

Abstract
Hepatitis viral infection is a leading cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC). Over one million people are estimated to be persistently infected with hepatitis C virus (HCV) worldwide. As capsid core protein is the key element in spreading HCV; hence, it is considered to be the superlative target of antiviral compounds. Novel drug inhibitors of HCV are in need to complement or replace the current treatments such as pegylated interferon’s and ribavirin as they are partially booming and beset with various side effects. Our study was conducted to predict 3D structure of capsid core protein of HCV from northern part of India. Core, the capsid protein of HCV, handles the assembly and packaging of HCV RNA genome and is the least variable of all the ten HCV proteins among the six HCV genotypes. Therefore, we screened four phytochemicals inhibitors that are known to disrupt the interactions of core and other HCV proteins such as (a) epigallocatechin gallate (EGCG), (b) ladanein, (c) naringenin, and (d) silybin extracted from medicinal plants; targeted against active site of residues of HCV-genotype 3 (G3) (Q68867) and its subtypes 3b (Q68861) and 3g (Q68865) from north India. To study the inhibitory activity of the recruited flavonoids, we conducted a quantitative structure–activity relationship (QSAR). Furthermore, docking interaction suggests that EGCG showed a maximum number of hydrogen bond (H-bond) interactions with all the three modeled capsid proteins with high interaction energy followed by naringenin and silybin. Thus, our results strongly correlate the inhibitory activity of the selected bioflavonoid. Finally, the dynamic predicted capsid protein molecule of HCV virion provides a general avenue to target structure-based antiviral compounds that support the hypothesis that the screened inhibitors for viral capsid might constitute new class of potent agents but further confirmation is necessary using in vitro and in vivo studies.
Keywords: hepatitis C virus, hepatocellular carcinoma, capsid protein, docking, inhibitors
Introduction
About 750,000 people worldwide are diagnosed with liver cancer.1 It has been reported that number of people suffering from this disease are strongly increasing in Europe followed by Japan and USA.2 Number of people chronically affected with hepatitis C virus (HCV) and its persistence are linked with different liver disorders such as cirrhosis, steatosis, and finally hepatocellular carcinoma (HCC).3 HCV is a positive strand RNA virus which encodes a polyprotein composing of 3000 amino acids (AAs). This polyprotein is cleaved and translated by multiple viral proteases to form mature protein components. N-terminal portion of polyprotein contains structural proteins including core protein and glycoproteins: E1 and E2. The C-terminal portion of polyprotein encompasses non-structural viral proteins such as NS3, necessary for RNA replication; NS4 A, involved in helicase and protease activity; NS4B, cofactor of NS3 protease; NS5 A, polytopic membrane protein; and NS5B, required for RNA-dependent RNA polymerase.4 The capsid protein of HCV is a valuable target for drug development. Since the core is a key component that assembles and packages HCV RNA genome through high-order oligomers linked with lipid droplets and endoplasmic reticulum with various other proteins and forms essential particles for viral assembly makeup.5 Core is known for its least variable component of all other HCV proteins and is well conserved among six HCV genotypes observed worldwide.6 This capsid core protein plays a major role in cell proliferation by inducing virus transformation through controlling RNA binding activity and its capability to form proteins homo-multimers.7,8 Therefore, inhibitors of viral capsid assembly interfere with uncoating viral elements upon infection and formation of new particles resulting in destabilization of the assembled HCV virion.9 Chronic HCV is eradicated from infected patients through antiviral treatments. However, combined therapy of pegylated interferon alpha (IFN-α) as well as ribavirin10 is not only expensive but also have side effects.11 Currently, it is believed that an effective anti-HCV therapy can be received by direct acting antivirals (DAAs).12 Combination of DAAs without interferon might reduce the side effects of currently used therapy.13 Therefore, effective and cheap therapy to cure HCV infected population in underdeveloped and developing countries is necessary to prevent chronic virus transmission. Since centuries, medicinal plants have been known and used for treating diverse viral diseases. Many modern drugs are also developed from molecules of plant origin.14 To search new compounds from various plants against infectious diseases, metabolic disorders, and immunosuppression, oncology is still required at a large scale.15 Dozens of natural compounds indicate an antiviral activity toward various kinds of viruses. Most promising natural molecules are screened to inhibit HCV polyprotein containing core protein of viral capsid.13
This work reports sequence alignment and phylogenetic study of various genotypes followed by complex based pharmacophore modeling of HCV-G3 and its subtype 3b and 3g core proteins from north India to find out the important pharmacophoric active sites essential for inhibition of this viral core protein activity by screening drugs, predicting drug-likeness, computational docking, protein ligands binding, H-bond interactions, and binding energy calculations. Finally, identification of best fit binding flavonoid target is sufficient to enable active inhibition against core protein, which might contribute in modeling the appropriate drug therapy for future studies.
