

Abstract
Obesity and metabolic syndrome are linked to an increased prevalence of breast cancer among postmenopausal women. A common feature of obesity, metabolic syndrome, and a Western diet rich in saturated fat is a high level of circulating cholesterol. Epidemiological reports investigating the relationship between high circulating cholesterol levels, cholesterol-lowering drugs, and breast cancer are conflicting. Here, we modeled this complex condition in a well-controlled, preclinical animal model using innovative isocaloric diets. Female severe combined immunodeficient mice were fed a low-fat/no-cholesterol diet and then randomized to four isocaloric diet groups: low-fat/no-cholesterol diet, with or without ezetimibe (cholesterol-lowering drug), and high-fat/high-cholesterol diet, with or without ezetimibe. Mice were implanted orthotopically with MDA-MB-231 cells. Breast tumors from animals fed the high-fat/high-cholesterol diet exhibited the fastest progression. Significant differences in serum cholesterol level between groups were achieved and maintained throughout the study; however, no differences were observed in intratumoral cholesterol levels. To determine the mechanism of cholesterol-induced tumor progression, we analyzed tumor proliferation, apoptosis, and angiogenesis and found a significantly greater percentage of proliferating cells from mice fed the high-fat/high-cholesterol diet. Tumors from hypercholesterolemic animals displayed significantly less apoptosis compared with the other groups. Tumors from high-fat/high-cholesterol mice had significantly higher microvessel density compared with tumors from the other groups. These results demonstrate that hypercholesterolemia induces angiogenesis and accelerates breast tumor growth in vivo.
Breast cancer is the most frequent and second most deadly cancer in women, with >230,000 new cases and nearly 40,000 estimated annual deaths in the United States alone.1 Most breast cancers are invasive ductal carcinomas that have penetrated through the ductal wall and have invaded surrounding breast tissue, often leading to metastasis to other sites, including the lung, liver, and bone, resulting in significant morbidity, suffering, and death.
Breast cancer rates differ significantly among nations worldwide, with developed countries in North America and Northern and Western Europe reporting the highest incidence of breast cancer globally. The incidence of breast cancer in these regions is three to four times higher than the incidence of breast cancer in parts of Asia and Africa.1 More important, relocation studies have shown that women who move from an area of low incidence of breast cancer (Japan) to a high-incidence area (Los Angeles, CA) develop breast cancer at rates similar to that of U.S. women, with the rate of breast cancer incidence associated with the time of life at immigration.2 For Japanese women who immigrated early in life, the rate of breast cancer was significantly higher than in women who moved from Japan to Los Angeles later in life, with both groups demonstrating a significantly greater incidence of breast cancer compared with women in their homeland.2 These findings point to a possible dietary factor in a traditional Western diet that may be contributing to increased breast cancer incidence.2 Although specific dietary factors that increase the rate of breast cancer among U.S. women are not well understood, some evidence suggests a significant, positive association between breast cancer incidence and intake of saturated fat in postmenopausal women.3 Moreover, both adult weight gain and metabolic syndrome are linked to an increased prevalence of breast cancer.4–6 A common feature shared by adult obesity, metabolic syndrome, and a Western diet rich in fat is a high level of circulating cholesterol.
Similar to other mammalian cells, breast ductal epithelial cells synthesize cholesterol endogenously via the mevalonate pathway and absorb cholesterol-containing lipoproteins from the circulation. Consequently, control of cellular cholesterol content is achieved by regulating systemic cholesterol concentration and by balancing intrinsic cellular metabolic processes.7,8 Yet, despite the sophisticated mechanisms for maintaining the appropriate cholesterol balance, it has been reported that ductal epithelium of the breast may be sensitive to systemic changes in serum cholesterol levels.9 These changes may result in aberrant cellular responses to specific growth factors and/or cytokines, resulting in altered patterns of cellular growth, apoptosis, morphological characteristics, and intercellular communication.
Retrospective epidemiological studies have suggested that elevated serum cholesterol and/or hyperlipidemia increase the rate of breast cancer among women of developed countries.4,5,10 These case-controlled, cohort, or comparative studies also reported that statin drugs, which inhibit the rate-limiting step in cholesterol synthesis11 or the hydrophobic subclass of statins, decrease overall breast cancer prevalence,12–15 reduce the rate of developing an estrogen receptor/progesterone receptor–negative cancer, and promote a downward shift in breast cancer grade and stage.16 However, results from several large, prospective, epidemiological studies examining the relationship between serum cholesterol levels, cholesterol-lowering drugs, and breast cancer incidence have been inconclusive.14,17,18 For example, it has been shown that women with clinically high (≥240 mg/dL) fasting total serum cholesterol have a slight, but statistically significant, increased incidence of breast cancer.18 A separate study demonstrated that untreated, self-reported high cholesterol was not associated with an increased rate of breast cancer, even among obese and postmenopausal women.14 A third study found that the hydrophobic class of statins significantly reduced the incidence of invasive breast cancer among older women.13,15
In contrast to the previously described studies,17 which suggested an association between statin use and decreased incidence of breast cancer, a prospective study examining the effect of cholesterol-lowering drug use on many cancers showed that the incidence of breast cancer was no different among women reporting long-term statin use compared with women who reported never having used statins. This latter report adjusted for age and history of high cholesterol levels but did not separate women on the basis of menopausal status.
Although the effect of statins on the incidence of primary breast cancer remains unclear, epidemiological studies report that lipophilic statin use among women diagnosed with stage I to III breast cancer was associated with a reduced rate of cancer recurrence.19–21 Taken together, these numerous, inconsistent, and sometimes contradicting epidemiological results highlight an important need for studies that model the complex relationship between serum cholesterol, cholesterol-lowering drugs, and breast cancer using well-controlled, preclinical models.
As a function of the suggested links between postmenopausal weight gain, metabolic syndrome, and breast cancer,4–6 prior work describing an effect of elevated cholesterol and statin drugs on breast cancer risk,4,5,12–14 and our own reports demonstrating a role for cholesterol in prostate cancer progression,22–25 we chose to determine whether hypercholesterolemia would promote breast cancer progression in an in vivo model that tightly controlled other metabolic variables associated with a Western diet.
It has been previously reported that a high-fat Western diet promoted the growth of breast tumors in the MMTV-PyMT breast cancer model in which tumor formation is driven by polyoma middle T antigen26 and in the apolipoprotein E (ApoE) knockout mouse.27 Although these studies suggested that a Western diet promotes breast cancer growth, a specific role for cholesterol was not established because isocaloric diets were not compared and cholesterol was not specifically targeted (as we have done in the current study).
By using novel diet and feeding strategies, we have generated an innovative isocaloric diet approach in which we are able to determine the specific effects of cholesterol in mice. This approach provides the opportunity to study the effects of hypercholesterolemia on tumor growth. In the current study, we apply this approach to an orthotopic xenograft model of human estrogen receptor–negative breast cancer and demonstrate that a hypercholesterolemic diet promotes the growth of breast cancer. This effect is blocked by the hypocholesterolemic drug ezetimibe. Histological and biochemical analyses indicate that hypercholesterolemia increased cell proliferation and tumor cell apoptosis, and significantly increased tumor angiogenesis. To our knowledge, this is the first report of a specific role for cholesterol in promoting breast cancer growth.
Materials and Methods
Antibodies
The following antibodies were used: anti-CD31 monoclonal antibody (rat anti-mouse); anti–Ki-67 primary antibody (Abcam, Cambridge, MA); anti-smooth muscle actin monoclonal antibody (Sigma, St. Louis, MO); Alexa Fluor 488–conjugated goat anti-rat; Alexa Fluor 488–conjugated goat anti-mouse; Alexa Fluor 568–conjugated goat anti-rabbit; Alexa Fluor 568–conjugated goat anti-mouse (Invitrogen, Carlsbad, CA); and Cy3-conjugated AffiniPure goat anti-rabbit IgG, Fc fragment specific (Jackson ImmunoResearch, West Grove, PA).
