testis weight (Fig. 2A), DSP (Fig. 2B), cauda epididymal sperm concentration (Fig. 2C), or plasma FSH (Fig. 2D) between Leydig Smad4 cKO mice and controls. Body weight or androgen-sensitive endpoints such as seminal vesicle weight were not statistically different from strain-specific controls (Table 1). Testis histology was indistinguishable between control (Fig. 2E) and Leydig Smad4 cKO (Fig. 2F) adult testes, as was the case at E19.5. Based on these data, we conclude that deletion of Smad4 within Leydig cells does not alter Leydig cell function or impair testis development.
FIG. 2.

Testis parameters and histology in young adult (12- to 16-wk old) control and Leydig Smad4 cKO mice. A–D) Testis weight (A), daily sperm production (DSP) per milligram testis weight (B), cauda epididymal sperm concentration (C), and plasma FSH levels (D) in control and Leydig Smad4 cKO mice. Values are given as mean ± standard deviation; n = 7 for both control and Leydig Smad4 cKO. E, F) H&E stained histological sections from control (E) and Leydig Smad4 cKO (F) testes shown at ×10 magnification. Bars = 250 μm.
TABLE 1.
Body weight and seminal vesicle weight of control, Leydig cell-specific, and Sertoli/Leydig cell-specific Smad4 conditional knockout mice.
Testis Dysgenesis Persists into Adulthood in Sertoli/Leydig Smad4 cKO Male Mice
In contrast to our observations in Leydig Smad4 cKO mice, the testes of 12- to 16-wk-old Sertoli/Leydig Smad4 cKO males were significantly smaller than those of strain-specific controls (Fig. 3A). Despite this striking difference in testis size, DSP (Fig. 3B), cauda epididymal sperm concentration (Fig. 3C), plasma FSH (Fig. 3D), and body weight and seminal vesicle weight (Table 1) did not statistically differ between Sertoli/Leydig Smad4 cKO and control mice at 12- to 16-wk of age. In light of the large standard deviation in DSP of Sertoli/Leydig Smad4 cKO mice, we performed statistical tests for outlier values (including Grubbs test and the extreme Studentized deviate method) and did not identify any significant outlier values. Additionally, statistical analysis utilizing a repeated measurement design for limited sample size indicated that increasing our sample size would decrease standard deviation but would not significantly affect our statistical P value and thus would not change our conclusion that DSP was not statistically different between Sertoli/Leydig Smad4 cKO mice and their age-matched controls. Because we did not have any justifiable scientific basis for excluding any of the cKO males from our analysis, we have herein reported DSP for the entire cohort despite the large standard deviation value.
FIG. 3.

Testis parameters and histology in young adult (12- to 16-wk old) control and Sertoli/Leydig Smad4 cKO mice. A–D) Testis weight (A), daily sperm production (DSP) per milligram testis weight (B), cauda epididymal sperm concentration (C), and plasma FSH levels (D) in control and Sertoli/Leydig Smad4 cKO mice. Asterisk indicates P < 0.0001. Values are given as mean ± standard deviation; n = 6 for controls; n = 5 for Sertoli/Leydig Smad4 cKO. Representative images of control (left) and Sertoli/Leydig Smad4 cKO (right) testes are shown at identical magnification in A. E–G) H&E stained histological sections from control (E) and Sertoli/Leydig Smad4 cKO (F, G) testes shown at ×20 magnification. Bars = 100 μm. Yellow asterisk indicates areas of Leydig cell hyperplasia. Yellow arrowhead indicates enlarged blood vessel.
Histological analysis of Sertoli/Leydig Smad4 cKO testes revealed a mixture of seminiferous tubules actively completing spermatogenesis and dysgenic tubules (Fig. 3E–G). The dysgenic tubules were variously characterized by vacuolization and loss of spermatogenic cells (Fig. 3F). Additionally, Sertoli/Leydig Smad4 cKO testes contained visibly larger areas of interstitial cells between tubules (asterisk in Fig. 3F) compared to control testes (Fig. 3E). Our findings indicate the testis cord abnormalities in Sertoli/Leydig Smad4 cKO embryos carry over into structural deficits of seminiferous tubules and patchy disruption of spermatogenesis in young adulthood. Interestingly, the hemorrhagic phenotype we observed in newborn Sertoli/Leydig Smad4 cKO testes (Fig. 1, G and H) appears to resolve after birth as we did not observe hemorrhages in 12- to 16-wk-old Sertoli/Leydig Smad4 cKO testes (Fig. 3A). However, some blood vessels within Sertoli/Leydig Smad4 cKO testes were enlarged (Fig. 3G, representative blood vessel measuring ∼200 μm × 140 μm), indicating ongoing abnormalities of testis vasculature.