Methodology
Protein sequence alignment
Nucleotide sequence of HCV-G3 and its subtypes 3b and 3g from North India was retrieved from UniProtKB/Swissprot with accession numbers: Q68867, Q68861, and Q68865 respectively.16 The retrieved FASTA sequences for the target three capsid cores were submitted to protein basic local alignment search tool (pBLAST) to determine the template protein. Unique identifiers between template sequence and target sequence were compared to determine homology within the sequence by using multiple sequence alignment available in Command Line Calculator (CLC) drug discovery workbench.17 Parameters set in creating a sequence alignment in CLC are as follows: gap open cost, 10; gap extension cost, 1.0; and very accurate progressive alignment algorithm is used.
Phylogenetic alignment
Robust phylogenetic tree was analyzed through the software Phylogeny.fr (Phylogeny.fr.com).18 The proteome tree was processed through default parameters including both multiple alignment: MUSCLE and alignment curation: G-blocks.19 Curation parameters used for minimum number of sequences for conserved position, 19; minimum number of sequences for flanking position, 31; and maximum number of contiguous non-conserved positions, 8. Construction and visualization of phylogenetic tree was done by using Phylogenetic estimation using Maximum Likelihood (PhyML) and TreeDyn. PhyML was used for statistical tests for tree constructions and branch support was determined by approximate Likelihood-Ratio Test (aLRT), SH-Like.20–23
Homology modeling
Protein structures for the three strains are not available; therefore comparative modeling was done to resolve the 3D protein structure of capsid core protein for the northern HCV strains. Template sequence 1XCQ and 1N64 was chosen along with multiple templates to predict the 3D protein models by using homology modeling software called Modeller v9.12, a python based protein modeling tool that constructs 3D model by satisfying spatial constraints as well as performs additional tasks including de novo modeling of protein loops in the predicted 3D structures and optimizing various models with respect to flexibility, multiple sequence, structure alignment, and clustering.
Ligand generation
Ligands selected for protein interactions were based on the natural plant ingredients screened for anti-HCV activity. Medicinal plants derived phytochemicals with their antiviral activity against HCV core protein are summarized in Table 1. Structure of compounds were drawn in Advanced Chemistry Development’s (ACD) Chemsketch, converted into 3D (Program Database file – PDB) structure by using Online Smiles Translator and employed for docking analysis.
Table 1.
Medicinal plants derived phytochemicals with their antiviral activity against HCV core protein.