Orthotopic Breast Tumor Model
Five-week-old female severe combined immunodeficient (SCID) mice, obtained from the Massachusetts General Hospital (Boston), were fed a low-fat/no-cholesterol diet (LFNC) (10% fat, 0% cholesterol, and 0% cholic acid; diet D12102; Research Diets, New Brunswick, NJ) (Table 1) for 3 weeks, followed by a blood draw from the saphenous tail vein and serum cholesterol determination using the Infinity Cholesterol Liquid Stable Reagent (ThermoFisher Scientific, Waltham, MA) (Figure 1 and Table 1). The mice were then divided into high-fat/high-cholesterol diet (HFHC) (40% fat, 1.25% cholesterol, and 0% cholic acid; diet D12109; Research Diets) (Table 1) and LFNC diet groups, with or without 30 mg/kg per day ezetimibe (Merck, Whitehouse Station, NJ), added to powdered food, and the mice continued on their respective isocaloric diets for 4 weeks before tumor implantation (Table 1). MDA-MB-231 breast cancer cells were obtained in October 2005 from American Type Culture Collection (ATCC; Manassas, VA) (catalog number HTB-26, lot number 3576801), grown as recommended and frozen within two passages for long-term storage. On resuscitation, MDA-MB-231 cells were expanded for <10 passages and were maintained in culture for <6 weeks before in vivo implantation. These cultured MDA-MD-231 cells were most recently authenticated in February 2013 by short tandem repeat DNA profiling (Genetica DNA Laboratories, Burlington, NC). Comparative analysis of our MDA-MB-231 DNA profile matched the known reference DNA profile of ATCC MDA-MD-231 (American National Standards Institute/ATCC ASN-0002-2011), with 100% identity confirming the authentication of our MDA-MB-231. A total of 2 × 106 MDA-MD-231 cells were orthotopically injected per site, with 1:1 volume of Matrigel (BD BioSciences, San Jose, CA) into the fourth mammary fat pad of each animal (two tumors per mice).28,29 To eliminate any injection bias, the mice were randomized before implantation and the implanter (K.P. or C.M.C.) was blinded to which group each mouse was assigned. All animal procedures were done in compliance with Boston Children's Hospital (Boston, MA) animal care and use policies. Tumors were measured every other day from the initiation of the first palpable tumors, and the mice were sacrificed before reaching the maximum tumor burden (87 ± 1 day after implantation). Terminal bleeds for serological analysis were taken via cardiac puncture. Serum insulin and estradiol tests were performed by the Vanderbilt Hormone Assay and Analytical Core (Nashville, TN). Triglyceride, bilirubin, and other liver function tests were performed by the Yale University Mouse Phenotyping Center (New Haven, CT). Tumors were removed and either placed in optimal cutting temperature (OCT) solution (Tissue-Tek, Torrance, CA) or snap frozen for analysis.
Table 1.
Diet Formulations
| Diet component | HFHC |
LFNC |
||
|---|---|---|---|---|
| g% | kcal% | g% | kcal% | |
| Protein (casein, 30 mesh l-cysteine) | 23 | 20 | 19 | 20 |
| Carbohydrate (corn starch, maltodextrin, sucrose, cellulose) | 45 | 40 | 67 | 70 |
| Fat (soybean oil, cocoa butter) | 20 | 40 | 4 | 10 |
| Cholesterol | 1.25 | 0 | 0 | 0 |
| Vitamin, mineral, other | ∼10 | 0 | 10 | 0 |
Figure 1.

Study design. A and B: A total of 120 female SCID mice (A) were maintained on a LFNC diet (B) for 3 weeks. C: Blood was drawn to measure serum cholesterol concentration. Any mouse with serum cholesterol levels that did not normalize (means ± SD) was removed from the study. D: Mice were then randomized into four different diet/treatment groups: LFNC, with or without ezetimibe, and high fat/high cholesterol, with or without ezetimibe. E: Mice were maintained on these diets for 4 weeks, and their serum cholesterol levels were monitored biweekly, at which point 2 × 106 MDA-MB-231 cells were orthotopically injected into the fourth mammary fat pads (two tumors per mouse, approximately 28 to 30 mice per group). F: Mice were maintained on their respective diets and were checked daily for tumor formation, and their blood was drawn every several weeks for serum cholesterol determination. After 4 weeks, when tumors appeared, tumor volume was measured three to four times per week until reaching maximum tumor volume. G: Mice were sacrificed, and tumors were harvested and processed for analysis.
Tumor Cholesterol Analysis
Tumors were finely minced in PBS on ice and cholesterol concentration was determined as described previously.30
Apoptosis Analysis
The percentage of apoptotic cells in tumors divided into sections was determined by TUNEL assay using the In Situ Cell Death Detection Kit (Roche Diagnostics Corp, Indianapolis, IN). Briefly, frozen tumor sections were fixed in 4% paraformaldehyde and permeabilized, and the DNA was stained with fluorescein following the manufacturer's protocol. Nuclei were counterstained with DAPI (Vector Labs, Burlingame, CA). Images were captured using a Zeiss microscope (Carl Zeiss Microscopy, Jena, Germany), and the positive cells and nuclei were counted using Axiovision 4.0 (Carl Zeiss Microscopy) and ImageJ software version 1.4.3.67 (NIH, Bethesda, MD).
Immunofluorescence
Frozen tumor samples in OCT were divided into sections (3 to 20 μm thick), mounted on Superfrost slides (ThermoFisher Scientific), and air dried for 30 minutes. Sections were fixed with cold acetone (5 minutes), followed by 1:1 acetone:chloroform (5 minutes), and then acetone (5 minutes), or by using 4% paraformaldehyde at room temperature (30 minutes), followed by 0.025% Triton X-100 (Sigma) in PBS (5 minutes) to permeabilize the cells. Sections were washed with cold PBS 3× for 5 minutes, incubated in a blocking solution of PBS/Tween (0.1%) containing 5% bovine serum albumin (Sigma) at room temperature (30 minutes), and then washed in cold PBS 3× for 5 minutes each. The appropriate primary antibodies diluted 1:200 to 1:1000 in blocking solution were incubated with the sections overnight at 4°C. Sections were then washed 3× for 5 minutes in cold PBS and incubated in blocking solution (10 minutes). Sections were then incubated with the appropriate fluorescent reporter antibodies diluted 1:500 to 1:5000 in blocking solution at room temperature (30 minutes), followed by washing 3× in PBS. Nuclei were counterstained with DAPI. Ki-67– and TUNEL-stained sections were imaged on a Zeiss microscope at ×20 magnification and quantified using ImageJ software. Double staining of CD31 with smooth muscle actin (SMA) was performed sequentially, as previously described.23 Blood vessels were imaged at ×10 magnification on a Zeiss microscope, and blood vessel analyses were conducted using IPLab software version 3.6.3 (BD Biosciences Bioimaging, Rockville, MD), as previously described.31
Statistical Analysis
A two-way repeated-measures analysis of variance was applied to assess the effects of diet and drug on tumor volume over time. The F-test was used to test changes in tumor growth for each diet and drug subgroup and to compare the slopes between groups over the time course (equivalent to area under the curve) after injection. Post hoc Bonferroni comparisons were used to test mean differences in tumor volume between the groups at specific days. A mixed-model analysis of variance analysis using a diagonal covariance structure was used to assess tumor characteristics (TUNEL, Ki-67, microvessel density, and vessel stability with SMA), and Bonferroni correction was used to adjust for multiple pairwise comparisons. Serological measurements, intratumoral cholesterol levels, and body mass were analyzed using one-way analysis of variance and Bonferroni's multiple-comparison tests or t-test. Data are reported in terms of the means ± SEM. Statistical analyses were performed using SPSS, version 19.0 (SPSS Inc./IBM, Chicago, IL), and GraphPad Prism version 5 for Windows (GraphPad Software, San Diego, CA). Two-tailed values of P < 0.05 were considered statistically significant.