Loss of Smad4 in Sertoli and Leydig Cells Leads to Seminiferous Tubule Degeneration, Leydig Cell Adenomas, and Testicular Tumors in Aged Mice
Based on the disorganized appearance of the seminiferous tubules in 12- to 16-wk-old Sertoli/Leydig Smad4 cKO testes, we hypothesized that testicular dysgenesis would worsen with age in this mouse model. We analyzed testes from 56- to 62-wk-old cKO and control mice (Fig. 4). As observed at 12–16 wk of age, testis size in aged Sertoli/Leydig Smad4 cKO mice was significantly smaller than in strain-specific age-matched controls (Fig. 4, A and E). Despite the difference in testis size, body weight and seminal vesicle weight did not statistically differ between aged controls and Sertoli/Leydig Smad4 cKO mice (Table 1). Anogenital distance was significantly increased (P < 0.01) in Sertoli/Leydig Smad4 cKO males compared to control males at 56–62 wk of age (Table 1). Thus, loss of Smad4 expression in Sertoli and Leydig cells beginning in fetal life did not compromise body size or decrease androgen-sensitive parameters such as seminal vesicle weight and anogenital distance.
FIG. 4.

Testis parameters and histology in aged (56- to 62-wk old) control and Smad4 Sertoli/Leydig cKO mice. A–D) Testis weight (A), daily sperm production (DSP) per milligram testis weight (B), cauda epididymal sperm concentration (C), and plasma FSH levels (D) in control and Sertoli/Leydig Smad4 cKO mice. Single asterisk indicates P < 0.0001; double asterisk indicates P < 0.02. Values are given as mean ± standard deviation; n = 6 for controls; n = 4 for Sertoli/Leydig Smad4 cKO. E) Representative Sertoli/Leydig Smad4 cKO testes exhibiting visible tumors at ×0.6 magnification with control testis (left) for size comparison. Asterisk indicates the knockout testis without visible tumor formation. F, G) H&E stained histological sections from aged Sertoli/Leydig Smad4 cKO testes with tumor formation at ×4 magnification. Black arrows indicate large blood vessels within hemorrhagic regions. Yellow arrowheads indicate areas containing degenerated seminiferous tubules. H) H&E stained histological sections from aged Sertoli/Leydig Smad4 cKO testis without tumor formation, denoted by an asterisk in E, at ×4 magnification.
Deletion of Smad4 in Sertoli and Leydig cells did, however, manifest as testicular pathology at 56–62 wk of age. Seven of eight (87.5%) Sertoli/Leydig Smad4 cKO testes were misshapen and exhibited obvious tumors, hemorrhages, and other abnormalities (Fig. 4E–H). Histological sections of the tumorous aged Sertoli/Leydig Smad4 cKO testes revealed the majority of these testes consisted of hemorrhagic tissues within a fibrous capsule (Fig. 4, F and G). The presence of abnormally large blood vessels (arrows in Fig. 4, F and G) and fibrous tissues suggested a process of chronic hemorrhaging followed by revascularization. Seminiferous tubules in tumorous Sertoli/Leydig Smad4 cKO testes were relegated to the outer edge of the testis; no seminiferous tubules were observed within the fibrous capsule or hemorrhagic regions (Fig. 4, F and G). The single Sertoli/Leydig Smad4 cKO testis that had no visible tumors (marked by an asterisk in Fig. 4E) did not develop any widespread hemorrhages or fibrosis but the seminiferous tubules were severely degenerated (Fig. 4H). Not surprisingly, we did not observe any sperm from aged Sertoli/Leydig Smad4 cKO mice via either DSP analysis or caudal epididymal sperm collection (Fig. 4, B and C). Plasma FSH levels were significantly reduced in aged Sertoli/Leydig Smad4 cKO mice compared to strain-specific controls (Fig. 4D; P < 0.02).