| PHYTOCHEMICALS | PROPERTIES | CHEMICAL STRUCTURE | FUNCTIONS | REFERENCES |
|---|---|---|---|---|
| EGCG | Molecular Weight: 458.37172 [g/mol] Molecular Formula: C22H18O11 XLogP3: 1.2 H-Bond Donor: 8 H-Bond Acceptor: 11 |
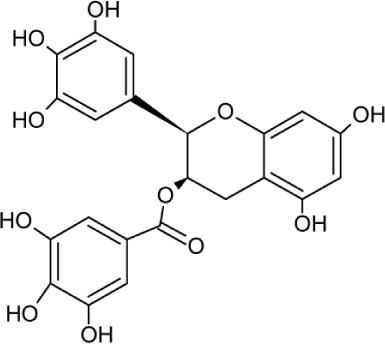
|
Glycoprotein attachment and replication | 30–32 |
| Ladanein | Molecular Weight: 314.28946 [g/mol] Molecular Formula: C17H14O6 XLogP3: 2 H-Bond donor: 2 H-Bond Acceptor: 6 |
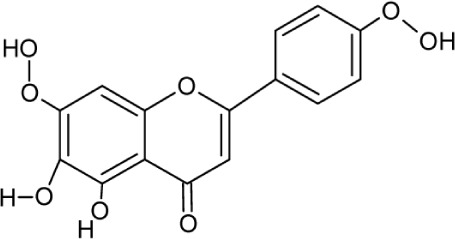
|
HCV entry | 33 |
| Naringenin | Molecular Weight: 272.25278 [g/mol] Molecular Formula: C15H12O5 XLogP3-AA: 2.4 H-Bond donor: 3 H-Bond Acceptor: 5 |
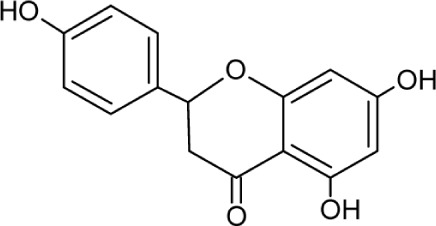
|
Assembly and secretion from core and HCV RNA | 34,35 |
| Silybin | Molecular Weight: 482.43618 [g/mol] Molecular Formula: C25H22O10 XLogP3-AA: 2.4 H-Bond Donor: 5 H-Bond Acceptor: 10 |
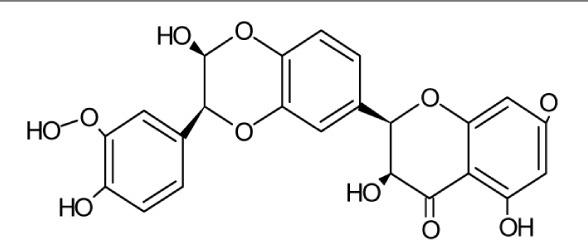
|
Entry, replication, Cell to cell spread of secreted viral proteins | 27–29,36 |
Quantitative structure–activity relationship (QSAR)
Virtual models for property evaluation of chemicals within global architecture-QSAR (VEGA-QSAR) program analyzed the selected ligands to determine the relationship of physiochemical properties and biological activities of descriptor molecules in various classified QSAR models. Toxicity, ecotoxicity, and physiochemical predicted properties of ligands such as log P (version 1.1.2), bioconcentration factor (BCF) (CAESAR-version 2.1.13), carcinogenicity model (CAESAR 2.1.8), mutagenicity model (CAESAR version 2.1.12), skin sensitization model (CAESAR-version 2.1.5), developmental toxicity model (CAESAR-version 2.1.6), fathead minnow LC50 96 hour (lethal concentration to 50% of the test animals) (Environmental Protection Agency (EPA)-version1.0.6), daphnia magna LC50 48 hour (EPA-version 1.0.6), fish LC50 classification (version 1.0.1), BCF read across (version−1.0.2), and ready biodegradability model (version 1.0.8) were determined.24,25 VEGA-QSAR models were initially derived from CAESAR models and other models were added to stimulate the models available, one such model is EPA (US Environmental Protection Agency). The used input formats were SMILES and SDF files.26
Protein and ligand optimization
Protein and ligand optimization was performed by molecular simulation software called Chemistry at Harvard Macromolecular Mechanics (CHARMm) force field using CHARMMing online tool. Potential energy, electrostatic energy, van der Waals energy, and root mean square deviation (RMSD) gradient were checked for the core protein before and after minimization. Bond energy, dihedral energy, electrostatic energy, initial RMSD, and potential energy for the ligands were also calculated.
Validation of 3D model
The 3D structure of capsid protein from HCV-G3 and its subtypes 3b and 3g was validated and evaluated by Ramachandran plot using structural analysis and verification server (SAVS) PROCHECK version 4, which determines the stereo-chemical quality of modeled protein structure by analyzing residue–residue geometry as well as overall structural geometry. The evaluated Ramachandran plot for the modeled proteins were also compared to the template proteins (1XCQ and 1 N64) to check the distribution of residues in favored and disallowed regions between each target and template models.