Results
The design of our cholesterol-targeted experimental approach combines a diet regimen with a pharmaceutical agent, ezetimibe (Zetia; Merck, Whitehouse Station, NJ), which specifically targets cholesterol. In this protocol, we used an LFNC diet, with or without 30 mg/kg per day ezetimibe, and an HFHC diet (without sodium cholate), with or without 30 mg/kg per day ezetimibe (Figure 1 and Table 1). Because studies using statins cannot be interpreted as demonstrating a cholesterol effect because statins do not target cholesterol specifically,22,32 because cholesterol metabolism and turnover is substantially different in the mouse versus humans,32,33 and since statins cannot lower serum cholesterol levels in normal mice,34,35 we used ezetimibe to reduce serum cholesterol. Ezetimibe is a U.S. Food and Drug Administration–approved drug that blocks dietary cholesterol uptake in the gut, thereby lowering serum cholesterol levels.36–38 Ezetimibe is a specific antagonist of NPC1L1, the bona fide gut cholesterol transporter.36 Unlike statins, which do not specifically target cholesterol, ezetimibe has no known target other than NPC1L1, a transporter expressed exclusively in the jejunum and hepatocytes in humans and only in the jejunum in mice.39
Female SCID mice were fed the LFNC diet for 3 weeks and were then randomized to the four diets previously described, with calorie intake set at 21.18 calories per day in all groups. This produced four cohorts of mice with significantly distinct cholesterol levels of 89.8 ± 15.1, 95.9 ± 15.9, 109.2 ± 14.2, and 139.9 ± 28.9 mg/dL, for the LFNC + ezetimibe, the LFNC, the HFHC + ezetimibe, and the HFHC cohorts, respectively (Figure 2A). As we have described,24,26 both the diet and ezetimibe had statistically significant effects on murine serum cholesterol levels.
Figure 2.

Increased serum cholesterol levels are associated with accelerated tumor growth. A: Serum cholesterol levels. Mice were fed either an HFHC or an LFNC diet, with or without ezetimibe (Z), and bled for cholesterol determination every 3 to 4 weeks throughout the course of the study. Serum cholesterol levels just before the first measurable tumors appeared are presented as serum cholesterol (mg/dL) versus diet cohort ± SEM. All groups were statistically different from one another (F = 72.70, P < 0.0001), and this difference was maintained throughout the study. An analysis of variance was used to determine statistical significance. Values <0.01 were considered significant (n = 28 per group). B: Serum insulin levels. Serum insulin levels are plotted as insulin levels (ng/mL) ± SEM versus diet group. There was no difference in serum insulin levels between diet and ezetimibe (Z) treatment groups at sacrifice (P = 0.278, analysis of variance; n = 15 per group). C: Serum estradiol levels. Serum estradiol levels are plotted as estradiol levels (ng/mL) ± SEM versus diet group. There was no difference in serum estradiol levels between groups at sacrifice (P = 0.332, analysis of variance; n = 15 per group). D: Body mass. There was no effect of diet or drug on body weight. Average body mass is plotted as mass (g) ± SEM versus diet group. There was no difference in animal body mass at the time of tumor implantation (P = 0.135, analysis of variance), and no differences in body weight between groups were observed throughout the study, as indicated by the average body weight at tumor end point presacrifice (P = 0.206, analysis of variance; n = 27 to 30 animals per group). E–G: Tumor growth. Line graphs show mean tumor volume for each of the four groups after first appearance. E: Significant mean differences were observed between tumor growth in mice fed an HFHC compared with the LFNC diet (asterisks). The mice in the LFNC group had a reduced rate of tumor growth (slope test: F = 2.05, P = 0.04). F: Significant differences were also measured between tumor growth in mice fed an HFHC diet, with and without ezetimibe (asterisks), where the ezetimibe-treated group demonstrated a reduced rate of tumor growth (slope test: F = 6.97, P < 0.001). G: No significant differences were measured between tumor growth in the mice fed the LFNC versus LFNC + ezetimibe diets. Data are presented as mean volume (mm3) ± SEM versus days. Statistical significance was determined by analysis of variance. Two-tailed values of P < 0.05 were considered statistically significant. H: Tumor cholesterol. Cholesterol was extracted from tumor tissue and assayed (as described in Materials and Methods). Tumor cholesterol is plotted as the average cholesterol (mg) tumor tissue (g) ± SEM versus diet group. Analysis of variance indicated no significant difference in the intratumoral cholesterol levels between groups (P = 0.825) (n = 11 tumors per group).
Prior in vivo studies of dietary fat and cholesterol often control for caloric effects of high-fat diets by balancing the control diets with additional carbohydrates,40 making the animals hyperglycemic and increasing insulin levels, or in other cases, do not control for energy at all.27,41 Given that carbohydrate restriction and the resultant lower serum insulin levels can also slow tumor growth,42 most diet studies cannot separate the effects of insulin from the effects of cholesterol on tumor growth. In the present study, we observed normal serum insulin levels in mice at sacrifice and there was no effect of diet or drug treatment on insulin levels (P = 0.702, analysis of variance) (Figure 2B). Given that cholesterol is a precursor to steroid hormone synthesis, including estrogen, which has a clear role in normal mammary gland function and breast cancer, we verified that ezetimibe- and diet-induced changes in serum cholesterol levels did not alter serum estradiol levels (P = 0.442, analysis of variance) (Figure 2C). In addition, we confirmed that the isocaloric diet used in this study resulted in significantly different serum cholesterol levels without leading to increased body mass in mice fed the high-fat diet (with or without ezetimibe) (Figure 2D). Average body mass did not differ between diet and drug groups at any point in the study, as demonstrated by body weight data presented at the time of tumor implantation (P = 0.136, analysis of variance) (Figure 2D) and at the time of tumor end point and sacrifice (P = 0.206, analysis of variance) (Figure 2D). Moreover, all of the mice in the study maintained a normal body weight throughout the study.
After 4 weeks on the diets, mice were implanted with 2 × 106 MDA-MB-231 cells mixed in Matrigel into the fourth mammary fat pad of the breast (two tumors per mouse). Once tumors appeared (approximately 4 weeks; day 1) (Figure 2, E–G), they were measured every other day for 25 days, at which time the mice were sacrificed and the tumors removed for analysis. Tumors from animals fed the HFHC diet progressed fastest, whereas significantly reduced tumor growth rates were observed in the LFNC diet group and the ezetimibe-treated HFHC diet group (Figure 2, E and F). Repeated-measures analysis of variance indicated highly significant increases in tumor volume from baseline for each of the four groups (all P < 0.01). In addition, the area under the curve for the HFHC + ezetimibe diet was significantly less, reflecting a slower rate of tumor growth, than the HFHC diet (F = 6.97, P < 0.01), where mean tumor volume was significantly lower with ezetimibe on day 13 (P = 0.026), day 16 (P = 0.004), day 19 (P < 0.001), day 22 (P < 0.001), and day 25 (P < 0.01) (Figure 2F). In addition, the rate of change assessed by the slope test indicated significantly more rapid tumor growth in the HFHC group compared with the LFNC group (F = 2.05, P = 0.04), with mean tumor volume significantly higher on day 19 (P = 0.008), day 22 (P = 0.003), and day 25 (P = 0.004) (Figure 2E). There was no synergistic effect of ezetimibe treatment and LFNC diet on tumor growth (Figure 2G). Tumor take was >95% in all groups, and no significant differences in tumor take between groups were observed.
We expected that long-term maintenance on the HFHC diet might increase serum triglycerides; conversely, ezetimibe has been reported to exert a modest reduction of triglyceride levels in humans.38 However, in the present study, triglyceride levels were within the normal range for all groups, and the treatment of ezetimibe did not result in any changes in serum triglycerides. Notably, triglyceride levels were significantly lower in the HFHC cohorts (data not shown). Serological testing (aspartate aminotransferase, alkaline phosphatase, and bilirubin) indicated no liver dysfunction in any of the mice (data not shown). We expected that intratumoral cholesterol levels would reflect the different serum cholesterol levels of the diet and treatment groups; however, when assayed, no difference in intratumoral cholesterol levels was observed (P = 0.825, analysis of variance) (Figure 2H).