A closer pathological analysis in aged Sertoli/Leydig Smad4 cKO testes revealed a multitude of abnormalities not found in age- and strain-matched controls (Fig. 5). Whereas aged control testes were still undergoing normal spermatogenesis (Fig. 5A), none of the seminiferous tubules in Sertoli/Leydig Smad4 cKO testes had active spermatogenesis (Fig. 5B). In fact, Sertoli/Leydig Smad4 cKO seminiferous tubules lacked spermatogenic cells, and Sertoli cells could be observed clumped together in tubule lumens, having detached from the basement membrane (arrows in Fig. 5B). Enlarged blood vessels, seen in 12- to 16-wk-old Sertoli/Leydig Smad4 cKO males, were also observed in aged cKO testes (black arrowhead in Fig. 5C). In addition to the seminiferous tubule pathology, interstitial abnormalities were visible, including the presence of eosinic fluid between remaining tubules (Fig. 5C) and Leydig cell adenomas (Fig. 5, C and D). Multinucleated Leydig cells were observed within adenomas (yellow arrowheads in Fig. 5D). We did observe a teratoma containing chondrocytes and osteoblasts in one out of eight aged Sertoli/Leydig Smad4 cKO testes (Fig. 5E).
FIG. 5.

H&E stained sections from aged (56- to 62-wk old) control (A) and Sertoli/Leydig Smad4 cKO mice (B–E). Black arrows in B indicate degenerated seminiferous tubules with Sertoli cells sloughed off into the lumen. Black arrowhead in C indicates enlarged blood vessels within hemorrhagic regions. Yellow arrowheads in C and D indicate Leydig cell hyperplasia and multinucleated Leydig cells, respectively. E) Testicular teratoma containing chrondrocytes and osteoblasts was found in one of the 56-wk-old knockout testes. Bars = 100 μm (A–C) and 50 μm (D, E).
DISCUSSION
Leydig Cell Expression of Smad4 Is Not Required for Normal Testis Development
As the central component of canonical signaling for the largest known group of growth factors (TGFβ superfamily), SMAD4 is required for normal formation and function of a vast array of tissues. In particular, genetic manipulation studies in mice have revealed roles for a large number of TGFβ superfamily ligands and receptors in testes [19]. Despite the importance of TGFβ superfamily signaling for male reproduction, we did not observe testicular deficits in mice lacking Smad4 expression in Leydig cells from E11.5 onward. Although fetal and adult Leydig cells arise from two different precursor populations, both cell types have been shown to express Amhr2-Cre; therefore, our mouse model should lack SMAD4 in Leydig cells throughout life [20]. Our findings of normal testis histology and functions in Leydig Smad4 cKO mice suggest that although SMAD4 is reported to be highly expressed in Leydig cells, these cells may utilize SMAD4-independent pathways to transduce signals from critical TGFβ superfamily proteins [14]. Leydig cells are established targets of AMH and TGFβ, both of which negatively regulate proliferation of Leydig cell progenitors [7, 21]. The absence of excessive Leydig cell proliferation in Leydig Smad4 cKO mice suggests AMH and TGFβ activities remain intact and that these factors likely inhibit Leydig cell proliferation through a noncanonical, SMAD4-independent mechanism. On the other hand, the formation of Leydig cell adenomas observed in Sertoli/Leydig Smad4 cKO mice suggests loss of SMAD4 in Sertoli cells may indirectly disrupt normal Leydig cell behavior.
As with any study utilizing the Cre/loxP system to produce tissue- or cell type-specific cKO mice, concern remains that the floxed gene of interest was perhaps not efficiently removed by Cre recombinase. While the lack of gross phenotypic changes in our Leydig cell cKO model could be the result of inefficient recombination of the floxed allele, the Amhr2-Cre mouse strain has been effectively utilized to knock out a variety of genes in the gonads in more than 50 peer-reviewed scientific articles; a list of reference articles is available courtesy of Mouse Genome Informatics [22]. In addition, studies by our lab (including the Sertoli/Leydig Smad4 cKO mouse model presented here) and by a multitude of other groups have demonstrated phenotypes in a large number of tissue contexts utilizing the Smad4 flox/flox mouse strain crossed to various Cre-expressing mouse lines, resulting in more than 80 publications using this mouse strain to date [23]. As with our previous studies, we attempted to maximize the efficiency of the Cre recombinase by utilizing a breeding scheme requiring removal of only a single floxed allele (the other allele being a null allele) in order to produce a cKO animal [12, 24–29]. The two Cre recombinase lines in this study (Sf1-Cre and Amhr2-Cre) are both active in the fetal testis, allowing enough time for the Cre recombinase to exert its action on a single allele of floxed Smad4. Although we are not able to provide direct evidence of loss of Smad4, we feel our conclusion is justified based on the evidence in the literature and the design of genetic models.
