

Abstract
Desmosterolosis is an autosomal recessive disorder of cholesterol biosynthesis caused by biallelic mutations of DHCR24 (homozygous or compound heterozygous), which encodes 3-β-hydroxysterol Δ-24-reductase. We report two sisters homozygous for the 571G>A (E191K) DHCR24 mutation. Comparison of the propositae to other reported individuals shows that psychomotor developmental delay, failure to thrive, dysgenesis of the corpus callosum, cerebral white matter atrophy and spasticity likely constitute the minimal desmosterolosis phenotype. The nonspecific features of desmosterolosis make it difficult to suspect clinically and therefore screening for it should be entertained early in the diagnostic evaluation.
Keywords: DHCR24, Desmosterol, Intellectual disability, Cholesterol biosynthesis, Exome sequencing
Findings
Background
Desmosterolosis is an infrequently reported disorder of cholesterol biosynthesis causing syndromic intellectual disability (ID) arising from biallelic mutations (homozygous or compound heterozygous) in DHCR24. DHCR24 encodes 3-β-hydroxysterol Δ-24-reductase (DHCR24) [1,2], which catalyzes the C-24 NADPH-dependent reduction of the 24–25 double bond of cholesterol precursors [3,4].
Case report
We present two sisters with syndromic ID and desmosterolosis. Following uncomplicated pregnancies, they were born at term with normal growth parameters. Each had transient neonatal seizures. Their family history was noncontributory.
Beginning in infancy, they manifested growth restriction and delayed milestones for speech, fine and gross motor, and adaptive development. Patient 1 was able to walk with support and communicate with short phrases by 6 years; Patient 2 developed these skills by 8 years. Patient 1 had an IQ of 42 at 10.5 years. Patient 2 had an IQ of 46 at 5.5 years. Neither lost skills although Patient 1 had progressive sensorineural hearing loss.
On examination at 13.8 and 9.1 years, respectively, each had similar dysmorphic features. Patient 1 had short stature (<1st centile) with proportionate limbs, a weight of 41 kg (13th centile), and a head circumference of 55.2 cm (86th centile). Patient 2 had a height of 117.8 cm (1st centile), a weight of 21.3 kg (3rd centile), and a head circumference of 49.5 cm (4th centile). They had mild generalized hirsutism, facial dysmorphism, high arched hard palates, optic atrophy, nystagmus, spasticity, deep tendon hyperreflexia, camptodactyly of the fingers and toes, and muscle wasting, particularly of the thenar and hypothenar muscles (Figure 1A, B, C, F, G and H). Patient 1 also had a midline cleft of the soft palate as well as a short neck and large ears (>97th centile). Patient 2 also had myopia.
Figure 1.
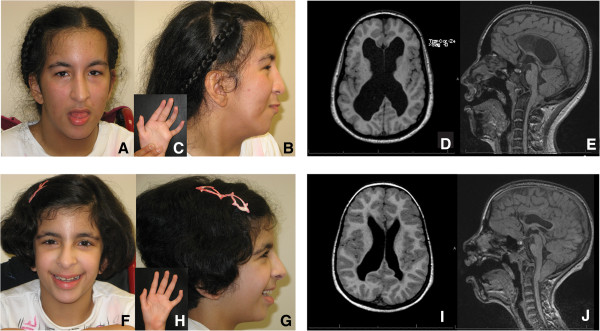
Clinical and MRI features of siblings with Desmosterolosis. A to C: Patient 1 craniofacial and hand features at age 14.8 years. Her dysmorphic features show scaphocephaly, tall forehead with bitemporal narrowing, short palpebral fissures, long nose, hypoplastic nasal alae, prominent columella, and low-set posteriorly rotated ears. D, E (Axial T1, Sagittal FLAIR): Patient 1 brain MRI showing white matter volume loss, dilated ventricles, thin corpus callosum, and peg-like cerebellar tonsils displaced into the upper cervical canal through the foramen magnum (Chiari I malformation). F to H: Patient 2 craniofacial and hand features at age 10.1 years, similar to Patient 1. I, J (Axial T1 and Sagittal FLAIR): Patient 2 brain MRI showing prominent and irregular ventricles, thin corpus callosum, and Chiari I malformation.
