

Abstract
Because scavenger receptor class B type 1 is the cholesterol uptake liver receptor, whereas peroxisome proliferator–activated receptor γ coactivator-1β (PGC-1β) and PGC-1α are critical for lipid synthesis and degradation, we investigated the roles of these signaling molecules in the actions of ethanol-polyunsaturated fatty acids and betaine on hepatosteatosis and steatohepatitis. Ethanol-polyunsaturated fatty acid treatment caused the following: i) hepatosteatosis, as evidenced by increased liver cholesterol and triglycerides, lipid score, and decreased serum adiponectin; ii) marked inhibition of scavenger receptor class B type 1 glycosylation, its plasma membrane localization, and its hepatic cholesterol uptake function; and iii) moderate steatohepatitis, as evidenced by histopathological characteristics, increased liver tumor necrosis factor α and IL-6, decreased glutathione, and elevated serum alanine aminotransferase. These actions of ethanol involved up-regulated PGC-1β, sterol regulatory element-binding proteins 1c and 2, acetyl-CoA carboxylase, and HMG-CoA reductase mRNAs/proteins and inactive non-phosphorylated AMP kinase; and down-regulated silence regulator gene 1 and PGC-1α mRNA/proteins and hepatic fatty acid oxidation. Betaine markedly blunted all these actions of ethanol on hepatosteatosis and steatohepatitis. Therefore, we conclude that ethanol-mediated impaired post-translational modification, trafficking, and function of scavenger receptor class B type 1 may account for alcoholic hyperlipidemia. Up-regulation of PGC-1β and lipid synthetic genes and down-regulation of silence regulator gene 1, PGC-1α, adiponectin, and lipid degradation genes account for alcoholic hepatosteatosis. Induction of proinflammatory cytokines and depletion of endogenous antioxidant, glutathione, account for alcoholic steatohepatitis. We suggest betaine as a potential therapeutic agent because it effectively protects against adverse actions of ethanol.
Cholesterol uptake by the liver is crucial for maintaining cholesterol homeostasis in peripheral tissues of mammals, as exemplified by early onset of atherosclerosis in familial hypercholesterolemia.1,2 In this process, scavenger receptor class B type 1 (SR-B1) plays a major role in the direct uptake of high-density lipoprotein (HDL) cholesterol by the liver.3 Although the nascent SR-B1 is a glycosylated 57-kDa protein, the mature glycosylated SR-B1 is an approximately 82-kDa membrane glycoprotein from the CD36 transmembrane protein family.4 The mature SR-B1 mediates the hepatic uptake of HDL-derived cholesterol.5,6
Ethanol exposure affects hepatic protein trafficking pathways, leading to marked accumulation of nascent proteins, which then leads to ballooning of the hepatocyte.7–9 This is due to altered assembly of newly synthesized proteins into the plasma membrane and/or their secretion.10–12 One mechanism for this defect is due to the impairment of asialoglycoprotein receptor synthesis and its inactivation, presumably due to hyperphosphorylation of the receptor.13,14
We have shown that chronic ethanol causes defective glycosylation and impaired intracellular trafficking of both the resident and secretory proteins, such as transferrin and lipid transport proteins, apolipoprotein E, and apolipoprotein J, leading to their hepatic accumulation.15–17 Because SR-B1 is also a highly glycosylated protein, we speculate that ethanol-mediated impaired glycosylation of SR-B1 in the liver may affect its trafficking to the plasma membrane, and thereby affect its cholesterol uptake function.
Numerous studies have established that chronic alcohol exposure leads to the following: i) increased adipose fat mobilization into the liver due to increased adipose lipoprotein lipase18; ii) increased fat synthesis due to up-regulation of lipogenic genes via peroxisome proliferator–activated receptor γ coactivator-1β (PGC-1β) and sterol regulatory element-binding proteins (SREBPs)19,20; iii) decreased fat oxidation due to down-regulation of fatty acid oxidation genes via PGC-1α and peroxisome proliferator–activated receptor α (PPARα)19,20; and iv) impaired synthesis of apolipoprotein B and very-low-density lipoprotein secretion,8 the major lipoprotein for the export of hepatic lipids to peripheral tissues. Significantly, PPARα and SREBPs are tightly controlled by two transcription coactivators, PGC-1α and PGC-1β, respectively.21–23 Silence regulator gene (SIRT) inactivates PGC-1α by deacetylation, whereas histone acetyltransferases activate PGC-1α by acetylation,24 which, in concert with PPARα, increases fatty acid oxidation. On the contrary, SREBPs are stabilized by histone acetyltransferases by acetylation and destabilized by SIRT by deacetylation. Dietary saturated fat up-regulates PGC-1β and SREBPs, which coactivates liver X receptor families of transcription factors, leading to increased fat and cholesterol synthesis, lipoprotein transport, and very-low-density lipoprotein secretion.25
Betaine, a potent lipotropic nutrient, plays an important role in reducing fatty liver,26 and has been reported to restore the decreased liver glutathione (GSH) level in ethanol-treated guinea pigs.27 Therefore, it is reasonable that betaine may prevent the deleterious effects of heavy alcohol and high omega-3 polyunsaturated fatty acids (ω-3 PUFAs) on SR-B1, plasma lipids, and hepatic lipid metabolizing pathway, and lipid homeostasis by altering hepatic GSH and reactive oxygen species (ROS).