Fetal Testis Dysgenesis in Sertoli/Leydig Smad4 cKO Mice Progresses to Cessation of Spermatogenesis in Adulthood
Testes from Sertoli/Leydig Smad4 cKO mice differed substantially from control testes at all the stages analyzed. At the newborn stage, Sertoli/Leydig Smad4 cKO testes displayed severe retardation of testis cord expansion, frequently coupled with abnormal testis shape and visible hemorrhages. This underdevelopment of the fetal testis cords gave way to significantly reduced testis size and a trend of decreasing sperm production in young adult Sertoli/Leydig Smad4 cKO males. By 1-yr of age, testes of Sertoli/Leydig Smad4 cKO mice had degenerated to consist largely of chronically hemorrhaged fibrous tissue and Leydig cell adenomas surrounded by a few residual seminiferous tubules that completely lack spermatogenic cells.
Consideration of the two Smad4 cKO mouse models presented here and our previous report [12] of a Sertoli cell-specific Smad4 cKO model leads to some intriguing theories regarding the cell-specific requirements for SMAD4 (and by extension TGFβ superfamily signaling) during testis morphogenesis. Loss of Smad4 expression in Sertoli cells alone resulted in decreased testis cord elongation/coiling during fetal life followed by decreased testis size and sperm production in young adulthood, phenotypes shared with the Sertoli/Leydig Smad4 cKO mouse model [12]. However, aged mice lacking Smad4 expression in both Sertoli and Leydig cells exhibit testicular dysgenesis well beyond that of mice lacking Smad4 in Sertoli cells alone. One possibility is that loss of Smad4 in both Sertoli and Leydig cells at the same time disrupts some yet-to-be-characterized SMAD4-dependent cross-talk between these two important cell lineages during testis development. While the study of cell-cell communication in the testis often centers on Sertoli cell-derived factors acting upon Leydig cells (Desert Hedgehog, AMH, etc.), Leydig cells modulate Sertoli cell function via secretion of activin A and androgens [12, 30]. Thus, although both Sertoli and Leydig cells secrete TGFβ superfamily ligands, it remains to be determined whether a SMAD4-dependent cross-talk exists involving these factors.
Another, perhaps simpler, explanation for the phenotypic differences between Sertoli-only and Sertoli/Leydig Smad4 cKO mouse models may relate to the timing of Smad4 deletion. Activation of the Sf1-Cre we utilized to remove Smad4 in both Sertoli and Leydig cell precursors occurs early in gonad development (E10.5) compared to the later expression of Sertoli cell-specific Amh-Cre model (∼E15 onward) [15, 31]. Fetal testes are a highly dynamic organ, meaning that the difference in onset of Cre expression/Smad4 deletion by even a few days could explain the phenotypic variability between mice lacking Smad4 in Sertoli cells alone or in Sertoli and Leydig cells simultaneously.
Loss of Smad4 in Sertoli and Leydig Cells Leads to Changes in Testis Vasculature and Tumor Development
Following sex determination, endothelial cell migration and establishment of a vasculature network are critical for the progression of testis morphogenesis [26]. Given that disruption of Smad4 in Sertoli/Leydig cKO mouse model occurs right around the time of sex determination (E10.5), it is possible that loss of Smad4 in Sertoli and fetal Leydig precursor cells somehow affects vasculature development resulting in the hemorrhages we observed in many newborn cKO testes. However, this vascular dysfunction would have to be mild enough so as not to disrupt testis cord formation as has been reported in a mouse model lacking platelet-derived growth factor receptor α (Pdgfrα), a key regulator of fetal testis vasculogenesis [32]. We do not know the direct mechanism by which loss of Smad4 in Sertoli and Leydig cells would lead to aberrant fetal testis vasculogenesis, but abnormal blood accumulation within the fetal testis interstitium has been reported in a mouse model lacking expression of Fkhl18, a member of the forkhead (Fox) transcription factor family [28]. This blood leakage was thought to occur as a result of gaps between endothelial cells within testicular blood vessels [33]. Interestingly, Fox family proteins are known to be downstream components of TGFβ superfamily signal transduction pathways in other tissue contexts [34–37]. Fkhl18 expression was detected in both Sertoli cells and periendothelial cells, raising the possibility that Smad4 and Fkhl18 could both influence vasculature formation via their transcriptional effects within Sertoli cells [33].