Given that the intratumoral cholesterol levels did not differ between diet/drug groups, we further examined whether hypercholesterolemia was inducing tumor progression, independent of diet and drug treatment. We defined hypercholesterolemia as serum cholesterol levels 2 SDs higher than the mean cholesterol level of age-matched, sex-matched mice on a normal (not high-fat) diet. In this study, the mean cholesterol value plus 2 SDs was ≥124 mg/dL. Therefore, mice having terminal serum cholesterol levels of ≥124 mg/dL were characterized as being hypercholesterolemic. Blinded to diet and drug treatment, we separated hypercholesterolemic mice from mice with normal serum cholesterol levels and compared the tumor volume between these two groups. A t-test of the means revealed that the final tumor volume of hypercholesterolemic mice was significantly greater than the tumor volume in mice with normal serum cholesterol levels, independent of treatment group (P = 0.007, t-test) (Figure 3A). The mean difference between the two subgroups was 168 mm3. The strength of this finding is represented by the 95% CI (48 to 298 mm3), indicating that animals with hypercholesterolemia are expected to have significantly larger tumor volumes in the range of 48 to 298 mm3 larger. In addition, in an analysis blinded to treatment, the Pearson correlation showed the relationship between serum cholesterol and total tumor volume is positive (Pearson r = 0.23, P = 0.018) and clearly reveals an association that animals with higher serum cholesterol have high tumor volumes.
Figure 3.

Hypercholesterolemia, independent of diet/drug, is associated with increased tumor volume. A: Bar graph shows the average final tumor volume (mm3) ± SEM versus serum cholesterol group. Hypercholesterolemic mice, blinded to drug/diet group, were defined as animals possessing serum cholesterol levels of 2 SDs higher than the mean of age-matched, sex-matched mice receiving a normal diet (a value of ≥124 mg/dL cholesterol). These hypercholesterolemic mice demonstrated a significantly greater average terminal tumor volume than animals with cholesterol levels <124 mg/dL cholesterol (751 ± 372 mm3 versus 583 ± 22 mm3; P = 0.007, t-test) (n = 25 mice in ≥124 mg/dL cholesterol group and 81 mice in <124 mg/dL cholesterol group). The Pearson correlation, blinded to treatment, between serum cholesterol and total tumor volume is positive (Pearson r = 0.23, P = 0.018) and clearly reveals an association that animals with higher serum cholesterol tend to have high tumor volumes. B: Hypercholesterolemic mice do not have high serum insulin levels. Bar graph shows the average serum insulin levels ± SEM versus cholesterol. Mice with the highest serum cholesterol levels (≥124 mg/dL), independent of treatment group, do not have increased serum insulin levels. Mean serum insulin levels did not differ between mice with hypercholesterolemia (≥124 mg/dL) and mice with normal serum cholesterol levels (<124 mg/dL) (0.67 ± 0.33 versus 0.73 ± 0.31 ng/mL; P = 0.48, t-test) (n = 15 to 43 per group). No relationship between serum cholesterol and serum insulin exists (Pearson r = −0.01, P = 0.93). C: Serum insulin levels are not associated with tumor volume. Animals with the highest serum insulin levels, defined as serum insulin levels 2 SDs above the mean, a value of ≥1.0 ng/mL, had a final average tumor volume that was the same tumor volume of animals with normal serum insulin levels of <1.0 ng/mL (P = 0.377, t-test) (n = 8 to 49 animals per group). A Pearson correlation analysis blinded to treatment group showed that no association exists between serum insulin levels and tumor volume (Pearson r = −0.140, P = 0.30).
Notably, a comparison of insulin levels observed between these two cholesterol groups (normal versus hypercholesterolemia, <124 versus ≥124 mg/dL) revealed that serum insulin levels did not differ (P = 0.477, t-test) (Figure 3B). In addition, in an analysis blinded to treatment, no relationship between serum cholesterol and serum insulin existed (Pearson r = −0.01, P = 0.93). This further demonstrates that the isocaloric diet strategy used in this study induces hypercholesterolemia that is not associated with hyperinsulinemia and does not increase serum insulin levels (Figure 3B).
Next, we asked whether insulin levels are associated with tumor volume. Independent of diet and drug treatment, we grouped the mice with the highest serum insulin levels (defined as 2 SDs higher than the mean or ≥1.0 ng/mL) from those mice with normal cholesterol levels (<1.0 ng/mL) and found that those animals possessing the highest insulin levels did not have the highest tumor volume. A comparison of the mean tumor volume of high insulin mice (those with ≥1.0 ng/mL) versus those with normal insulin levels (<1.0 ng/mL) revealed no difference (P = 0.377, t-test) (Figure 3C). A regression analysis of insulin (blinded to diet and drug treatment group) revealed that serum insulin level has no association with tumor volume (F = 0.047, P = 0.828) and, therefore, high serum insulin level itself did not predict increased tumor volume in this study (Pearson r = −0.140, P = 0.30).
Taken together, these data demonstrate that high serum cholesterol levels (independent of diet and drug treatment) are predictive of greater mean tumor volume, whereas insulin levels are not. In addition, high and low serum cholesterol groups do not differ with respect to mean insulin levels. These data demonstrate that high serum cholesterol is exerting the observed effects independent of insulin on tumor volume and supports the conclusion that hypercholesterolemia is associated with higher tumor volume.
To investigate the mechanisms underlying the tumor-promoting effect of elevated cholesterol, we analyzed the tumors using a variety of approaches. Apoptosis and cell proliferation were evaluated quantitatively in the tumors. Apoptosis, quantified by TUNEL staining, was significantly suppressed in tumors from mice in the HFHC group, which had 39% apoptotic cells compared with 49% and 52% apoptotic cells in tumors from the LFNC, with or without ezetimibe, groups and 58% apoptotic cells in the HFHC + ezetimibe group (F = 9.67, P < 0.01) (Figure 4, A and B). However, no synergy was observed between LFNC diet and ezetimibe use, and tumors from HFHC + ezetimibe treated mice had significantly more apoptotic cells than tumors from LFNC + ezetimibe treated mice (P = 0.012) (Figure 4B).
Figure 4.

Tumor apoptosis is significantly reduced in mice fed the HFHC diet. A: TUNEL staining. Top row: Representative tumor sections for each group with TUNEL staining (green). Bottom row: Merged image of tumor section with TUNEL stain (green) and nuclei counterstained with DAPI (blue). B: Quantification of tumor cell apoptosis. The level of tumor apoptosis plotted as average percentage of TUNEL-positive cells ± SEM versus diet/drug group. Tumors from animals fed an HFHC diet had less apoptotic tumor cells compared with the other diet/drug groups (F = 9.67, P < 0.001, repeated-measures analysis of variance). ∗P ≤ 0.007 (Bonferroni's multiple-comparison test), significant difference between the HFHC group and the HFHC + ezetimibe (Z), LFNC, and LFNC + Z groups; †P = 0.012 (Bonferroni's multiple-comparison test), significant difference between the HFHC + Z and LFNC + Z groups. No difference in tumor apoptosis was seen between the LFNC + Z and LFNC groups (P = 0.357) (n = 70 images per group).
Importantly, cell proliferation, as measured by Ki-67 staining, was significantly greater in the tumors grown in the HFHC-fed mice (51%) (Figure 5A), whereas a significantly decreased percentage of proliferating tumor cells was observed in the groups with lower circulating cholesterol [ie, those receiving ezetimibe (33% and 33%) and/or the LFNC diet (32.5%)] (F = 17.62, P < 0.001) (Figure 5B).