In view of the previous descriptions, we have explored the possible action of chronic alcohol/high PUFA fish oil diet and the protection by betaine on the following: i) hepatic SR-B1 expression, relative glycosylation rate, and its localization on the plasma membrane; ii) the expression of various lipogenic and lipid-oxidizing signaling pathway genes and activities, and hepatic lipid status versus serum adiponectin; and iii) liver injury status, as evidenced by depletion of hepatic endogenous antioxidant, GSH, proinflammatory cytokines, tumor necrosis factor α (TNFα) and IL-6, and serum alanine aminotransferase (ALT).
Materials and Methods
Animals
Experimental animals were female Wistar rats (130 to 150 g) (Charles River Laboratories, Inc., Wilmington, MA) housed in groups of two per cage in plastic cages (40 × 24 × 18 cm), in a temperature-controlled room at 25°C with a 12-hour light-dark cycle. Experiments were performed according to the approved Institutional Animal Care and Use Committee protocol. After the first week of acclimation on a pelleted commercial diet (Purina Rodent Chow, number 500; TMI Nutrition, St. Louis, MO), the animals were randomly divided into the following three groups, and pair fed their respective high-fat diets (Dyets Inc., Bethlehem, PA). The high-fat control (HFC) group had a high menhaden fish oil liquid diet (35% of total calories came from ω-3 PUFA); the high-fat ethanol (HFE) group had 35% ethanol calories in place of carbohydrate; and in the HFEB group, HFE was supplemented with 10 g betaine/L of the diet for 8 weeks. This amounted to a consumption of 3.5 g betaine per kg body weight per day on the basis of daily consumption of approximately a 70 mL diet per rat per day. This dosage of betaine was chosen because of its beneficial effects in many alcoholic steatosis studies.28,29
Liver Lipids
Liver cholesterol and triglycerides were determined using enzymatic reagents, as described by the manufacturer (Wako Pure Chemicals, Richmond, VA), adapted to a microtiter plate assay.30,31
H&E Staining
For histological examination, paraffin-embedded sections (5 μm thick) were processed by routine histological protocol for H&E staining.
Liver TNFα, IL-6, and Circulating Adiponectin and ALT Assays
Commercially available enzyme-linked immunosorbent assay kits specific to rat liver TNFα, IL-6, and adiponectin (R&D Systems, Inc., Minneapolis, MN) were used to determine these parameters according to manufacturer's instructions, except that the plates were incubated overnight at 4°C to completely capture the respective target proteins. Standard curves for the respective rat recombinant cytokines were included in each assay; all samples from each group were assayed in triplicate. In the initial experiments, we determined that the optimal concentration of liver post-mitochondrial protein for the assay of IL-6 was 250 μg per well, whereas it was 100 μg per well for the assay of TNFα. To measure circulating adiponectin in plasma, we diluted each sample 1000 times in the diluent provided by the manufacturer. The ALT assay kit was used to measure plasma ALT activity, as per the manufacturer's instructions (Teco Diagnostic, Anaheim, CA).
Isolation of Plasma HDL and Its Labeling with [3H] Cholesteryl Oleate
HDL was isolated from pooled HFC or HFE rat plasma, according to Gidez et al.32 Protein concentration was determined colorimetrically.33 HDL cholesterol content was measured according to Zlatkis and Zak.34 HDL labeling with [3H] cholesteryl oleate was performed according to Basu et al,35 and the specific activity is expressed as dpm/mg HDL cholesterol.
Hepatic HDL-Cholesterol Uptake Capacity and Relative Glycosylation Rate of SR-B1 in Various Groups
Hepatocytes were isolated from the HFC, HFE, and HFEB groups, and their cholesterol uptake capacity [3H] cholesteryl oleate–labeled HDL was determined, as we described.36,37 Similarly, the relative hepatic glycosylation rates of SR-B1 in various groups were determined as we described.12 Briefly, the hepatocytes (approximately 100 mg wet wt./mL; 10 mL final volume) from the respective groups were incubated for 120 minutes with either 100 μCi of [3H] ManNAc or 20 μCi of [35S]methionine, and the incorporations of [3H]ManNAc relative to that of [35S]methionine into immunoprecipitable SR-B1 were determined.
RNA Isolation
The total RNA was isolated from each liver using the Tri-Reagent (Molecular Research Center, Cincinnati, OH), following the manufacturer's instructions, as we described.38 Isolated RNA was used immediately or stored at −80°C until use.
Real-Time Quantitative PCR
PGC-1α, PGC-1β, SREBP1c, acetyl-CoA carboxylase (ACC), SIRT1, carnitine palmitoyltransferase (CPT)-1, and β-actin mRNAs were measured by real-time PCR using their corresponding primer pairs, as we described previously,39 using the housekeeping gene, β-actin (Ambion, Austin, TX), as the internal control. The relative expression level of the specific gene of interest was calculated by subtracting the CT of the β-actin gene from the CT of the gene of interest and raising 2 to the power of this difference. CT values are defined as the number of PCR cycles at which the fluorescence signal during the PCR reaches a fixed threshold.
Immunohistochemistry
Immunohistochemistry of all liver sections was performed as we described,39 except that the sections were incubated overnight at 4°C with goat anti–SR-B1 (Novus Biologicals, Littleton, CO) at 1:500 dilution, and mouse anti-Na+/K+ATPase1, a plasma membrane marker, at 1:200 dilution, in 1% bovine serum albumin/PBS with Tween 20 (Abcam, Cambridge, MA). The liver sections were mounted with Prolong Gold Antifade mounting medium (Life Technologies, Carlsbad, CA). Confocal microscopy images were obtained with a Zeiss LSM 710 Zeiss Confocal microscope (Carl Zeiss Micro-Imaging, Inc., Thornwood, NY). Digital imaging software (Axio Vision Rel version 4.8.2; Carl Zeiss Micro-Imaging, Inc.) was used to generate the colocalization measurements using Pearson's correlation coefficient, describing the correlation of intensity distribution, and colocalization, which is denoted as the percentage of relative area of SR-B1 colocalized with Na+/K+ATPase1.