Investigations
Each had extensive non-diagnostic laboratory testing. This included normal profiles for urine organic acids, urine purines and pyrimidines, plasma amino acids, and plasma very long chain fatty acids as well as urine mucopolysaccharide and oligosaccharide screens, liver function studies, transferrin isoelectric focusing and levels for lactate, ammonia, uric acid, albumin, creatinine phosphokinase, and thyroid stimulating hormone. Each had a normal karyotype and no evidence of a genomic deletion or duplication detectable by array comparative genomic hybridization. Patient 1 also had normal nucleotide excision repair assays, electromyography and nerve conduction studies. Patient 2 had normal complete blood counts and levels for copper, ceruloplasmin and blood acylcarnitines; her EEG identified no focal seizure activity. The patients were unavailable for additional testing, specifically plasma sterols, which were not performed on initial assessment.
Radiological assessment showed that both had dislocated radial heads and bilateral equinovarus. In addition, Patient 1 had a small and deformed pelvis, lumbar scoliosis, and moderate osteopenia. Patient 2 had parietal foramina.Magnetic resonance imaging (MRI) identified mild brain atrophy, asymmetric ventriculomegaly, a thin corpus callosum, and a Chiari I malformation (Figure 1D, E, I and J) In addition, Patient 1 had a sacral cyst suggestive of a meningocele or dural/perineural cyst.
Exome sequencing and biochemical confirmation of desmosterolosis
Exome sequencing (for methods see Additional file 1) [5] identified a homozygous DHCR24 mutation (NM_014762.3:c.571G>A; p.E191K), a recognized cause of desmosterolosis (Additional file 1, Supplementary methods and Additional file 2: Figure S1A) [2]. Sanger sequencing confirmed this and the carrier state of the parents (Additional file 2: Figure S1B). As predicted, gas chromatography-mass spectroscopy analysis of lysates from cultured lymphoblastoid cells [6] from the propositae detected an increased ratio of desmosterol to total sterols (Figure 2). All other sterols measured (Supplementary methods) including cholesterol and 7-dehydrocholesterol were within normal range compared to healthy controls.
Figure 2.
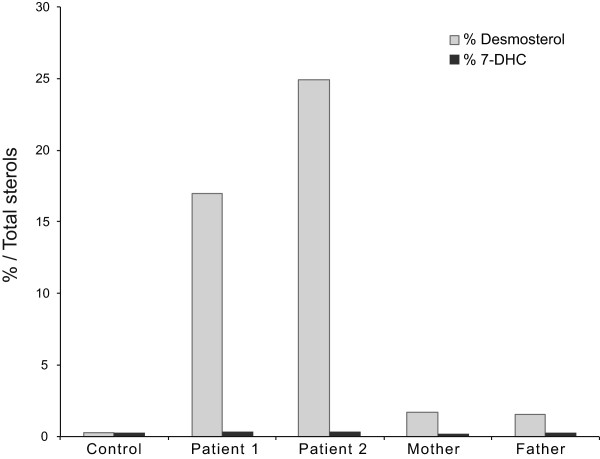
Biochemical confirmation of desmosterolosis. Comparative sterol profiles for the patients (second and third bar sets), heterozygous parents (fourth and fifth bar sets), and unaffected control (first bar set). Sterols were measured in lysates from lymphoblasts cultured in delipidated medium for 3 days and showed a 17–25 fold increased ratio of desmosterol to total sterols in comparison to controls, whereas as heterozygous parents present a 1.7 and 1.5 increase, respectively. Each bar represents the average of 3 technical replicates. 7-DHC is represented as an internal control.
Conclusions
We present two sisters with the biallelic mutation NM_014762.3:c.571G>A in DHCR24. The recurrence of this mutation [2] in a different ethnic group implies that this mutation arose independently and suggests that mutations altering only certain amino acids give rise to a viable human with desmosterolosis (Table 1).
Table 1.