Figure 5.

Tumor cell proliferation is significantly increased in mice fed the HFHC diet. A: Ki-67 staining. Top row: Representative images of tumor sections for each group stained for Ki-67 (red), a proliferation marker. Bottom row: Images of Ki-67 staining (red) merged with nuclei counterstained with DAPI (blue). B: Quantification of tumor cell proliferation. The level of tumor cell proliferation plotted as average percentage of Ki-67–positive cells ± SEM versus diet/drug group. Tumors from the animals fed the HFHC have a significantly greater percentage of proliferating cells compared with tumors from all other groups (F = 17.62, P < 0.001, analysis of variance and Bonferroni's multiple-comparison tests, P < 0.001). The asterisk indicates significant difference compared with the HFHC group, no difference in tumor proliferation was seen between HFHC + ezetimibe (Z), LFNC, and LFNC + Z groups (Bonferroni's multiple-comparison tests, P ≥ 0.665) (n = 50 images per group).
Microvessel density (MVD) in the breast tumors was quantified using an antibody for CD31 (platelet endothelial cell adhesion molecule 1), an endothelial cell marker. MVD was significantly suppressed by both ezetimibe and by the LFNC diet when compared with the HFHC diet (F = 18.06, P < 0.001) (Figure 6, A and B). MVD was not a reflection of tumor size because tumors of similar mass were examined for each treatment group.43,44
Figure 6.

Tumor angiogenesis is significantly greater in tumors from mice fed an HFHC diet. A: CD31 staining. Representative immunofluorescent images of tumor sections from each group stained for the endothelial cell marker, CD31 (green). Images of the CD31-stained panel (green) merged with nuclei counterstained with DAPI (blue). B: Quantification of MVD. Data are plotted as relative level of CD31 staining versus diet/ezetimibe (Z) group ± SEM. Data were analyzed by mixed-model analysis of variance, which indicates a significantly greater vessel area in tumors from mice fed an HFHC diet compared with animals receiving ezetimibe (Z) and/or receiving an LFLC diet (F = 18.06, P < 0.001). ∗∗P < 0.01 (Bonferroni's multiple-comparison test), significant difference compared with HFHC group, whereas no difference in MVD was observed between LFNC + Z and HFHC + Z groups (Bonferroni's multiple comparison test, P = 0.502) (n = 80 images per group).
Given the significant effect of high cholesterol on tumor microvessel density and given our previous finding that thrombospondin (TSP-1), the endogenous, angiogenesis inhibitor, was decreased in prostate tumors from mice fed an HFHC diet,24 we asked whether TSP-1 expression might be suppressed by hypercholesterolemia in breast tumors as well. After conducting a thorough quantitative analysis of TSP-1 expression in eight tumors per group (>80 images) and after calculating the areas of positive staining and corrected total cell fluorescence, we found, interestingly, that although significant differences could be observed in TSP-1 expression between tumors in the HFHC and LFNC + Z diet groups (the groups with the lowest and highest MVD, respectively), no significant overall correlation between TSP-1 expression and tumor microvessel density was detected (data not shown).
Blood vessels undergoing rapid angiogenesis in tumors tend to exhibit poor vascular morphological characteristics, characterized by low perivascular cell coverage.45 Vessel stability was quantified in tumor sections by comparing the perivascular cell marker, SMA staining, to total vessel staining (CD31). A significant increase in perivascular cell coverage was seen in the ezetimibe-treated groups (Figure 7, A and B), suggesting a stabilizing effect of ezetimibe on tumor vascular structure. There was no synergistic effect of low-fat diet and ezetimibe on blood vessel stability (Figure 7B). Collectively, these results indicate a pronounced effect of circulating cholesterol on tumor neovascularization.
Figure 7.
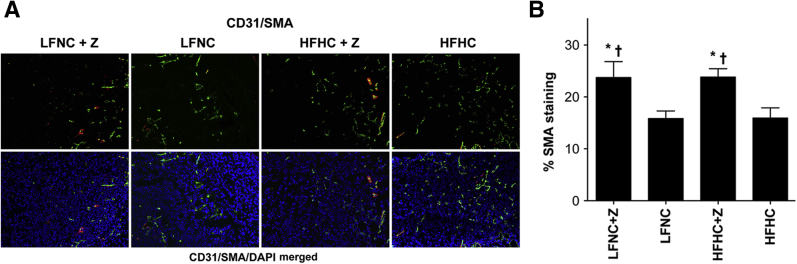
Tumors from animals treated with ezetimibe have significantly greater perivascular cell coverage. A: SMA staining. Representative immunofluorescent images of tumor sections from each group stained for the endothelial cell marker, CD31 (green), and for the perivascular cell marker SMA (red). Images of CD31 (green) and SMA (red) stained tumors merged with the nuclei counterstained with DAPI (blue). B: Quantification of perivascular cell coverage. Data are plotted as the average percentage of SMA coverage of CD31-positive microvessels ± SEM versus diet/drug group and demonstrate that ezetimibe (Z)–treated tumors had significantly greater SMA staining (greater vessel stability) compared with untreated tumors (F = 5.916, P = 0.001, analysis of variance). There was no effect of diet on vessel stability (no difference in % SMA staining), mixed-model analysis of variance, P = 0.842. ∗P ≤ 0.01 (Bonferroni's multiple-comparison test), significant difference from the HFHC group; †P ≤ 0.018 (Bonferroni's multiple-comparison test), significant difference from the LFNC group (n = 50 images per group).
Discussion
Herein, we demonstrate that a high-fat, high-cholesterol, Western diet that significantly increases circulating cholesterol levels promotes the growth of human breast tumors in an orthotopic mouse model of breast cancer. We also demonstrate that targeting cholesterol by use of a low-fat, no-cholesterol diet or use of ezetimibe, a specific hypocholesterolemic agent, reduces circulating cholesterol and results in slower tumor growth. Our conclusion that these effects can be attributed to cholesterol is supported by the following evidence: i) The fastest-growing tumors were found in the cohort with the highest serum cholesterol levels. ii) We used isocaloric diets and body mass did not differ significantly between groups, eliminating any potential effect of energy imbalance on tumor growth. iii) Cholesterol was specifically targeted and lowered with the drug ezetimibe (Zetia). iv) The diet interventions did not affect circulating estrogen or insulin levels; consequently, insulin levels were not associated with tumor volume. v) All liver function tests were within the normal range for all treatment groups. vi) The cohort of mice with the highest cholesterol levels had tumors with significantly less apoptotic cells, significantly more proliferating cells, and significantly higher levels of MVD. To our knowledge, our results are the first to conclusively implicate hypercholesterolemia in breast cancer progression.
The current study examines the effect of cholesterol on breast cancer progression in an orthotopic adult mouse model. Although several interesting reports have investigated the relationship between a Western diet and breast cancer, our report is the first, to our knowledge, to experimentally control for calorie intake, obesity, dyslipidemia, serum insulin, and triglyceride levels, and to directly implicate the effects of dietary cholesterol by pharmacologically targeting and reducing cholesterol.27 By initiating the Western diet after 8 weeks of age, after development of the mature mammary gland is complete, the role of dietary cholesterol on breast cancer can be separated from the effect of diet on mammary gland development in our study, in contrast to prior reports.27 Another recent report using orthotopic injection of mouse mammary carcinoma cells into wild-type and ApoE−/− transgenic mice examined the effect of dyslipidemia on mammary cancer growth and metastasis.28 This study used a high-fat, high-cholesterol diet that resulted in weight gain in both the wild-type mouse and the ApoE−/− mice and high serum cholesterol levels in the wild-type and severe, lethal, hypercholesterolemic (approximately 2000 mg/dL), and increased plasma triglyceride levels in the ApoE−/− mice. To unequivocally determine the specific role of cholesterol on breast cancer progression, we chose to compare an HFHC diet that raised cholesterol significantly but modestly with an LFNC diet and to specifically target cholesterol while maintaining normal mouse weight, normal serum insulin, and normal plasma triglyceride levels.