Western Blot Analysis
The indicated proteins of interest were determined in liver extracts and subcellular fractions from various groups by using Western blot analyses, as we described,39 and the following primary antibodies were used: PGC-1α, ACC, phosphorylated (p)ACC, AMP kinase (AMPK), and pAMPK (Cell Signaling Technology, Inc., Danvers, MA), SREBP1c (Abcam), SIRT1 (Santa Cruz Biotechnology, Santa Cruz, CA), PGC-1β (Genway Biotech, San Diego, CA), and SR-B1 (Novus Biologicals). Lamin B1 (Abcam), β-actin (Sigma, St. Louis, MO), and calreticulin (Abcam) were used as the housekeeping nuclear, cytosolic, and endoplasmic reticulum proteins, respectively, to normalize the results. The specificity of each antibody was verified before use for the previously described analyses.
Apoptosis Assay
Caspase 3 activity in liver lysate was measured with a colorimetric assay kit that relies on caspase-mediated cleavage of para-nitroanilide from a synthetic caspase-specific substrate peptide containing amino acid motif. The kit was used in accordance with the manufacturer's guidelines (R&D Systems Inc.).
Assessment of Mitochondrial Integrity
Liver mitochondria integrity was assessed by measuring its component enzyme, citrate synthase (CS) activity, in various groups, according to Srere et al,40 using 5,5′-dithiobis-(2-nitrobenzoic acid) in a 96-well plate of 0.1-cm light path with a SpectroMAX 190 spectrophotometer (Molecular Devices Co, Sunnyvale, CA).
Liver GSH Measurement
Liver GSH was determined in samples from various groups using the GSH assay kit (Sigma) at 412 nm using a SpectroMAX 190 spectrophotometer.
Isolation of Liver Subcellular Fractions
Fresh livers from experimental groups were immediately homogenized in ice-cold buffer containing 10 mmol/L HEPES (pH 7.4), 250 mmol/L mannitol, 75 mmol/L sucrose, 100 μmol/L EDTA, 500 μmol/L EGTA, and protease inhibitor cocktail (Sigma). Cell lysates were centrifuged at 1000 × g for 10 minutes to remove nuclear pellets. A supernatant fraction was centrifuged at 10,000 × g for 15 minutes to isolate the mitochondrial pellet. This pellet was washed twice in homogenizing buffer and then solubilized in the same buffer. The supernatant solution was centrifuged at 100,000 × g for 1 hour to collect the microsomal pellet. The protein estimation was performed with the Bradford method (Bio-Rad, Hercules, CA).
Statistical Analysis
All data are expressed as means ± SD using Microsoft Excel 2010 software (Microsoft Inc., Redmond, WA) and the significance of variance (analysis of variance), followed by the Tukey's test.41
Results
Effect of Alcohol and Betaine on Liver Structure, Liver Lipids, and Hepatic Lipid Score
Histopathological analysis revealed a remarkable fat accumulation in the HFE group, but no visible hepatosteatosis was observed in other groups (Figure 1A). Liver cholesterol and triglycerides increased markedly by 258% (P < 0.01) (Figure 1B) and 186% (P < 0.01) (Figure 1C), respectively, in the HFE group, with a concomitant 260% (P < 0.01) increase in hepatic lipid score (Figure 1, D and E), which were significantly blunted in the HFEB groups.
Figure 1.

Effect of alcohol and betaine on liver structure, liver lipids, and hepatic lipid score. Six rats per group were pair fed their respective HFC, HFE, HFCB, and/or HFEB liquid diets for 8 weeks, after which they were sacrificed for measuring the following lipid parameters, as described in Materials and Methods. H&E staining shows structural tissue changes (A), total liver cholesterol (B), liver triglycerides (C), histological features of liver sections showing oil red O–stained lipid droplets (D), and a bar graph representing percentage of relative levels of oil red O–stained lipid pixel values to total red pixels, calculated to show the extent of hepatosteatosis (E). The data are expressed as means ± SD in each group. Statistical significance of variance was calculated using the Tukey's test: ∗∗P < 0.01 versus HFC, ††P < 0.01 versus HFE.
Effect of Alcohol and Betaine on Serum Adiponectin, ALT, and Liver TNFα and IL-6
Plasma adiponectin level was significantly decreased by 37% in the HFE group compared with the HFC group (P < 0.01), whereas its level was markedly increased by 27% in the HFEB group, which was similar to the HFC group (Figure 2A). We next investigated the protective effect of betaine on hepatic proinflammatory cytokines: both IL-6 and TNFα showed an increasing trend in the HFE group compared with the HFC group (Figure 2, B and C); however, these differences were not statistically significant. Surprisingly, betaine significantly lowered both IL-6 and TNFα by 18% (Figure 2B) and 40% (Figure 2C), respectively, compared with the HFE group (P < 0.01). Concomitantly, compared with the HFC group, plasma ALT activity was significantly increased by 86% (P < 0.01) in the HFE group, which was significantly lowered by 70% (P < 0.01) in the HFEB group (Figure 2D).
Figure 2.