Clinical features of reported patients with Desmosterolosis
| Feature |
Present family |
Previously reported cases of desmosterolosis |
Frequency (n = 9) | ||||
|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 1 | Patients 4-7 2 | Patient 8 3 | Patient 9 4 | ||
| Mutation |
c.[571G>A] + [571G>A] p.[E191K] + [E191K] |
c.[571G>A] + [571G>A] p.[E191K] + [E191K] |
c.[307C>T] + [307C>T] p.[R103C] + [R103C] |
c.[281G>A] + [1438G>A] p.[R94H] + [E480K] |
c.[1412A>C] + [881A > C;918G>C] p.[Y471S] + [N294T; K306N] |
|
|
| Protein domain |
FAD-binding domain |
FAD-binding domain |
FAD-binding domain |
FAD-binding domain + C terminal cytoplasmic domain |
C terminal cytoplasmic domain |
|
|
| Ancestry |
Middle Eastern |
European |
Israeli Bedouin |
|
European |
|
|
| Failure to thrive |
1 |
1 |
1 |
4/4 |
1 |
n.a. |
8/8 |
| Short stature |
1 |
1 |
1 |
n.a. |
n.a. |
0 |
3/4 |
| Microcephaly |
0 |
0 |
1 |
4/4 |
0 |
0 |
5/9 |
| Macrocephaly |
0 |
0 |
0 |
0/4 |
1 |
1 |
2/9 |
| Microretrognatia |
1 |
0 |
1 |
4/4 |
1 |
1 |
8/9 |
| Cleft palate |
1 |
0 |
1 |
0 |
0 |
1 |
3/9 |
| Facial features |
Dolicocephaly; bitemporal narrowing; low set ears; short downslanting PF; prominent columella; cleft palate |
Dolicocephaly; bitemporal narrowing; low set ears; short downslanting PF; prominent columella; |
Downslanting PF; bilateral epicanthal folds |
|
Prominent forehead; Short nose; anteverted nares; telecanthus; |
Frontal bossing; hypoplastic nose; low set ears; cleft palate |
|
| ID/DD |
1 |
1 |
1 |
4/4 |
1 |
n.a. |
8/8 |
| Spasticity |
1 |
1 |
1 |
4/4 |
n.a. |
n.a. |
7/7 |
| Distal arthrogryposis |
1 |
1 |
1 |
4/4 |
1 |
0 |
8/9 |
| Large joint contractures |
0 |
1 (talipes) |
1 (talipes) |
n.a. |
1 |
1 |
4/5 |
| Shortening of the limbs |
0 |
0 |
0 |
n.a. |
1 |
1 |
2/5 |
| ACC (partial/full) |
1 |
1 |
1 |
4/4 |
1 |
1 |
9/9 |
| Ventriculomegaly |
1 |
1 |
0 |
4/4 |
1 |
1 |
8/9 |
| Cerebral WM atrophy |
1 |
1 |
1 |
4/4 |
1 |
n.a. |
8/8 |
| Cerebellar WM atrophy |
1 |
1 |
n.a. |
2/2 |
n.a. |
n.a. |
4/4 |
| Nystagmus /strabismus |
1 |
1 |
0 |
3/4 |
0 |
n.a. |
5/8 |
| Seizures |
1 |
1 |
0 |
3/4 |
n.a. |
n.a. |
5/7 |
| Other features |
SNHL; Hirsutism |
Parietal foramina. Hirsutism |
Cutis aplasia; Limb anomalies; PDA; |
|
Hydrocephalus |
Osteosclerosis; ambiguous genitalia; anomalous pulmonary venous drainage; renal hypoplasia; death at 1 h |
|
| Functional assays | Expressed both mutations in c.cerevisiae (separately) with significant ⇩enzyme activity | Expressed mutations in c.cerevisiae with significant ⇩enzyme activity (including compound het) | |||||
References: 1: Waterham et al. [2], Andersson et al. [8]; 2: Zolotushko et al. [7]; 3: Schaaf et al. [9]; 4: FitzPatrick et al. [1], Waterham et al. [2].
Abbreviations: ACC agenesis of the corpus callosum, DD developmental delay, ID intellectual disability, n.a. not available, PF palpebral fissures, PDA patent ductus arteriosus, SNHL sensorineural hearing loss, WM white matter, h hour, het heterozygote.
Assessing genotype-phenotype correlation, the propositae were discordant for microretrognathia, cleft palate, large joint contractures, deafness, and skull foramina (Table 1). In contrast, the previously reported four cousins of a consanguineous family were discordant for oculomotor abnormalities and seizures (Patients 4–7, Table 1). This might suggest either that the genetic backgrounds of the propositae are significantly different or that because of their consanguinity, the four family members reported by Zolotushko et al. [7] share other genomic or epigenetic variants modifying the expressivity of desmosterolosis.