It is not unexpected that neither our current study nor those of others27 did not find increasing levels of cholesterol in the membranes of breast tumor cells as serum cholesterol levels increase. Although it has been shown that circulating lipoproteins and lipid rafts can alter growth and intracellular signaling properties of breast cells,9 there is no direct evidence that loss of normal cholesterol homeostasis occurs in the ductal epithelia of the breast. This is in contrast to our prior study of prostate cancer, where prostate tumor cholesterol levels increase with increasing serum cholesterol levels,24–26 a finding that is consistent with the literature showing that both aged normal and malignant prostate ductal epithelial cells lose their ability to regulate cholesterol levels normally.46
As mentioned previously, cholesterol homeostasis differs in mouse and humans [namely, most of the circulating cholesterol in the mouse is high-density lipoprotein (HDL) versus low-density lipoprotein (LDL) in humans]. This difference is unlikely to affect our study in such a manner as to alter our conclusions because it is not a reflection of functional species distinctions, because LDL (which transports cholesterol into cells) and HDL (which transports cholesterol away from cells) function similarly in mice and humans. Rather, this difference is simply a product of different cholesterol turnover rates in mice versus humans.33 More important with respect to our study is the fact that the major apolipoproteins of LDL (apolipoprotein B) and HDL (apolipoprotein A1) are highly conserved between mouse and human, as is the LDL receptor (76% homology on the DNA level).47 It is also widely appreciated that both human and mouse LDL receptor can bind and uptake both mouse and human LDL.48,49 In addition, we have performed experiments in which we increased murine serum cholesterol levels and achieved substantial increases in cellular cholesterol in human cells that exhibit poor cholesterol efflux, demonstrating that cross-species cholesterol transport can successfully occur in our hybrid model(s).24
Although our findings that cholesterol regulation of some of the growth properties of breast cancer are robust, there is an absence of strong, consistent epidemiological data demonstrating that cholesterol-lowering drugs reduce the incidence of breast cancer. One potential reason for this lack of conclusive epidemiological evidence is that the median age of breast cancer diagnosis is 61 years, whereas the mean age of statin-using women is >60 years.50 Therefore, far fewer women with breast cancer have used statins for >5 years compared with subjects in similar studies focusing on prostate cancer and statin use among men.51,52 Moreover, in general, fewer women are prescribed a statin than men.53 Taken together, these data suggest that the absence of robust epidemiological data demonstrating an effect of statins on breast cancer may simply be because fewer women use statins for shorter periods than men, and there are insufficient patients to derive statistically significant results.
In these studies, we demonstrate that ezetimibe blocks the accelerated tumor growth stimulated by the HFHC diet. Ezetimibe works by blocking intestinal uptake of dietary cholesterol and bile-derived cholesterol; therefore, the drug is expected to reduce circulating cholesterol levels when cholesterol is both present and absent from the diet.36–38 Unlike statins, which block cholesterol synthesis at an early step in the mevalonate pathway and, thus, suppress production of upstream intermediates (including isoprenoids), ezetimibe blocks cholesterol uptake and likely has no direct effect on other components of the pathway.
An important effect elicited by the manipulation of circulating cholesterol levels was its impact on tumor angiogenesis. Our studies revealed that reducing cholesterol levels reduces the amount of blood vessels, and that ezetimibe increases vessel perivascular cell coverage (suggesting a more stable vascular structure). These results demonstrate that a major biological effect of hypercholesterolemia on breast tumors is increased angiogenesis. Our finding that the endogenous angiogenesis inhibitor, TSP-1, although expressed at lowest levels in tumors from the HFHC diet group (where MVD was the highest), did not significantly correlate with tumor MVD (data not shown) suggests that regulation of angiogenesis in breast cancer may be more complex than that of prostate and other cancers and opens up the exciting possibility that other regulators of angiogenesis may be playing an important role. This observation, although beyond the scope of the current study, merits further investigation. Our finding that ezetimibe-treated animals had improved perivascular cell coverage of vessels highlights a novel potential role for ezetimibe in the stabilization of tumor vasculature. Vessels of untreated animals receiving the LFNC diet, with circulating cholesterol levels similar to ezetimibe-treated animals, did not exhibit the extent of perivascular cell coverage and vessel stability that was observed in either of the ezetimibe-treated groups, suggesting that this result may be independent of the cholesterol-lowering properties of ezetimibe. The potential for ezetimibe to contribute to the normalization of tumor vasculature opens the possibility of ezetimibe synergizing with the effect of chemotherapeutic and tumor-targeting agents, a phenomenon that could have important clinical implications.
One potential mechanism to explain how cholesterol may be inducing tumor progression in the absence of intratumoral accumulation of cholesterol would be through the up-regulation of cholesterol-derived steroid hormones, such as androgens within the tumor. Although we demonstrated that estrogen levels are not altered by diet or by drug treatment, examination of androgen levels and androgen receptor expression and subcellular localization within the tumor warrant further examination, especially in light of recent reports demonstrating increased androgen signaling in triple-negative breast cancers.54 Given that the MDA-MB-231 breast cancer cells used in this study are triple negative, this potential androgen-associated mechanism of cholesterol-induced tumor progression is distinct from others that have been proposed55 because the MDA-MB-231 cells are estrogen receptor negative. The enhanced tumor angiogenesis observed in mice fed an HFHC diet in the current study suggests that monocyte and macrophage recruitment into the tumor may be one plausible mechanism to explain how hypercholesterolemia is inducing tumor progression in the absence of increased intratumoral cholesterol. This is an intriguing potential mechanism, especially given that hypercholesterolemia is known to increase monocyte and macrophage adhesion to vascular endothelium56 and given that it has been shown that tumor-associated macrophages contribute to breast tumor angiogenesis and progression.57
Our results demonstrating that treatment of hypercholesterolemia with diet and/or with ezetimibe suppresses the growth of breast tumors in vivo are consistent with, and complement, the most recent prospective epidemiological studies reporting an association between statin use and decreased incidence of breast cancer recurrence in women diagnosed with stage I to III breast cancer.19–21 They also suggest the possibility that chemopreventive intervention to inhibit breast cancer progression by lowering cholesterol pharmacologically, possibly in combination with diet, may be clinically effective. This study raises the possibility that cholesterol reduction, which is routinely accomplished through pharmacological and non-pharmacological means in humans, may reduce tumor neovascularization, ultimately leading to less aggressive tumors.
Footnotes
Supported by The Breast Cancer Research Foundation, The Fortin Foundation, and NIH grant P01 CA045548 (M.A.M.).
K.P. and C.M.C. contributed equally to this work.
K.R.S. and M.A.M contributed equally to this work as senior authors.
The author C.P.S is deceased.
Disclosures: None declared.