While we did not observe visible hemorrhages in the testes of young adult Sertoli/Leydig Smad4 cKO mice, the presence of unusually large testicular blood vessels and eosinophilic fluid within the testis interstitium indicated ongoing problems with the vasculature. The testes of aged adult Sertoli/Leydig Smad4 cKO displayed fibrous encapsulation of hemorrhagic tissue coupled with revascularization of abnormal regions. Interestingly, enlarged blood vessels and increased angiogenesis following skin wounding were reported in mice lacking Smad4 within the epidermis, indicating SMAD4 is involved in a critical cross-talk between epidermal keratinocytes and the stromal compartment [38]. Loss of Smad4 also promotes angiogenesis in a number of cancers, leading to the hypothesis that SMAD4 functions as an angiogenic switch within tumors [39]. Like many epithelial lineages, Sertoli cells produce a number of angiogenic growth factors, and Sertoli cell-conditioned media is even known to enhance the formation of capillary-like structures in vitro [40]. In addition to the likely role of Sertoli cell-derived growth factors in the abnormal vasculature observed in our Sertoli/Leydig Smad4 cKO mouse model, Leydig cells have been shown to secrete factors such as VEGF that increase endothelial cell proliferation and vascular permeability [41]. The Leydig cell adenomas in aged adult Sertoli/Leydig Smad4 cKO males may increase levels of these vasoactive factors and directly contribute to the abnormal vasculature appearance and function. This disruption of normal vasculature may also be directly tied to the loss of spermatogenic cells in aged Sertoli/Leydig Smad4 cKO males because spermatogonial stem cells are thought to require a vasculature-associated niche [42]. While our results indicate Sertoli cell SMAD4 is a suppressor of Leydig cell adenoma formation and aberrant angiogenesis, further studies are required to determine the downstream transcriptional targets that lead to these suppressive effects.
Despite the presence of Leydig cell adenomas in Sertoli/Leydig Smad4 cKO mice, the tumors present in these testes are not Leydig cell tumors. Based on tumor histology, we believe they result from chronic fibrosis followed by revascularization of the fibrotic region. Our observations of hemorrhages in newborn testes and eosinophilic fluid in the testicular interstitium of young adults leads us to speculate that vasculature dysregulation is the major driving force behind the widespread fibrosis in aged Sertoli/Leydig Smad4 cKO testes. However, dysgenesis of the seminiferous epithelium may be a precipitating factor in the development of these fibrotic tumors. Signs of Sertoli cell dysfunction, including vacuolization and loss of spermatogenic cells, were evident in 12- 16-wk-old Sertoli/Leydig Smad4 cKO males, and the seminiferous tubules were completely degenerated in ∼1-yr-old mice. In normal testes, Sertoli cells regulate not only spermatogenesis but also immune function; therefore, disruption of critical Sertoli cell functions in Sertoli/Leydig Smad4 cKO testes may have set the stage for the fibrotic response and eventual tumor development observed in this mouse model. The presence of Leydig cell adenomas and their secreted factors may have additionally contributed to fibrosis and/or revascularization of Sertoli/Leydig Smad4 cKO testes. Testicular tumors have previously been reported in a mouse model in which SMAD1 and SMAD5 were simultaneously knocked out using Amhr2-Cre; specifically, Smad1-Smad5 double knockout (dKO) mice developed metastatic sex cord stromal tumors with Sertoli and Leydig cell differentiation and hemorrhagic ascites [43]. Initially, the milder tumor phenotype in our Sertoli/Leydig Smad4 cKO mice might seem counterintuitive given the fact that SMAD4 is required for canonical SMAD1/5/8 and SMAD2/3 signaling. However, Pangas et al. [43] found upregulation of components of the TGFβ/SMAD2/SMAD3 signaling pathway in their Smad1 Smad5 dKO mice, indicating that the SMAD1/5/8 pathway likely modulates TGFβ function under normal conditions. In our Sertoli/Leydig Smad4 cKO mouse model, canonical signaling through both SMAD2/3 and SMAD1/5/8 is disrupted by loss of SMAD4, thus eliminating the imbalance of TGFβ function that likely led to metastatic tumor formation in the Smad1 Smad5 dKO mouse model.
In the current study, we observed a single teratoma out of eight Sertoli/Leydig Smad4 cKO testes analyzed at ∼1 yr of age. Given the small sample size, we cannot confirm whether this teratoma is related to the genetic manipulation of SMAD4 in our mouse model. While SMAD4 is an important tumor suppressor in many tissue contexts, testicular teratomas are reported to occur spontaneously in inbred mice [44]. In our study, the observed teratoma may have arisen from a germ cell or abnormal activation of a mesenchymal stem cell, either of which would indicate a SMAD4-independent mechanism because SMAD4 deletion was targeted to Sertoli and Leydig cells [45]. However, the abnormal testicular environment in our Sertoli/Leydig Smad4 cKO mouse model may have indirectly permitted or even promoted teratoma development.