Comparison of the propositae to Patient 3 (Table 1), who has the same DHCR24 mutation, provides an assessment of interfamilial genotype-phenotype concordance [2]. They were discordant for dysmorphic facial features, oculomotor abnormalities, seizures, brain ventriculomegaly, cutis aplasia, limb anomalies, and congenital heart defects. They were concordant for ID, failure to thrive, short stature, spasticity, distal arthrogryposis, dysgenesis of the corpus callosum, and cerebral white matter atrophy. Comparison of the propositae to all reported in individuals with desmosterolosis (Table 1) [1,2,7-9] identifies ID, failure to thrive, spasticity, dysgenesis of the corpus callosum, and cerebral white matter atrophy as the minimal clinical phenotype for desmosterolosis. Distal arthrogryposis occurred in 8 of 9 individuals (Table 1). This minimal phenotype, which is not distinctive and the absence of sterol testing, explains the decade-long diagnostic odyssey of the propositae.
Review of all reported patients suggests a minimal genotype-phenotype correlation for desmosterolosis. Only individuals with mutations affecting the 3-β-hydroxysterol Δ-24-reductase cytoplasmic domain had rhizomelia (Patients 8 and 9, Table 1). Sharing this feature with the knockout mice [10], might suggest that mutations in the cytoplasmic domain disrupt enzyme function more severely than mutations in the FAD-binding domain (protein features of UniProt Q15392 [11]).
The neurological features of desmosterolosis might arise from either deficiency of cholesterol biosynthesis or the toxic effects of sterols accumulating upstream of 3-β-hydroxysterol Δ-24-reductase. Both mechanisms contribute to other disorders of cholesterol biosynthesis and thus likely apply here [12]. The non-progressive neuropathology in desmosterolosis is in keeping with the primary impact occurring during brain development.
In summary, the pleiotropy and nonspecificity of desmosterolosis explain the long diagnostic odyssey of the propositae. Also, findings of developmental delay, CNS malformation, spasticity (with or without distal arthrogryposis), short stature with and without limb anomalies are sufficient indication to screen for disorders of cholesterol biosynthesis.
Patient consent and ethics approval
Individuals enrolled in the study gave informed consent for protocol H07-02142 (Vancouver, BC, Canada), approved by the University of British Columbia Research Ethics Board. Written informed consent was provided for the collection of samples, subsequent analysis and use of photographs by the parents of the children.
Abbreviations
bp: base pair; CNS: Central nervous system; IQ: Intelligence quotient; DHCR24: 3-β-hydroxysterol Δ-24-reductase; EBP: Emopamil binding protein; FISH: Fluorescence in situ hybridization; ID: Intellectual disability; MRI: Magnetic resonance imaging; 7-DHC: 7-dehidrocholesterol; PE: Paired-end.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CD, RR and CFB interpreted the data and drafted and revised the manuscript. CD, BM, KC and LEK performed experimental work and interpreted the data. CFB, MD and RR provided clinical data. CD and MM performed and interpreted the bioinformatic analysis. All authors read and approved the final manuscript.
Supplementary Material
Supplementary methods. Exome sequencing and Gas chromatography-mass spectroscopy analysis.
Molecular confirmation of Desmosterolosis through exome sequencing and Sanger sequencing. NextGene (Softgenetics, Pennsylvania) view of the exome sequencing reads for one affected child (A) and dideoxy nucleotide sequencing validation (B) for the mother, father, and both affected children. The mother and father were heterozygous for NM_014762.3:c.571G > A (p.E191K) mutation. The propositae have the mutation in homozygosity.
Contributor Information
Cristina Dias, Email: cdias@cfri.ca.
Rosemarie Rupps, Email: rrupps@cw.bc.ca.
Benjamin Millar, Email: benwmillar@hotmail.com.
Kunho Choi, Email: khchoi@cfri.ca.
Marco Marra, Email: mmarra@bcgsc.ca.
Michelle Demos, Email: mdemos@cw.bc.ca.
Lisa E Kratz, Email: kratz@kennedykrieger.org.
Cornelius F Boerkoel, Email: nboerkoel@cfri.ca.
Acknowledgement
We thank the family for their generous participation and Dr. Rui F. Santos for interpretation of the radiological data. This work was supported in part by the Rare Disease Foundation. CD was supported by the Canadian Child Health Clinician Scientist Program and the Child and Family Research Institute.