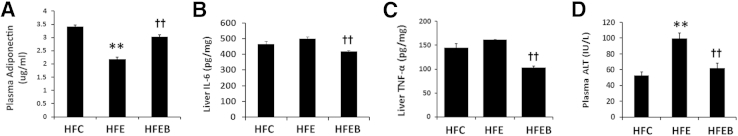
Effect of alcohol and betaine on proinflammatory and liver injury markers. Six rats per group were pair fed their respective HFC, HFE, and HFEB liquid diets for 8 weeks, after which they were sacrificed and plasma adiponectin (A), liver IL-6 (B), liver TNFα (C), and plasma ALT (D) were measured, as described in Materials and Methods. The data are expressed as means ± SD in each group. Statistical significance of variance was calculated using the Tukey's test: ∗∗P < 0.01 versus HFC, ††P < 0.01 versus HFE.
Effects of Alcohol and Betaine on Hepatic SR-B1 mRNA Expression, Plasma Membrane 57- and 82-kDa SR-B1 Species, Relative Glycosylation Rate, and Correlation with Cholesterol Uptake Function of Hepatocytes
SR-B1 mRNA expression was markedly decreased by 58% (P < 0.05) in the HFE group compared with the HFC group, whereas it was increased by 56% (P < 0.05) in the HFEB group compared with the HFE group (Figure 3A). The mature 82-kDa SR-B1 species in plasma membrane was decreased markedly by 43% (P < 0.05) in the HFE group, which was restored nearly to the control level in the HFEB group (Figure 3B). There was a reciprocal increase in the 57-kDa unglycosylated SR-B1 species in HFE that was restored nearly to the control level in the HFEB group. In immunoprecipitated extracts of hepatocytes, the incorporation of labeled N-AcMnNH2 relative to labeled methionine incorporation into newly synthesized SR-B1 was decreased by 42% (P < 0.01) in the HFE group, which was restored nearly to the control level in the HFEB group (Figure 3C). Compared with HFC, the uptake of cholesterol by hepatocytes from the HFE group was significantly inhibited by 48% (P < 0.05), whereas it was stimulated to 154% (P < 0.001) in the HFEB group (Figure 3D).
Figure 3.

Effects of alcohol and betaine on hepatic SR-B1 mRNA expression, plasma membrane 57- and 82-kDa SR-B1 species, relative glycosylation rate, and correlation with cholesterol uptake function of hepatocytes. Six rats per group were pair fed their respective HFC, HFE, and HFEB liquid diets for 8 weeks, after which they were sacrificed and SR-B1 mRNA expression (A), plasma membrane 57- and 82-kDa SR-B1 species (B), relative glycosylation rate (C), and correlation with cholesterol uptake function of hepatocytes (D) were measured as described in Materials and Methods. The data are expressed as means ± SD in each group. Statistical significance of variance was calculated using Tukey's test: ∗P < 0.05, ∗∗P < 0.01 versus HFC; †P < 0.05, ††P < 0.01, and †††P < 0.001 versus HFE.
Confirmation of the Influence of Chronic Ethanol and Betaine on the Localization of SR-B1 in Liver Plasma Membrane
The intensity of the colocalized SR-B1 with ATPase in the plasma membrane was markedly decreased by 34% (P < 0.05) in the HFE group compared with the HFC group, which was completely reversed in the HFEB group (Figure 4).
Figure 4.

Effects of alcohol and betaine on hepatic colocalization of SR-B1 with Na+/K+ ATPase1. Six rats per group were pair fed their respective HFC, HFE, and HFEB liquid diets for 8 weeks, after which the liver sections were obtained and stained for SR-B1 and Na+/K+ ATPase1, as described in Materials and Methods. A: Representative images of the individual channels for SR-B1 (green), Na+/K+ ATPase1 (red), nucleus (blue), and the merged channels, indicating regions of colocalization in yellow. B: The co-occurrence and quantification of SR-B1 with the plasma membrane marker protein, Na+/K+ ATPase1, is expressed as the average percentage of pixels of SR-B1 relative to the pixels of Na+/K+ ATPase1. C: Pearson's correlation coefficient was calculated to describe the colocalization of the intensity distribution between SR-B1 and Na+/K+ ATPase1. Data are expressed as means ± SD in each group. Statistical significance of variance was calculated using Tukey's test: ∗P < 0.05 versus HFC, †P < 0.05 versus HFE. Scale bar = 20 μm (A).
Effects of Chronic Ethanol and Betaine on Liver GSH, Mitochondrial Cholesterol, Mitochondrial Integrity, and the Extent of Apoptosis
Liver mitochondrial cholesterol (Figure 5A) was markedly elevated by 61% (P < 0.01), accompanied by a significant 34% (P < 0.05) decrease in mitochondrial GSH (Figure 5B) in the HFE group, which was partially prevented in the HFEB group. Furthermore, the relative leakage of cytochrome c (Figure 5C) into the cytosolic fraction was increased by 30% (P < 0.05), accompanied by 75% (P < 0.01) elevation of caspase 3 activity in the HFE group (Figure 5D), which were partially blocked in the HFEB group (P < 0.05). Compared with the HFC group, liver CS activity in the HFE group (Figure 5E) was significantly decreased by 41% (P < 0.01), which was significantly restored in the HFEB group (P < 0.05).
Figure 5.