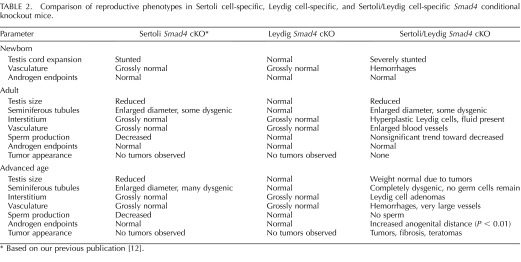
The current study investigates testicular phenotypes in two mouse models—one lacking Smad4 in the Leydig cell population and the other lacking Smad4 in both Sertoli and Leydig cells. Taken together with our previous report of testicular development in a mouse model with Sertoli cell-specific deletion of Smad4, the requirement for SMAD4 in Sertoli and Leydig cells can be explored in terms of physiological outcomes (Table 2 and Supplemental Fig. S1; available online at www.biolreprod.org) [12]. In all three of our mouse models, physiological endpoints of androgen action such as testicular descent and seminal vesicle weight were comparable to control mice. Anogenital distance, also under androgen control, was similar between cKO and control mice with the exception of statistically increased anogenital distance in aged Sertoli/Leydig cKO mice (Table 2). The increase in anogenital distance coupled with the trend toward decreased plasma FSH in aged Sertoli/Leydig cKO males suggests an increase in androgen production at this advanced time point, which could be related to the extensive Leydig cell adenomas present in cKO testes (Fig. 5D). These results indicate that loss of SMAD4 expression in Sertoli and/or Leydig cells does not decrease androgen synthesis or signaling to the point of reducing androgen-sensitive endpoints. In fact, we did not detect any obvious abnormalities in testicular development or function in mice lacking SMAD4 in Leydig cells alone. The lifelong changes in testis biology we observed in the mouse models lacking Sertoli cell expression of Smad4 indicate SMAD4 is important for not only normal testis morphogenesis during embryonic life but also for optimum sperm production in adulthood. Whether due to deletion of SMAD4 in both Sertoli and Leydig cells or due to the timing of Smad4 deletion, our Sertoli/Leydig cKO mouse strain proved to be a more severe model of testicular dysgenesis than our Sertoli Smad4 cKO strain. Further study of why deletion of Smad4 in Sertoli and Leydig cells results in vasculature abnormalities, tumors, and complete degeneration of the seminiferous tubules whereas deletion of Smad4 in either cell type alone does not will surely reveal new complexities in communication between these two essential players in testis functions.
TABLE 2.
Comparison of reproductive phenotypes in Sertoli cell-specific, Leydig cell-specific, and Sertoli/Leydig cell-specific Smad4 conditional knockout mice.
Based on our previous publication [12].
Supplementary Material
ACKNOWLEDGMENT
We would like to thank the late Dr. Keith L. Parker (University of Texas Southwestern Medical Center) for the Sf1cre/+ mouse strain; Dr. Chuxia Deng (National Institute of Diabetes and Digestive and Kidney Disease) for the Smad4fl/fl mouse strain; Sam Marion and Dr. Patricia Hoyer (University of Arizona) for FSH measurement; and Drs. Rex Hess, Susan Ball-Kell, and Kuldeep Singh (University of Illinois) for their histological expertise. We also appreciate all of the Yao lab members for their assistance and support.
Footnotes
Supported by National Institute of Health Grants HD046861 and NIEHS Intramural Research Fund ES102965 (to H.H.Y.) and T32 ES07326 (to D.R.A.).