References
- FitzPatrick DR, Keeling JW, Evans MJ, Kan AE, Bell JE, Porteous MEM, Mills K, Winter RM, Clayton PT. Clinical phenotype of desmosterolosis. Am J Hum Genet. 1998;75(2):145–152. doi: 10.1002/(SICI)1096-8628(19980113)75:2<145::AID-AJMG5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Waterham HR, Koster J, Romeijn GJ, Hennekam RCM, Vreken P, Andersson HC, FitzPatrick DR, Kelley RI, Wanders RJA. Mutations in the 3β-Hydroxysterol Delta24-Reductase Gene Cause Desmosterolosis, an Autosomal Recessive Disorder of Cholesterol Biosynthesis. Am J Hum Genet. 2001;69(4):685–694. doi: 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SH, Paik YK. Cholesterol biosynthesis from lanosterol: development of a novel assay method and characterization of rat liver microsomal lanosterol delta 24-reductase. Biochem J. 1997;326(2):609–616. doi: 10.1042/bj3260609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerenturk EJ, Sharpe LJ, Ikonen E, Brown AJ. Desmosterol and DHCR24: Unexpected new directions for a terminal step in cholesterol synthesis. Prog Lipid Res. 2013;52(4):666–680. doi: 10.1016/j.plipres.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Dias C, Sincan M, Cherukuri PF, Rupps R, Huang Y, Briemberg H, Selby K, Mullikin JC, Markello TC, Adams DR, Gahl WA, Boerkoel CF. An analysis of exome sequencing for diagnostic testing of the genes associated with muscle disease and spastic paraplegia. Hum Mutat. 2012;33(4):614–626. doi: 10.1002/humu.22032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RI. Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clin Chim Acta. 1995;236(1):45–58. doi: 10.1016/0009-8981(95)06038-4. [DOI] [PubMed] [Google Scholar]
- Zolotushko J, Flusser H, Markus B, Shelef I, Langer Y, Heverin M, Bjorkhem I, Sivan S, Birk OS. The desmosterolosis phenotype: spasticity, microcephaly and micrognathia with agenesis of corpus callosum and loss of white matter. Eur J Hum Genet. 2011;19(9):942–946. doi: 10.1038/ejhg.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson HC, Kratz L, Kelley R. Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am J Med Genet. 2002;113(4):315–319. doi: 10.1002/ajmg.b.10873. [DOI] [PubMed] [Google Scholar]
- Schaaf CP, Koster J, Katsonis P, Kratz L, Shchelochkov OA, Scaglia F, Kelley RI, Lichtarge O, Waterham HR, Shinawi M. Desmosterolosis—phenotypic and molecular characterization of a third case and review of the literature. Am J Med Genet. 2011;155(7):1597–1604. doi: 10.1002/ajmg.a.34040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza R, Hayasaka S, Takagishi Y, Kambe F, Ohmori S, Maki K, Yamamoto M, Murakami K, Kaji T, Zadworny D, Murata Y, Seo H. DHCR24 Gene Knockout Mice Demonstrate Lethal Dermopathy with Differentiation and Maturation Defects in the Epidermis. J Invest Dermatol. 2006;126(3):638–647. doi: 10.1038/sj.jid.5700111. [DOI] [PubMed] [Google Scholar]
- Consortium TU. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42(D1):D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarren KW, Severson TM, du Souich C, Stockton DW, Kratz LE, Cunningham D, Hendson G, Morin RD, Wu D, Paul JE, An J, Nelson TN, Chou A, DeBarber AE, Merkens LS, Michaud JL, Waters PJ, Yin J, McGillivray B, Demos M, Rouleau GA, Grzeschik KH, Smith R, Tarpey PS, Shears D, Schwartz CE, Gecz J, Stratton MR, Arbour L, Hurlburt J. et al. Hypomorphic Temperature-Sensitive Alleles of NSDHL Cause CK Syndrome. Am J Hum Genet. 2010;87(6):905–914. doi: 10.1016/j.ajhg.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary methods. Exome sequencing and Gas chromatography-mass spectroscopy analysis.
Molecular confirmation of Desmosterolosis through exome sequencing and Sanger sequencing. NextGene (Softgenetics, Pennsylvania) view of the exome sequencing reads for one affected child (A) and dideoxy nucleotide sequencing validation (B) for the mother, father, and both affected children. The mother and father were heterozygous for NM_014762.3:c.571G > A (p.E191K) mutation. The propositae have the mutation in homozygosity.