Docking studies
Molegro virtual docker (MVD) was used to study docking interaction; we used protocol for template docking available in MVD and determined MolDock; docking score, rerank score, and interaction energy scores between protein and ligand from MolDock generic region for information display (GRID) options. Template docking was based on chemical properties such as pharmacophore elements of target ligand bound in the active sites of molecule and the resulted information was used for docking analysis. Default parameter settings such as grid resolution of 0.30 Å for grid generation and a radius of 15 Å from the template molecule as the binding site were used. Setting such as MolDock optimizer was used to search algorithm; number of runs, 10; population size, 50; maximum iteration, 2000; scaling factor, 0.50; cross over rate, 90; and variation-based termination setup was used for parameter setting. Maximum number of poses to generate best model was increased to 20 from a default of 5, MVD works by following evolutionary algorithm, and therefore consecutive docking runs do not give the same fitness scores. In our experiment, we addressed this issue of inherent randomness by using ten consecutive runs and then the top ranked score for each run was used to calculate the average score for each of the targeted ligand; so as to correlate with the experimental binding values.
Results
BLAST alignment
We searched for the reference sequence for the three selected HCV capsid by expanding the pBLAST search. Only two types of sequences exhibited maximum homology to the target FASTA sequence, presented in Figure 1. One of them is 1XCQ, which is HCV core FAb Protein L mutant (isolate HC-J8, UniProtKB: P26661) with X-ray diffracted at 3.50 Å, and the other one is 1 N64, which is a crystal structure of immunodominant antigenic site on HCV protein bound to mAB (isolate Taiwan, Uniprot KB: P29846), X-ray diffracted at 2.34 Å. The former protein showed above 90% identity to the target sequence with only 27% query coverage whereas the latter showed up to 95% identity but only 10% query coverage. Figure 1 depicts the pBLAST search for each HCV subtypes from north India.
Figure 1.

Expanded BLAST search conducted for three HCV subtypes from north India. Maximum homology is denoted by linear line (green color) whereas poor identity with coverage is denoted in fragmented line (black color). (A) Protein BLAST of HCV-G3 (Q688867), (B) subtype 3b alignment (Q688861), and (C) subtype 3g alignment (Q688865).
Conserved sequence analysis
In order to understand the level of unique identifiers of conservation, we aligned the three target capsid proteins with the template proteins using CLC drug discovery workbench (Fig. 2). Sequence homology for 1XCQ showed only 36 residues unique when compared to HCV proteins whereas 1N64 denoted 16 residues homology to 1XCQ and the targeted HCV subtypes. This multiple sequence alignment showed a very few conservation with the target sequence modeled.
Figure 2.

Comparison of protein sequences and its conserved residues. Unique identifiers between target and template sequence were analyzed. Each AA is identified by specific color and consensus conserved residues are labeled in capital letters. 1XCQ and 1N64 shared only 15% and 35% homology with the target sequence, respectively.
Phylogenetic relationship
Template proteins 1XCQ and 1N64 showed similar homology about 100% identity among all the HCV genotypes with query coverage of 23% (1XCQ) and 8% (1N64) except for HCV isolate 6a (O39927), 1c (Q913D4), and 3b (Q81487) shown in Figure 3. HCV-6a represented only 93% identity with a query cover of 23% (1XCQ) and 94% identity with 8% query cover (1N64). Similarly, HCV-1c isolate showed 93% identity with 23% total query cover for 1XCQ followed by 8% query coverage and 94% identity for 1N64. Genotype-3b (Q81487) aligned 77% with 23% query coverage in both templates. AA substitution rate per site in core capsid protein of HCV was found to be 0.01%.
Figure 3.

Phylogram chart of capsid core proteins in HCV genotypes. Tree represents strong correlation among both template and subtypes of different HCV genotypes capsid protein sequence as their branch length leading to these nodes is very close. The unit change in AA sequence among the species is 0.1%.