Effects of alcohol and betaine on mitochondrial cholesterol, liver GSH, mitochondrial integrity, and the extent of apoptosis. Six rats per group were pair fed their respective HFC, HFE, and HFEB liquid diets for 8 weeks, after which they were sacrificed to obtain liver samples for measuring mitochondrial cholesterol (A), mitochondrial GSH (B), cytochrome c (C), caspase 3 activity (D), and CS activity (E), as described in Materials and Methods. Data are expressed as means ± SD in each group. Statistical significance of variance was calculated using Tukey's test: ∗P < 0.05, ∗∗P < 0.01 versus HFC; †P < 0.05, ††P < 0.01 versus HFE.
Effects of Chronic Ethanol and Betaine on PGC-1 Signaling Pathway Genes and the Corresponding Proteins Involved in Lipogenesis and Lipid Oxidation
PGC-1β mRNA expression was significantly increased in the HFE group (Figure 6A) by 1.6-fold (P < 0.01) compared with the HFC group, which was partially blocked in the HFEB group, accompanied by concomitant increased expressions of SREBP1c mRNA (Figure 6B) by 51% (P < 0.01), ACC mRNA (Figure 6C) by 83% (P < 0.05), SREBP2 mRNA (Figure 6D) by 30% (P < 0.05), and HMG-CoA reductase mRNA (Figure 6E) by 50% (P < 0.05). In contrast, PGC-1α mRNA (Figure 6F) and CPT-1 mRNA (Figure 6G) expressions were markedly reduced by 45% (P < 0.01) and 40% (P < 0.05), respectively, in the HFE group, with a parallel decrease by 38% (P < 0.05) in SIRT1 mRNA expression (Figure 6H) in the HFE group. These were partially prevented in the HFEB group, although restoration of SIRT1 mRNA was marginal in the HFEB group. Parallel changes were observed in the expressions of the corresponding proteins (Figure 7, A–E). Moreover, phosphorylated species of ACC (Figure 7F) was decreased by 42% (P < 0.001). Significantly, although the total AMPK did not change between the various groups (Figure 7G), pAMPK (Figure 7H) decreased by 43% (P < 0.001) in HFE. These were restored to the normal level in HFEB. Similarly, nuclear protein levels of SIRT1 (Figure 8A) and PGC-1α (Figure 8B) were also significantly reduced in the HFE group by 63% (P < 0.01) and 47% (P < 0.01), respectively, with a parallel decrease in the protein levels of CPT-1 (Figure 8C) by 20% (P < 0.05), which were restored partially to the control level in HFEB. These changes in the lipid-oxidizing pathway resulted in the corresponding 48% (P < 0.001) reduced rate of fatty acid oxidation by hepatocytes from the HFE group, which was restored to normal in the HFEB group (P < 0.001) (Figure 8D).
Figure 6.

Effects of alcohol and betaine on PGC-1 signaling pathway genes and corresponding proteins involved in lipogenesis and lipid oxidation. Six rats per group were pair fed their respective HFC, HFE, and HFEB liquid diets for 8 weeks, after which they were sacrificed and the hepatic mRNA expressions were determined by quantitative real-time PCR, as described in Materials and Methods for PGC-1β (A), SREBP1c (B), ACC (C), SREBP2 (D), HMG-CoA reductase (E), PGC-1α (F), CPT-1 (G), and SIRT1 (H). The results are expressed as means ± SD in each group. Statistical significance of variance was calculated using Tukey's test: ∗P < 0.05, ∗∗P < 0.01 versus HFC; †P < 0.05, ††P < 0.01 versus HFE.
Figure 7.

Effects of alcohol and betaine on PGC-1β signaling pathway proteins involved in lipogenesis. Six rats per group were pair fed their respective HFC, HFE, and HFEB liquid diets for 8 weeks, after which they were sacrificed and hepatic protein expressions were determined by using Western blot analysis, as described in Materials and Methods for PGC-1β (A), SREBP-1c (B), SREBP2 (C), HMG-CoA reductase (D), ACC (E), pACC (F), AMPK (G), and pAMPK (H). The results are expressed as means ± SD in each group. Statistical significance of variance was calculated using Tukey's test: ∗∗P < 0.01, ∗∗∗P < 0.001 versus HFC; †P < 0.05, ††P < 0.01, and †††P < 0.001 versus HFE.
Figure 8.
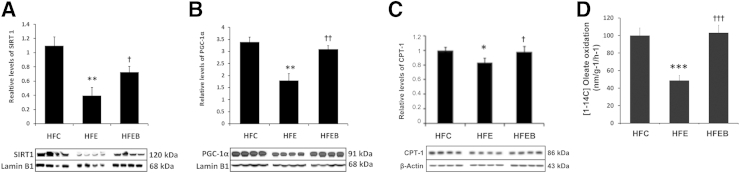
Effects of alcohol and betaine on PGC-1α signaling pathway proteins involved in lipid oxidation and hepatic fatty acid oxidation rate. Six rats per group were pair fed their respective HFC, HFE, and HFEB liquid diets for 8 weeks, after which they were sacrificed, and hepatic protein expressions were determined by using Western blot analysis, as described in Materials and Methods for SIRT (A), PGC-1α (B), and CPT-1 (C). D: Fatty acid oxidation rate in hepatocytes from each group was measured as described in Materials and Methods. The results are expressed as the means ± SD in each group. Statistical significance of variance was calculated using Tukey's test: ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 versus HFC; †P < 0.05, ††P < 0.01, and †††P < 0.001 versus HFE.