References
- 1.Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Shimizu H., Ross R.K., Bernstein L., Yatani R., Henderson B.E., Mack T.M. Cancers of the prostate and breast among Japanese and white immigrants in Los Angeles County. Br J Cancer. 1991;63:963–966. doi: 10.1038/bjc.1991.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bingham S.A., Luben R., Welch A., Wareham N., Khaw K.T., Day N. Are imprecise methods obscuring a relation between fat and breast cancer? Lancet. 2003;362:212–214. doi: 10.1016/S0140-6736(03)13913-X. [DOI] [PubMed] [Google Scholar]
- 4.Emaus A., Veierød M.B., Tretli S., Finstad S.E., Selmer R., Furberg A.S., Bernstein L., Schlichting E., Thune I. Metabolic profile, physical activity, and mortality in breast cancer patients. Breast Cancer Res Treat. 2010;121:651–660. doi: 10.1007/s10549-009-0603-y. [DOI] [PubMed] [Google Scholar]
- 5.Owiredu W.K., Donkor S., Addai B.W., Amidu N. Serum lipid profile of breast cancer patients. Pak J Biol Sci. 2009;12:332–338. doi: 10.3923/pjbs.2009.332.338. [DOI] [PubMed] [Google Scholar]
- 6.Eliassen A.H., Colditz G.A., Rosner B., Willett W.C., Hankinson S.E. Adult weight change and risk of postmenopausal breast cancer. JAMA. 2006;296:193–201. doi: 10.1001/jama.296.2.193. [DOI] [PubMed] [Google Scholar]
- 7.Simons K., Ikonen E. How cells handle cholesterol. Science. 2000;290:1721–1726. doi: 10.1126/science.290.5497.1721. [DOI] [PubMed] [Google Scholar]
- 8.Soccio R.E., Breslow J.L. Intracellular cholesterol transport. Arterioscler Thromb Vasc Biol. 2004;24:1150–1160. doi: 10.1161/01.ATV.0000131264.66417.d5. [DOI] [PubMed] [Google Scholar]
- 9.Danilo C., Frank P.G. Cholesterol and breast cancer development. Curr Opin Pharmacol. 2012;12:677–682. doi: 10.1016/j.coph.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 10.Kaye J.A., Meier C.R., Walker A.M., Jick H. Statin use, hyperlipidaemia, and the risk of breast cancer. Br J Cancer. 2002;86:1436–1439. doi: 10.1038/sj.bjc.6600267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desager J.P., Horsmans Y. Clinical pharmacokinetics of 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. Clin Pharmacokinet. 1996;31:348–371. doi: 10.2165/00003088-199631050-00003. [DOI] [PubMed] [Google Scholar]
- 12.Boudreau D.M., Gardner J.S., Malone K.E., Heckbert S.R., Blough D.K., Daling J.R. The association between 3-hydroxy-3-methylglutaryl coenzyme A inhibitor use and breast carcinoma risk among postmenopausal women: a case-control study. Cancer. 2004;100:2308–2316. doi: 10.1002/cncr.20271. [DOI] [PubMed] [Google Scholar]
- 13.Cauley J.A., McTiernan A., Rodabough R.J., LaCroix A., Bauer D.C., Margolis K.L., Paskett E.D., Vitolins M.Z., Furberg C.D., Chlebowski R.T., Women's Health Initiative Research Group Statin use and breast cancer: prospective results from the Women's Health Initiative. J Natl Cancer Inst. 2006;98:700–707. doi: 10.1093/jnci/djj188. [DOI] [PubMed] [Google Scholar]
- 14.Fagherazzi G., Fabre A., Boutron-Ruault M.C., Clavel-Chapelon F. Serum cholesterol level, use of a cholesterol-lowering drug, and breast cancer: results from the prospective E3N cohort. Eur J Cancer Prev. 2010;19:120–125. doi: 10.1097/CEJ.0b013e3283354918. [DOI] [PubMed] [Google Scholar]
- 15.Cauley J.A., Zmuda J.M., Lui L.Y., Hillier T.A., Ness R.B., Stone K.L., Cummings S.R., Bauer D.C. Lipid-lowering drug use and breast cancer in older women: a prospective study. J Womens Health (Larchmt) 2003;12:749–756. doi: 10.1089/154099903322447710. [DOI] [PubMed] [Google Scholar]
- 16.Kumar A.S., Benz C.C., Shim V., Minami C.A., Moore D.H., Esserman L.J. Estrogen receptor-negative breast cancer is less likely to arise among lipophilic statin users. Cancer Epidemiol Biomarkers Prev. 2008;17:1028–1033. doi: 10.1158/1055-9965.EPI-07-0726. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs E.J., Newton C.C., Thun M.J., Gapstur S.M. Long-term use of cholesterol-lowering drugs and cancer incidence in a large United States cohort. Cancer Res. 2011;71:1763–1771. doi: 10.1158/0008-5472.CAN-10-2953. [DOI] [PubMed] [Google Scholar]
- 18.Kitahara C.M., Berrington de González A., Freedman N.D., Huxley R., Mok Y., Jee S.H., Samet J.M. Total cholesterol and cancer risk in a large prospective study in Korea. J Clin Oncol. 2011;29:1592–1598. doi: 10.1200/JCO.2010.31.5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahern T.P., Pedersen L., Tarp M., Cronin-Fenton D.P., Garne J.P., Silliman R.A., Sørensen H.T., Lash T.L. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J Natl Cancer Inst. 2011;103:1461–1468. doi: 10.1093/jnci/djr291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chae Y.K., Valsecchi M.E., Kim J., Bianchi A.L., Khemasuwan D., Desai A., Tester W. Reduced risk of breast cancer recurrence in patients using ACE inhibitors, ARBs, and/or statins. Cancer Invest. 2011;29:585–593. doi: 10.3109/07357907.2011.616252. [DOI] [PubMed] [Google Scholar]
- 21.Kwan M.L., Habel L.A., Flick E.D., Quesenberry C.P., Caan B. Post-diagnosis statin use and breast cancer recurrence in a prospective cohort study of early stage breast cancer survivors. Breast Cancer Res Treat. 2008;109:573–579. doi: 10.1007/s10549-007-9683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solomon K.R., Freeman M.R. Do the cholesterol-lowering properties of statins affect cancer risk? Trends Endocrinol Metab. 2008;19:113–121. doi: 10.1016/j.tem.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 23.Solomon K.R., Pelton K., Boucher K., Joo J., Tully C., Zurakowski D., Schaffner C.P., Kim J., Freeman M.R. Ezetimibe is an inhibitor of tumor angiogenesis. Am J Pathol. 2009;174:1017–1026. doi: 10.2353/ajpath.2009.080551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhuang L., Kim J., Adam R.M., Solomon K.R., Freeman M.R. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115:959–968. doi: 10.1172/JCI200519935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mostaghel E.A., Solomon K.R., Pelton K., Freeman M.R., Montgomery R.B. Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors. PLoS One. 2012;7:e30062. doi: 10.1371/journal.pone.0030062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Llaverias G., Danilo C., Mercier I., Daumer K., Capozza F., Williams T.M., Sotgia F., Lisanti M.P., Frank P.G. Role of cholesterol in the development and progression of breast cancer. Am J Pathol. 2011;178:402–412. doi: 10.1016/j.ajpath.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alikhani N., Ferguson R.D., Novosyadlyy R., Gallagher E.J., Scheinman E.J., Yakar S., LeRoith D. Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene. 2013;32:961–967. doi: 10.1038/onc.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang J., Bielenberg D.R., Rodig S.J., Doiron R., Clifton M.C., Kung A.L., Strong R.K., Zurakowski D., Moses M.A. Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci U S A. 2009;106:3913–3918. doi: 10.1073/pnas.0810617106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roy R., Rodig S., Bielenberg D., Zurakowski D., Moses M.A. ADAM12 transmembrane and secreted isoforms promote breast tumor growth: a distinct role for ADAM12-S protein in tumor metastasis. J Biol Chem. 2011;286:20758–20768. doi: 10.1074/jbc.M110.216036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boucher K., Siegel C.S., Sharma P., Hauschka P.V., Solomon K.R. HMG-CoA reductase inhibitors induce apoptosis in pericytes. Microvasc Res. 2006;71:91–102. doi: 10.1016/j.mvr.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 31.Bielenberg D.R., Hida Y., Shimizu A., Kaipainen A., Kreuter M., Kim C.C., Klagsbrun M. Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J Clin Invest. 2004;114:1260–1271. doi: 10.1172/JCI21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Endo A., Tsujita Y., Kuroda M., Tanzawa K. Effects of ML-236B on cholesterol metabolism in mice and rats: lack of hypocholesterolemic activity in normal animals. Biochim Biophys Acta. 1979;575:266–276. [PubMed] [Google Scholar]