QSAR study
VEGA-QSAR method was applied to predict the diverse biochemical properties of ligand molecules. Results obtained by the computerized QSAR models are potentially effective in evaluating the chemical properties of the selected compounds by reducing the number of tests on animals. Different models were tested for each flavonoid descriptors as indicated in Table 2. The selected flavonoids showed positive prediction being non-mutagenic and non-carcinogenic. Fathead minnow LC50 was predicted less than 6.5 [−log (mol/L)] for all the compounds. Toxicity of the phytochemicals was determined lesser than one; except ladanein, all three ligands are non-ready biodegradable. Skin sensation model predicted sensitizes for ladanein and naringenin where fish LC50 classification predicted toxi-3 level in all the compounds ranging from 10 to 100 mg/L. The log P value is a useful parameter for understanding the behavior of drug molecules; the log P value is higher in ladanein (2.71 log units) and naringenin (2.52 log units) followed by epigallocatechin gallate (EGCG) (1.71 log units). Only silybin showed lesser log P value of 1.42 log units. It can be concluded from that the selected flavonoid can be interpreted reasonably and its inhibition ability is beneficial.
Table 2.
Classification of models and predicted values for various structure activity relationships.
| QSAR MODELS | PREDICTION AND APPLICABILITY DOMAIN ANALYSIS FOR MODELS | |||
|---|---|---|---|---|
| EGCG | LADANEIN | NARINGENIN | SILYBIN | |
| Fathead minnow LC50 (96 hour) [−log(mol/L)] | 6.42 | 4.95 | 4.58 | 5.7 |
| Bioaccumulation factor Log10-(Log10(mol/L) | 0.60 | 1.41 | 1.00 | 0.36 |
| Developmental toxicity value | 0.74 | 0.90 | 0.69 | 0.69 |
| Daphnia magna LC50 (48 hour) [−log(mol/L)] | 1.76 | 3.25 | 2.61 | 3.66 |
| Mutagenicity model (CAESAR) | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen |
| Mutagenicity sarPy model | Non-Mutagen | Non-Mutagen | Non-Mutagen | Non-Mutagen |
| Carcinogenicity model | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen | Non-Carcinogen |
| BCF model (logBCF) [log(L/kg)] | −0.01 | 0.53 | 0.53 | −0.15 |
| Ready biodegradability model | Non ready biodegradable | Ready biodegradable | Non ready biodegradable | Non ready biodegradable |
| Log P prediction [log units] | 1.71 | 2.71 | 2.52 | 1.42 |
| Skin sensitization model (CAESAR) | Non-sensitizer | Sensitizer | Sensitizer | Non-sensitizer |
| BCF read- across [log(L/kg)] | 0.97 | 2.25 | 2.18 | 1.3 |
| Fish LC50 classification | Toxic-3 (between 10 and 100 mg/l) | Toxic-3 (between 10 and 100 mg/l) | Toxic-3 (between 10 and 100 mg/l) | Toxic-3 (between 10 and 100 mg/l) |
Comparison of Ramachandran plot
The stereochemical quality of the template and the proposed homology model was evaluated using PROCHECK tool. Ramachandran plot for template 1XCQ exhibited only 64.3% AA residues in highly favorable regions, 28.3% and 7.1% in allowed and disallowed regions, respectively (Table 3). 1 N64 revealed higher percentage in favored region (87.1% and 12.6% in allowed region). Both the chosen templates indicated 0% in disallowed region. Therefore, three modeled capsid proteins exhibit good reliability of the structure predicted in Figure 4.
Table 3.
Comparison of Ramachandran plot between template and target sequence.
| PROTEIN MODELS | FAVORED REGIONS | ALLOWED REGION | GENEROUSLY ALLOWED REGION | DISALLOWED REGION | TOTAL NUMBER OF RESIDUES |
|---|---|---|---|---|---|
| 1XCQ | 64.3% | 28.3% | 7.1% | 0.0% | 474 |
| 1N64 | 87.1% | 12.6% | 0.0% | 0.3% | 454 |
| HCV-G3 model | 81.1% | 14.2% | 2.8% | 1.9% | 150 |
| subtype 3b model | 83.2% | 8.4% | 3.7% | 4.7% | 150 |
| subtype 3g model | 77.4% | 14.2% | 4.7% | 3.8% | 150 |
Figure 4.