Discussion
We show that chronic ethanol exposure causes profound hepatosteatosis (Figure 1), with concomitant significant increases in hepatic proinflammatory cytokines and liver injury marker that were blunted by betaine supplementation (Figure 2). We further show that chronic ethanol markedly inhibits the relative glycosylation of SR-B1 (Figure 3C), resulting in the decreased localization of the mature SR-B1 in the liver plasma membrane (Figure 3B). These results are further confirmed by chronic alcohol-mediated decreased colocalization of SR-B1 with Na+/K+ATPase, the plasma membrane marker (Figure 4). This explains why chronic ethanol markedly affects the hepatic cholesterol uptake capacity (Figure 3D) because normal SR-B1 synthesis, its post-translation modification, and localization on the liver plasma membrane are obligatory for it to perform this critical liver function. Significantly, betaine treatment completely protects against these deleterious effects of chronic ethanol by restoring the relative hepatic glycosylation of SR-B1, accompanied by increased colocalization of mature SR-B1 in the plasma membrane and consequent restoration of its cholesterol uptake function (Figures 3 and 4).
Because chronic ethanol is known to cause increased ROS,42 especially in the presence of high polyunsaturated fatty acids, the possible mechanism of this protective action of betaine seems to be due to its ability to restore the hepatic intracellular natural antioxidant, GSH, that is markedly decreased by chronic alcohol (Figure 5B).
Although ethanol-mediated impaired glycosylation of SR-B1 and its localization on plasma membrane can account for its defective reverse cholesterol transport function, it fails to explain why chronic alcohol leads to hepatosteatosis (Figure 1). In this regard, our findings on the action of chronic ethanol on the PGC-1 signaling pathways do shed some light on the possible underlying mechanisms. Thus, we clearly show that chronic ethanol up-regulates PGC-1β, SREBP1c, and the downstream lipogenic genes, and the respective proteins (Figures 6 and 8), with a concomitant increase in the active non-phosphorylated ACC (Figure 7E), because of a reciprocal decrease in the active phosphorylated AMPK (Figure 7H). In contrast, chronic ethanol down-regulates SIRT1, PGC-1α, and downstream lipid-oxidizing genes (Figure 6, F–H), resulting in decreased fatty acid oxidation (Figure 8D). These mechanistic findings of increased hepatic triglyceride and cholesterol syntheses, coupled with impaired lipid degradation leading to lipid accumulation, could justifiably account for the observed alcoholic hepatosteatosis (Figure 1) and are in accord with previous studies.19,20 More important, our new finding of the ability of betaine to prevent these hepatosteatotic actions of chronic alcohol by considerably down-regulating PGC-1β and lipogenic genes and up-regulating PGC-1α and lipid-oxidizing genes accounts for the near-normal hepatic lipid score found in betaine-fed rats (Figure 1), despite feeding a high PUFA diet. This is also reflected in parallel reciprocal changes in serum adiponectin (Figure 2), accompanied by marginal body weight gain of the betaine-fed group compared with the ethanol group alone or even the control group (data not shown). Significantly, in contrast to the mild liver injury observed after standard Lieber-Decarli ethanol treatment,43,44 our present study shows that chronic ethanol-high PUFA treatment causes moderate liver injury, as evidenced by increased serum ALT (Figure 2D) with concomitant changes in liver TNFα and IL-6 (Figure 2, B and C), although the extent of liver injury is not as severe as found in the chronic ethanol–binge ethanol model of Gao and colleagues.43,44
Furthermore, our finding of chronic alcohol-mediated increased liver mitochondrial cholesterol (Figure 5A) is in accord with those of Fernandez et al,45 who showed increased mitochondrial permeability with cholesterol loading. This may account for impaired mitochondrial integrity that is demonstrated by increased cytochrome c oxidase release from the mitochondria. Moreover, chronic ethanol is known to increase ROS production,45 which could induce apoptosis in liver due to stressed mitochondria and endoplasmic reticulum. This is demonstrated by increased caspase 3 after chronic ethanol exposure, accompanied by decreased mitochondrial CS activity (Figure 5, D and E). Again, the fact that betaine treatment essentially corrects these defects (Figure 5, D and E) suggests that membrane integrity of mitochondria is essentially restored by betaine treatment.
In summary, it is reasonable to conclude the following actions of chronic ethanol exposure: i) impaired post-translational modification, trafficking, and defective hepatic cholesterol uptake function of SR-B1 may account for alcoholic hyperlipidemia; ii) up-regulation of hepatic PGC-1β, and lipid synthetic genes, and down-regulation of SIRT1, PGC-1α, and lipid degradation genes account for alcoholic hepatosteatosis; and iii) induction of proinflammatory cytokines and depletion of endogenous antioxidant, GSH, account for alcoholic steatohepatitis. We, therefore, suggest that betaine may be a potential protective therapeutic agent to prevent alcoholic hepatosteatosis and steatohepatitis.
Acknowledgment
We thank Joseph Ibrahim for technical assistance.
Footnotes
Supported by NIH grants R21 AA017965 and RO1 AA020720 (M.R.L.).
Disclosures: None declared.
Current address of M.G., Department of Biochemistry and Molecular Biology, Howard University College of Medicine, Washington, DC.