- 33.Dietschy J.M., Turley S.D. Control of cholesterol turnover in the mouse. J Biol Chem. 2002;277:3801–3804. doi: 10.1074/jbc.R100057200. [DOI] [PubMed] [Google Scholar]
- 34.Endo A. Chemistry, biochemistry, and pharmacology of HMG-CoA reductase inhibitors. Klin Wochenschr. 1988;66:421–427. doi: 10.1007/BF01745510. [DOI] [PubMed] [Google Scholar]
- 35.Tang W., Ma Y., Yu L. Plasma cholesterol is hyperresponsive to statin in ABCG5/ABCG8 transgenic mice. Hepatology. 2006;44:1259–1266. doi: 10.1002/hep.21380. [DOI] [PubMed] [Google Scholar]
- 36.Altmann S.W., Davis H.R., Jr., Zhu L.J., Yao X., Hoos L.M., Tetzloff G., Iyer S.P., Maguire M., Golovko A., Zeng M., Wang L., Murgolo N., Graziano M.P. Niemann-Pick C1 like 1 protein is critical for intestinal cholesterol absorption. Science. 2004;303:1201–1204. doi: 10.1126/science.1093131. [DOI] [PubMed] [Google Scholar]
- 37.Davis H.R., Jr., Zhu L.J., Hoos L.M., Tetzloff G., Maguire M., Liu J., Yao X., Iyer S.P., Lam M.H., Lund E.G., Detmers P.A., Graziano M.P., Altmann S.W. Niemann-Pick C1 like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J Biol Chem. 2004;279:33586–33592. doi: 10.1074/jbc.M405817200. [DOI] [PubMed] [Google Scholar]
- 38.Knopp R.H., Gitter H., Truitt T., Bays H., Manion C.V., Lipka L.J., LeBeaut A.P., Suresh R., Yang B., Veltri E.P., Ezetimibe Study Group Effects of ezetimibe, a new cholesterol absorption inhibitor, on plasma lipids in patients with primary hypercholesterolemia. Eur Heart J. 2003;24:729–741. doi: 10.1016/s0195-668x(02)00807-2. [DOI] [PubMed] [Google Scholar]
- 39.Jia L., Betters J.L., Yu L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 2011;73:239–259. doi: 10.1146/annurev-physiol-012110-142233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lichtman A.H., Clinton S.K., Iiyama K., Connelly P.W., Libby P., Cybulsky M.I. Hyperlipidemia and atherosclerotic lesion development in LDL receptor-deficient mice fed defined semipurified diets with and without cholate. Arterioscler Thromb Vasc Biol. 1999;19:1938–1944. doi: 10.1161/01.atv.19.8.1938. [DOI] [PubMed] [Google Scholar]
- 41.Parhami F., Tintut Y., Beamer W.G., Gharavi N., Goodman W., Demer L.L. Atherogenic high-fat diet reduces bone mineralization in mice. J Bone Miner Res. 2001;16:182–188. doi: 10.1359/jbmr.2001.16.1.182. [DOI] [PubMed] [Google Scholar]
- 42.Freedland S.J., Mavropoulos J., Wang A., Darshan M., Demark-Wahnefried W., Aronson W.J., Cohen P., Hwang D., Peterson B., Fields T., Pizzo S.V., Isaacs W.B. Carbohydrate restriction, prostate cancer growth, and the insulin-like growth factor axis. Prostate. 2008;68:11–19. doi: 10.1002/pros.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez C.A., Yan L., Louis G., Yang J., Kutok J.L., Moses M.A. The matrix metalloproteinase-9/neutrophil gelatinase-associated lipocalin complex plays a role in breast tumor growth and is present in the urine of breast cancer patients. Clin Cancer Res. 2005;11:5390–5395. doi: 10.1158/1078-0432.CCR-04-2391. [DOI] [PubMed] [Google Scholar]
- 44.Fernandez C.A., Roy R., Lee S., Yang J., Panigrahy D., Van Vliet K.J., Moses M.A. The anti-angiogenic peptide, loop 6, binds insulin-like growth factor-1 receptor. J Biol Chem. 2010;285:41886–41895. doi: 10.1074/jbc.M110.166439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bergers G., Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–464. doi: 10.1215/S1152851705000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaffner C.P. Prostatic cholesterol metabolism: regulation and alteration. Prog Clin Biol Res. 1981;75A:279–324. [PubMed] [Google Scholar]
- 47.Hoffer M.J., van Eck M.M., Petrij F., van der Zee A., de Wit E., Meijer D., Grosveld G., Havekes L.M., Hofker M.H., Frants R.R. The mouse low density lipoprotein receptor gene: cDNA sequence and exon-intron structure. Biochem Biophys Res Commun. 1993;191:880–886. doi: 10.1006/bbrc.1993.1299. [DOI] [PubMed] [Google Scholar]
- 48.Masquelier M., Vitols S., Peterson C. Low-density lipoprotein as a carrier of antitumoral drugs: in vivo fate of drug-human low-density lipoprotein complexes in mice. Cancer Res. 1986;46:3842–3847. [PubMed] [Google Scholar]
- 49.Young S.G., Witztum J.L., Casal D.C., Curtiss L.K., Bernstein S. Conservation of the low density lipoprotein receptor-binding domain of apoprotein B: demonstration by a new monoclonal antibody, MB47. Arteriosclerosis. 1986;6:178–188. doi: 10.1161/01.atv.6.2.178. [DOI] [PubMed] [Google Scholar]
- 50.Walsh J.M., Pignone M. Drug treatment of hyperlipidemia in women. JAMA. 2004;291:2243–2252. doi: 10.1001/jama.291.18.2243. [DOI] [PubMed] [Google Scholar]
- 51.Flick E.D., Habel L.A., Chan K.A., Van Den Eeden S.K., Quinn V.P., Haque R., Orav E.J., Seeger J.D., Sadler M.C., Quesenberry C.P., Jr., Sternfeld B., Jacobsen S.J., Whitmer R.A., Caan B.J. Statin use and risk of prostate cancer in the California Men's Health Study Cohort. Cancer Epidemiol Biomarkers Prev. 2007;16:2218–2225. doi: 10.1158/1055-9965.EPI-07-0197. [DOI] [PubMed] [Google Scholar]
- 52.Platz E.A., Leitzmann M.F., Visvanathan K., Rimm E.B., Stampfer M.J., Willett W.C., Giovannucci E. Statin drugs and risk of advanced prostate cancer. J Natl Cancer Inst. 2006;98:1819–1825. doi: 10.1093/jnci/djj499. [DOI] [PubMed] [Google Scholar]
- 53.US Department of Health and Human Services. Health, United States, 2010: with special feature on death and dying. Hyattsville (MD): National Center for Health Statistics (US). DHHS Pub No. 2011-1232. Available from: US Government Printing Office, Washington, DC; 76-641496 [PubMed]
- 54.McGhan L.J., McCullough A.E., Protheroe C.A., Dueck A.C., Lee J.J., Nunez-Nateras R., Castle E.P., Gray R.J., Wasif N., Goetz M.P., Hawse J.R., Henry T.J., Barrett M.T., Cunliffe H.E., Pockaj B.A. Androgen receptor-positive triple negative breast cancer: a unique breast cancer subtype. Ann Surg Oncol. 2014;21:361–367. doi: 10.1245/s10434-013-3260-7. [DOI] [PubMed] [Google Scholar]
- 55.Nelson E.R., Wardell S.E., Jasper J.S., Park S., Suchindran S., Howe M.K., Carver N.J., Pillai R.V., Sullivan P.M., Sondhi V., Umetani M., Geradts J., McDonnell D.P. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094–1098. doi: 10.1126/science.1241908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu H., Gower R.M., Wang H., Perrard X.Y., Ma R., Bullard D.C., Burns A.R., Paul A., Smith C.W., Simon S.I., Ballantyne C.M. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119:2708–2717. doi: 10.1161/CIRCULATIONAHA.108.823740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leek R.D., Lewis C.E., Whitehouse R., Greenall M., Clarke J., Harris A.L. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56:4625–4629. [PubMed] [Google Scholar]