Comparison of Ramachandran plots between template and target proteins. (A) Plot for HCV G3, (B) model 3b, (C) model 3g, (D) template-1XCQ, and (E) template-1N64. Both the chosen templates indicated 0% in disallowed region. The three models exhibit good reliability of the predicted structure.
Site of binding of flavonoid inhibitors on core proteins
Interaction of EGCG as best fit ligand in the binding pockets of modeled capsid protein is shown in Figure 5. EGCG exhibited ten, nine, and eleven H-bond interactions within the active sites of HCV-G3, subtypes 3b and 3g. In HCV-G3, EGCG formed H-bonds with ARG101, ARF61, ARG112, and LYS118 and two H-bonds individually with SER103, GLU69, and ASP108 with a highest interaction energy (−153.142 kcal/mole). Naringenin also showed higher interaction energy (−129.636 kcal/mole), formed five H-bonds compared to ladanein and silybin. Similarly, in subtype 3b, EGCG spatially fits into the active site of the macromolecule with highest energy (−178.028 kcal/mole) and formed H-bond with active sites of LEU95, TRP93, ALA91, ASN18, VAL19, GLY87, and GLY25 whereas ladanein shows interaction of high energy compared to naringenin (−145.682 kcal/mole) and silybin (−134.336 kcal/mole). EGCG has remained best ranked in both HCV-G3 and subtype 3b and found to be a potential ligand upon all the surrounding interacting AAs particularly in subtype 3g and formed H-bonds with ARG6, ASN11, ASN85, TRP73, GLN75, and GLY77 with robust interaction energy (−204.166 kcal/mole). Followed by the potential binding of EGCG with subtype 3g, maximum number of nine H-bonds were in sighted from naringenin (H-bond energy: −4.3324 kJ/mole) and silybin (−7.6386 kJ/mole). Figure 6 shows the second best pose in each subtype of model core proteins within the binding pockets. Hence, EGCG can be considered as a potential drug target with the active site of HCV capsid protein, which coordinates the major role in assembly and structural protein synthesis that are easily manipulated leading to harvest high titters of HCV virus. Therefore, our hypothesis concerning the selected molecules should be verified further for in vitro and in vivo inhibitory activities for consideration as an effective drug (Table 4).
Figure 5.
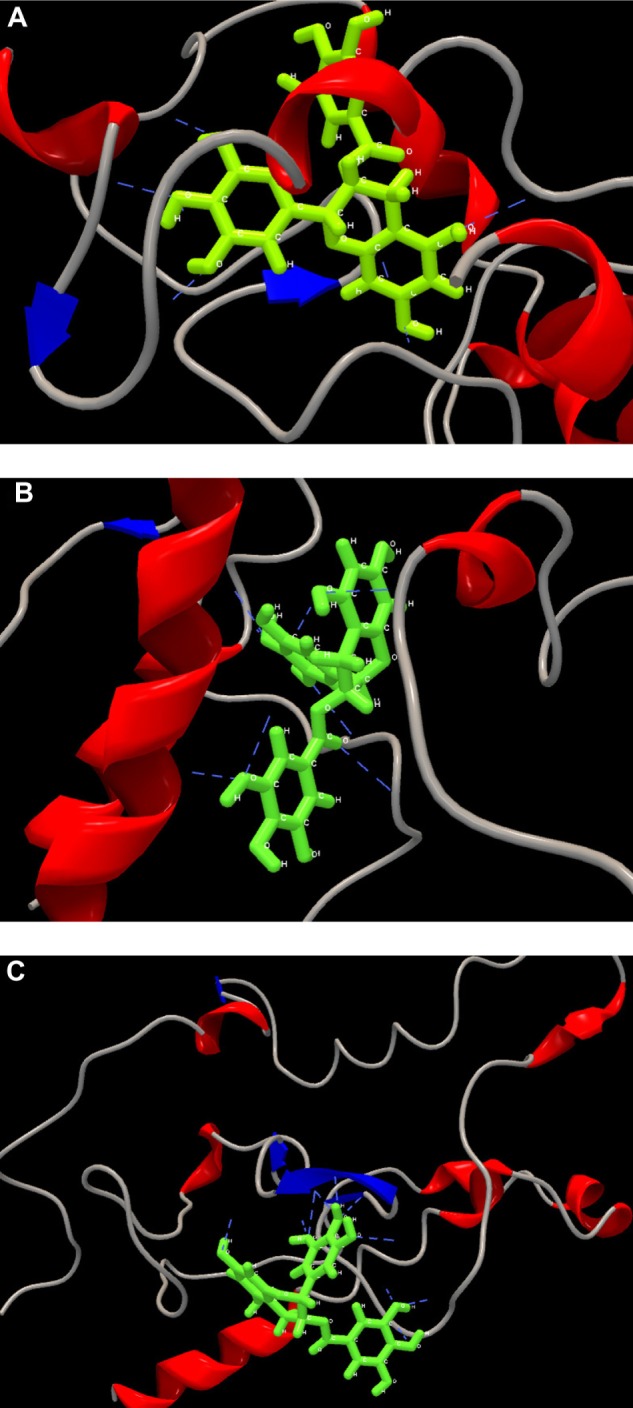
Graphical representations of the best poses within the potential drug binding site of the core protein. (A) Interaction of EGCG within the binding pockets of G3, (B) EGCG formed nine H-bond interactions with model 3b in its active binding sites, and (C) subtype 3g and its potential binding site for drug EGCG. The modeled protein is represented in ribbon like structure as backbone with alpha helix (blue color) and beta sheets (red color), ribbon like structure (grey color). Selected ligand is shown in ball and stick shape (green color) and hydrogen interactions in blue lines.
Figure 6.
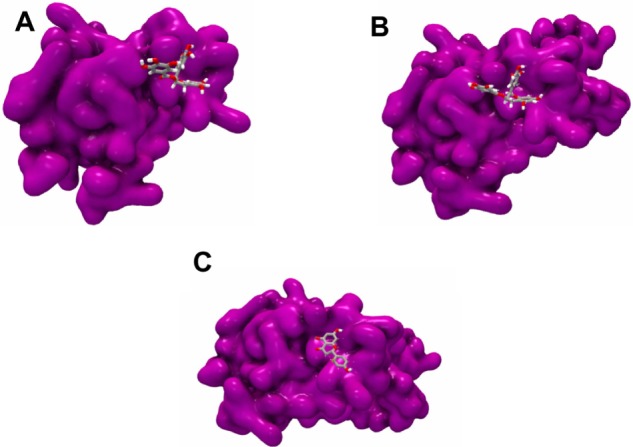
The 3D representations of the second best pose for each subtype cluster. (A) Naringenin formed nine H-bonds with the core of G3, (B) silybin interacting with core subtype 3b, and (C) docking of silybin to the core protein. Surface of the protein is colored in purple with ligand in the shape of ball and stick structure.
Table 4.
Calculated pose and estimated binding affinity with MVD module for the drugs EGCG, ladanein, naringenin, and silybin.