References
- 1.Hobbs H.H., Brown M.S., Goldstein J.L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat. 1992;1:445–466. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 2.Attie A.D., Kastelein J.P., Hayden M.R. Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J Lipid Res. 2001;42:1717–1726. [PubMed] [Google Scholar]
- 3.Silver D.L., Wang N., Xiao X., Tall A.R. High density lipoprotein (HDL) particle uptake mediated by scavenger receptor class B type 1 results in selective sorting of HDL cholesterol from protein and polarized cholesterol secretion. J Biol Chem. 2001;276:25287–25293. doi: 10.1074/jbc.M101726200. [DOI] [PubMed] [Google Scholar]
- 4.Acton S., Rigotti A., Landschulz K.T., Xu S., Hobbs H.H., Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 5.Hauser H., Dyer J.H., Nandy A., Vega M.A., Werder M., Bieliauskaite E., Weber F.E., Compassi S., Gemperli A., Boffelli D., Wehrli E., Schulthess G., Phillips M.C. Identification of a receptor mediating absorption of dietary cholesterol in the intestine. Biochemistry. 1998;37:17843–17850. doi: 10.1021/bi982404y. [DOI] [PubMed] [Google Scholar]
- 6.Out R., Hoekstra M., Spijkers J.A., Kruijt J.K., van Eck M., Bos I.S., Twisk J., Van Berkel T.J. Scavenger receptor class B type I is solely responsible for the selective uptake of cholesteryl esters from HDL by the liver and the adrenals in mice. J Lipid Res. 2004;45:2088–2095. doi: 10.1194/jlr.M400191-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Casey C.A., Kragskow S.L., Sorrell M.F., Tuma D.J. Chronic ethanol administration impairs the binding and endocytosis of asialo-orosomucoid in isolated hepatocytes. J Biol Chem. 1987;262:2704–2710. [PubMed] [Google Scholar]
- 8.Lakshman M.R., Chirtel S.J., Chambers L.C., Campbell B.S. Hepatic synthesis of apoproteins of very low density and high density lipoproteins in perfused rat liver: influence of chronic heavy and moderate doses of ethanol. Alcohol Clin Exp Res. 1989;13:554–559. doi: 10.1111/j.1530-0277.1989.tb00377.x. [DOI] [PubMed] [Google Scholar]
- 9.Tuma D.J., Sorrell M.F. Effects of ethanol on protein trafficking in the liver. Semin Liver Dis. 1988;8:69–80. doi: 10.1055/s-2008-1040529. [DOI] [PubMed] [Google Scholar]
- 10.Casey C.A., Kragskow S.L., Sorrell M.F., Tuma D.J. Ethanol-induced impairments in receptor-mediated endocytosis of asialoorosomucoid in isolated rat hepatocytes: time course of impairments and recovery after ethanol withdrawal. Alcohol Clin Exp Res. 1989;13:258–263. doi: 10.1111/j.1530-0277.1989.tb00323.x. [DOI] [PubMed] [Google Scholar]
- 11.Nagy L.E., Lakshman M.R., Casey C.A., Bearer C.F. Ethanol and membrane protein trafficking: diverse mechanisms of ethanol action. Alcohol Clin Exp Res. 2002;26:287–293. [PubMed] [Google Scholar]
- 12.Ghosh P., Lakshman M.R. Chronic ethanol induced impairment of hepatic glycosylation machinery in rat is independent of dietary carbohydrate. Alcohol Clin Exp Res. 1997;21:76–81. [PubMed] [Google Scholar]
- 13.Tworek B.L., Oka J.A., Casey C.A., Weigel P.H. Ethanol feeding causes inactivation of both state 1 and state 2 rat hepatic asialoglycoprotein receptors. Alcohol Clin Exp Res. 1997;21:1429–1434. [PubMed] [Google Scholar]
- 14.Tworek B.L., Tuma D.J., Casey C.A. Decreased binding of asialoglycoproteins to hepatocytes from ethanol-fed rats: consequence of both impaired synthesis and inactivation of the asialoglycoprotein receptor. J Biol Chem. 1996;271:2531–2538. doi: 10.1074/jbc.271.5.2531. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh P., Chirtel S.J., Lakshman M.R. Effect of chronic ethanol on apolipoprotein (Apo) E synthesis and glycosylation in rats. Alcohol Clin Exp Res. 1991;15:725–729. doi: 10.1111/j.1530-0277.1991.tb00586.x. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh P., Hale E.A., Lakshman R. Long-term ethanol exposure alters the sialylation index of plasma apolipoprotein J (Apo J) in rats. Alcohol Clin Exp Res. 1999;23:720–725. [PubMed] [Google Scholar]
- 17.Ghosh P., Liu Q.H., Lakshman M.R. Long-term ethanol exposure impairs glycosylation of both N- and O-glycosylated proteins in rat liver. Metabolism. 1995;44:890–898. doi: 10.1016/0026-0495(95)90242-2. [DOI] [PubMed] [Google Scholar]
- 18.Baraona E., Leo M.A., Borowsky S.A., Lieber C.S. Pathogenesis of alcohol-induced accumulation of protein in the liver. J Clin Invest. 1977;60:546–554. doi: 10.1172/JCI108806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lieber C.S., Leo M.A., Wang X., Decarli L.M. Effect of chronic alcohol consumption on hepatic SIRT1 and PGC-1alpha in rats. Biochem Biophys Res Commun. 2008;370:44–48. doi: 10.1016/j.bbrc.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 20.You M., Cao Q., Liang X., Ajmo J.M., Ness G.C. Mammalian sirtuin 1 is involved in the protective action of dietary saturated fat against alcoholic fatty liver in mice. J Nutr. 2008;138:497–501. doi: 10.1093/jn/138.3.497. [DOI] [PubMed] [Google Scholar]