REFERENCES
- Gordon KJ, Blobe GC. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta 2008; 1782: 197–228. [DOI] [PubMed] [Google Scholar]
- Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet 2009; 17: 860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MY, Hill SC. TGF-β superfamily signaling in embryonic development and homeostasis. Dev Cell 2009; 16: 329–343. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Shinozaki M, Hara T, Furuya T, Miyazono K. Two major Smad pathways in TGF-beta superfamily signaling. Genes Cells 2002; 7: 1191–1204. [DOI] [PubMed] [Google Scholar]
- Lagna G, Hata A, Hemmati-Brivanlou A, Massagué J. Partnership between DPC4 and SMAD proteins in TGF-β signaling pathways. Nature 1996; 383: 832–836. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Feng X, We RY, Derynck R. Receptor associated Mad homologues synergize as effectors of the TGF-β response. Nature 1996; 383: 168–172. [DOI] [PubMed] [Google Scholar]
- Behringer RR, Finegold MJ, Cate RL. Müllerian-inhibiting substance function during mammalian sexual development. Cell 1994; 79: 415–425. [DOI] [PubMed] [Google Scholar]
- Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. Activin/inhibin beta B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev 1994; 8: 414–427. [DOI] [PubMed] [Google Scholar]
- Matzuk MM, Kumar TR, Vassalli A, Bickenbach JR, Roop DR, Jaenisch R, Bradley A. Functional analysis of activins during mammalian development. Nature 1995; 374: 354–356. [DOI] [PubMed] [Google Scholar]
- Yao HH, Aardema J, Holthusen K. Sexually dimorphic regulation of inhibin beta B in establishing gonadal vasculature in mice. Biol Reprod 2006; 74: 978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memon MA, Anway MD, Covert TR, Uzumcu M, Skinner MK. Transforming growth factor beta (TGFβ1, TGFβ2, TGFβ3) null-mutant phenotypes in embryonic gonadal development. Mol Cell Endocrinol 2008; 294: 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambeault DR, Yao HHC., Activin A. a product of fetal Leydig cells, is a unique paracrine regulator of Sertoli cell proliferation and testis cord expansion. Proc Natl Acad Sci U S A 2010; 107: 10526–10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itman C, Loveland KL. SMAD expression in the testis: an insight into BMP regulation of spermatogenesis. Dev Dyn 2008; 237: 97–111. [DOI] [PubMed] [Google Scholar]
- Hussein N, Lu J, Casse H, Fontaniére S, Morera AM, Guittot SM, Calender A, Di Clemente N, Zhang CX. Deregulation of anti-Müllerian hormone/BMP and transforming growth factor-beta pathways in Leydig cell lesions developed in male heterozygous multiple endocrine neoplasia type 1 mutant mice. Endocr Relat Cancer 2008; 15: 217–227. [DOI] [PubMed] [Google Scholar]
- Jamin SP, Arango NA, Mishina Y, Hanks MC, Behringer RR. Requirement of Bmpr1a for Müllerian duct regression during male sexual development. Nat Genet 2002; 32: 408–410. [DOI] [PubMed] [Google Scholar]
- Bingham NC, Verma-Kurvari S, Parada LF, Parker KL. Development of a steroidogenic factor 1/Cre transgenic mouse line. Genesis 2006; 44: 419–424. [DOI] [PubMed] [Google Scholar]
- Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis 2002; 32: 80–81. [DOI] [PubMed] [Google Scholar]
- Joyce KL, Porcelli J, Cooke PS. Neonatal goitrogen treatment increases adult testis size and sperm production in the mouse. J Androl 1993; 14: 448–455. [PubMed] [Google Scholar]
- Itman C, Mendis S, Barakat B, Loveland KL. All in the family: TGF-beta family action in testis development. Reproduction 2006; 132: 233–246. [DOI] [PubMed] [Google Scholar]
- Jeyasuria P, Ikeda Y, Jamin SP, Zhao L, De Rooij DG, Themmen AP, Behringer RR, Parker KL. Cell-specific knockout of steroidogenic factor 1 reveals its essential roles in gonadal function. Mol Endocrinol 2004; 18: 1610–1619. [DOI] [PubMed] [Google Scholar]
- Mishina Y, Rey R, Finegold MJ, Matzuk MM, Josso N, Cate RL, Behringer RR. Genetic analysis of the Mullerian-inhibiting substance signal transduction pathway in mammalian sexual differentiation. Genes Dev 1996; 10: 2577–2587. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Informatics (MGI). Bar Harbor, ME: The Jackson Laboratory; http://www.informatics.jax.org/reference/allele/MGI:3042214. Accessed January 15, 2014. [Google Scholar]
- Mouse Genome Informatics (MGI). Bar Harbor, ME: The Jackson Laboratory; http://www.informatics.jax.org/reference/allele/MGI:2386393. Accessed January 15, 2014. [Google Scholar]