- 21.Canto C., Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemoto S., Fergusson M.M., Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha} J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 24.Lakshman M.R. Some novel insights into the pathogenesis of alcoholic steatosis. Alcohol. 2004;34:45–48. doi: 10.1016/j.alcohol.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 25.Sozio M., Crabb D.W. Alcohol and lipid metabolism. Am J Physiol Endocrinol Metab. 2008;295:E10–E16. doi: 10.1152/ajpendo.00011.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barak A.J., Beckenhauer H.C., Badakhsh S., Tuma D.J. The effect of betaine in reversing alcoholic steatosis. Alcohol Clin Exp Res. 1997;21:1100–1102. [PubMed] [Google Scholar]
- 27.Balkan J., Oztezcan S., Kucuk M., Cevikbas U., Kocak-Toker N., Uysal M. The effect of betaine treatment on triglyceride levels and oxidative stress in the liver of ethanol-treated guinea pigs. Exp Toxicol Pathol. 2004;55:505–509. doi: 10.1078/0940-2993-00347. [DOI] [PubMed] [Google Scholar]
- 28.Graf D., Kurz A.K., Reinehr R., Fischer R., Kircheis G., Häussinger D. Prevention of bile acid-induced apoptosis by betaine in rat liver. Hepatology. 2002;36:829–839. doi: 10.1053/jhep.2002.35536. [DOI] [PubMed] [Google Scholar]
- 29.Barak A.J., Beckenhauer H.C., Junnila M., Tuma D.J. Dietary betaine promotes generation of hepatic S-adenosylmethionine and protects the liver from ethanol-induced fatty infiltration. Alcohol Clin Exp Res. 1993;17:552–555. doi: 10.1111/j.1530-0277.1993.tb00798.x. [DOI] [PubMed] [Google Scholar]
- 30.Garige M., Gong M., Varatharajalu R., Lakshman M.R. Quercetin up-regulates paraoxonase 1 gene expression via sterol regulatory element binding protein 2 that translocates from the endoplasmic reticulum to the nucleus where it specifically interacts with sterol responsive element-like sequence in paraoxonase 1 promoter in HuH7 liver cells. Metabolism. 2010;59:1372–1378. doi: 10.1016/j.metabol.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 31.Shireman R.B., Durieux J. Microplate methods for determination of serum cholesterol, high density lipoprotein cholesterol, triglyceride and apolipoproteins. Lipids. 1993;28:151–155. doi: 10.1007/BF02535780. [DOI] [PubMed] [Google Scholar]
- 32.Gidez L.I., Miller G.J., Burstein M., Slagle S., Eder H.A. Separation and quantitation of subclasses of human plasma high density lipoproteins by a simple precipitation procedure. J Lipid Res. 1982;23:1206–1223. [PubMed] [Google Scholar]
- 33.Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 34.Zlatkis A., Zak B. Study of a new cholesterol reagent. Anal Biochem. 1969;29:143–148. doi: 10.1016/0003-2697(69)90017-7. [DOI] [PubMed] [Google Scholar]
- 35.Basu S.K., Goldstein J.L., Anderson G.W., Brown M.S. Degradation of cationized low density lipoprotein and regulation of cholesterol metabolism in homozygous familial hypercholesterolemia fibroblasts. Proc Natl Acad Sci U S A. 1976;73:3178–3182. doi: 10.1073/pnas.73.9.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lakshman M.R., Muesing R.A., LaRosa J.C. Regulation of cholesterol biosynthesis and 3-hydroxy-3-methylglutaryl coenzyme A reductase activity by chylomicron remnants in isolated hepatocytes and perfused liver. J Biol Chem. 1981;256:3037–3043. [PubMed] [Google Scholar]
- 37.Marmillot P., Munoz J., Patel S., Garige M., Rosse R.B., Lakshman M.R. Long-term ethanol consumption impairs reverse cholesterol transport function of high-density lipoproteins by depleting high-density lipoprotein sphingomyelin both in rats and in humans. Metabolism. 2007;56:947–953. doi: 10.1016/j.metabol.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gong M., Castillo L., Redman R.S., Garige M., Hirsch K., Azuine M., Amdur R.L., Seth D., Haber P.S., Lakshman M.R. Down-regulation of liver Galbeta1, 4GlcNAc alpha2, 6-sialyltransferase gene by ethanol significantly correlates with alcoholic steatosis in humans. Metabolism. 2008;57:1663–1668. doi: 10.1016/j.metabol.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 39.Luna L., editor. Manual of Histologic Staining Methods of the Armed Forces Institutes of Pathology. ed 3. McGraw-Hill; New York: 1968. [Google Scholar]
- 40.Srere P.A., Bhaduri A. The citrate cleavage enzyme, III: citryl coenzyme a as a substrate and the stereospecificity of the enzyme. J Biol Chem. 1964;239:714–718. [PubMed] [Google Scholar]
- 41.SAS Institute . SAS Institute; Cary, NC: 2000. SAS/STAT User's Guide, Version 6. [Google Scholar]
- 42.Cederbaum A.I., Lu Y., Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009;83:519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 43.Bertola A., Mathews S., Ki S.H., Wang H., Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ki S.H., Park O., Zheng M., Morales-Ibanez O., Kolls J., Bataller R., Gao B. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291–1300. doi: 10.1002/hep.23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fernandez A., Matias N., Fucho R., Ribas V., Montfort C.V., Nuno N., Baulies A., Martinez L., Tarrats N., Mari M., Colell A., Morales A., Dubuquoy L., Mathurin P., Bataller R., Caballeria J., Elena M., Balsinde J., Kaplowitz N., Garcia-Ruiz C., Fernandez-Checa J.C. Asmase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. J Hepatol. 2013;59:805–813. doi: 10.1016/j.jhep.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]