- Liu CF, Bingham N, Parker K, Yao HH. Sex-specific roles of beta-catenin in mouse gonadal development. Hum Mol Genet 2009; 18: 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Miyagawa S, Matsumaru D, Parker KL, Yao HH. Progenitor cell expansion and organ size of mouse adrenal is regulated by sonic hedgehog. Endocrinology 2010; 151: 1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Yao HH. Inactivation of Dicer1 in Steroidogenic factor 1-positive cells reveals tissue-specific requirement for Dicer1 in adrenal, testis, and ovary. BMC Dev Biol 2010; 10: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CF, Parker K, Yao HH. WNT4/beta-catenin pathway maintains female germ cell survival by inhibiting activin betaB in the mouse fetal ovary. PLoS One 2010; 5: e10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambeault DR, Tomaszewski J, Childs AJ, Anderson RA, Yao HH. Testicular somatic cells, not gonocytes, are the major source of functional activin A during testis morphogenesis. Endocrinology 2011; 152: 4358–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Liu C, Yao HH. Investigating the role of adrenal cortex in organization and differentiation of the adrenal medulla in mice. Mol Cell Endocrinol 2012; 361: 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Gendt K, Verhoeven G. Tissue- and cell-specific functions of the androgen receptor revealed through conditional knockout models in mice. Mol Cell Endocrinol 2012; 352: 13–25. [DOI] [PubMed] [Google Scholar]
- Lécureuil C, Fontaine I, Crepieux P, Guillou F. Sertoli and granulosa cell-specific Cre recombinase activity in transgenic mice. Genesis 2002; 33: 114–118. [DOI] [PubMed] [Google Scholar]
- Cool J, Capel B. Mixed signals: development of the testis. Semin Reprod Med 2009; 27: 5–13. [DOI] [PubMed] [Google Scholar]
- Brennan J, Tilmann C, Capel B. Pdgrf-alpha mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev 2003; 17: 800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Baba T, Zubair M, Miyabayashi K, Toyama Y, Maekawa M, Owaki A, Mizusaki H, Sawamura T, Toshimori K, Morohashi K, Katoh-Fukui Y. Importance of forkhead transcription factor Fkhl18 for development of testicular vasculature. Mol Reprod Dev 2008; 75: 1361–1371. [DOI] [PubMed] [Google Scholar]
- Chen X, Rubock MJ, Whitman M. A transcriptional partner for MAD proteins in TGF-beta signaling. Nature 1996; 383: 691–696. [DOI] [PubMed] [Google Scholar]
- Chen X, Weisberg E, Fridmacher V, Watanabe M, Naco G, Whitman M. Smad4 and FAST-1 in the assembly of activin-responsive factor. Nature 1997; 389: 85–89. [DOI] [PubMed] [Google Scholar]
- Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004; 117: 421–426. [DOI] [PubMed] [Google Scholar]
- Owens P, Engelking E, Han G, Haeger SM, Wang XJ. Epidermal Smad4 deletion results in aberrant wound healing. Am J Pathol 2010; 176: 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarte-Waldhoff I, Volpert OV, Bouck NP, Sipos B, Hahn SA, Klein-Scory S, Lüttges J, Klöppel G, Graeven U, Eilert-Micus C, Hintelmann A, Schmiegel W. Smad4/DPC4-mediated tumor suppression through suppression of angiogenesis. Proc Natl Acad Sci U S A 2000; 97: 9624–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golat BT, Cameron DF. Sertoli cells enhance formation of capillary-like structures in vitro. Cell Transplant 2008; 17: 1135–1144. [DOI] [PubMed] [Google Scholar]
- Collin O, Bergh A. Leydig cells secrete factors which increase vascular permeability and endothelial cell proliferation. Int J Androl 1996; 19: 221–228. [DOI] [PubMed] [Google Scholar]
- Yoshida S, Sukeno M, Nabeshima Y. A vasculature-associated niche for undifferentiated spermatogonia in the mouse testis. Science 2007; 317: 1722–1726. [DOI] [PubMed] [Google Scholar]
- Pangas SA, Li X, Umans L, Zwijsen A, Huylebroeck D, Gutierrez C, Wang D, Martin JF, Jamin SP, Behringer RR, Robertson EJ, Matzuk MM. Conditional deletion of Smad1 and Smad5 in somatic cells of the male and female gonads leads to metastatic tumor development in mice. Mol Cell Biol 2008; 28: 248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens LC, Jr, , Little CC. Spontaneous testicular teratomas in an inbred strain of mice. Proc Natl Acad Sci U S A 1954; 40: 1080–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay AM, Beck SC, Murphy JM, Barry FP, Chichester CO, Pittenger MF. Chondrogenic differentiation of cultured human mesenchymal stem cells from marrow. Tissue Eng 1998; 4: 415–428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.