| CAPSID STRAIN | LIGANDS | MOLDOCK SCORE (KCAL/MOLE) | DOCKING SCORE (KCAL/MOLE) | INTERACTION ENERGY (KCAL/MOLE) | H-BONDS | BINDING RESIDUES |
|---|---|---|---|---|---|---|
| HCV-3 | EGCG | 21.584 | −0.363055 | −153.142 | 10 | SER103(2), GLU69(2), ASP108(2), ARG101, ARG61, ARG112, LYS118 |
| Ladanein | −62.4713 | −66.4508 | −119.5555 | 3 | SER103(2), GLU69 | |
| Naringenin | −62.5971 | −72.3919 | −129.636 | 5 | GLU69, ASN115, SER103(3) | |
| Silybin | 105.996 | 82.9123 | −98.564 | 5 | ARG101, GLY99, ARG56, GLY77, IIE64 | |
| HCV-3b | EGCG | 4.83397 | −8.18517 | −178.028 | 9 | LEU95(2), TRP93, ALA91, ASN18, VAL19, GLY87(2), GLY25 |
| Ladanein | −86.1153 | −87.9091 | −155.472 | 4 | TRP90(2), ALA91, VAL19 | |
| Naringenin | −65.756 | −69.518 | −145.682 | 7 | TRP90(3), GLY-84, TRP-93, GLN-86(2) | |
| Silybin | 121.713 | 108.298 | −134.336 | 6 | GLY38(2), ALA128, ALA91, ASN18, GLY30 | |
| HCV-3g | EGCG | −45.1892 | −63.0404 | −204.166 | 11 | ARG6(2), ASN11, ASN85(2), TRP73(2), GLN75, GLY77(2) |
| Ladanein | −88.225 | −57.7694 | −98.8963 | 5 | GLY77(2), TRP73(2), ARG15 | |
| Naringenin | −83.5407 | −88.5536 | −159.483 | 9 | TRP73(2), ASN85, TYR78, TRP73, GLY77(2) | |
| Silybin | 84.1128 | 63.3348 | −171.023 | 9 | ASN11, GLY87, ASN85(2), GLY77, ARG15, TRP73(2), GLY89 |
Discussion
In recent years, many natural phytochemicals are identified with anti-HCV activity but many aspects concerning their mechanisms of actions are still to be predicted. It is only the replication step in viral life cycle that was investigated only based on the in vitro models.13 Flavonoids taken into our study have multiple effects on HCV. Prior studies showed that silybin inhibits HCV replication in cell culture27 and its inhibition is attributed toward NS5B RNA-dependent RNA polymerase.28,29 EGCG is an abundant flavonoid found in every green tea extract. Recently, three different studies showed inhibition of infection in dose dependent manner during infection of Huh-7 cells with HCV cell culture system.30–32 EGCG is a new anti-HCV molecule with unique properties that is not genotype specific and also prevents cell to cell transmission.13 Ladanein is also one of the most active plant extracts belonging to group of flavones.33 Compared to EGCG, ladanein did not show binding with viral protein but rather its inhibition activity appeared later in an uncharacterized viral entry.13 Naringenin is known for its dietary supplement that possesses anti-oxidant, anti-carcinogenic, and anti-inflammatory activities in both in vitro and in vivo.13 Inhibition activity of naringenin was observed which blocked the assembly of intracellular infectious viral molecules without interacting the intracellular levels of viral protein or RNA.34 A detailed study is clearly required to validate the effectiveness of the selected molecules in vivo. Therefore, understanding the protein docking experiments, which are done routinely to screen the modern drug design from the drug receptor interaction and binding strongly, is a helping hand to design a novel and a potent drug by disclosing its mechanism of drug receptor interaction.
Conclusion
This article evaluated the ability to enrich the importance of natural active compounds for three target capsid proteins from HCV-G3 and its subtypes. We hypothesized here the first evidence of effective binding of bioflavonoid to the HCV capsid protein especially EGCG, a medicinal plant derived compound to be the best fit inhibitor bound to the active sites core based on an in silico studies. Hence, we clinched to the fact that these bioflavonoid inhibitors might disrupt the core dimerization, a chief step necessary for core oligomers to form and stay stable. Therefore, our work demonstrates the derivatives of HCV inhibitor at the presumed site of viral particle assembly strongly supporting the validity of capsid core inhibitors as a useful molecular probes to understand the capsid assembly and to serve as a basis for development of potential new antiviral agents against HCV but in vitro and in vivo studies are necessary to further confirm these studies.
Acknowledgments
We acknowledge valuable discussions from Sujith Vadakkedathu Iype from Thangal Kunju Musaliar College of Engineering University at Kerala.
Footnotes
ACADEMIC EDITORS: Thomas Dandekar and JT Efird, Associate Editors
FUNDING: Authors disclose no funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
This paper was subject to independent, expert peer review by a minimum of two blind peer reviewers. All editorial decisions were made by the independent academic editor. All authors have provided signed confirmation of their compliance with ethical and legal obligations including (but not limited to) use of any copyrighted material, compliance with ICMJE authorship and competing interests disclosure guidelines and, where applicable, compliance with legal and ethical guidelines on human and animal research participants.
Author Contributions
SM and SM conceived and designed the experiments and analyzed the data. SM, SM, and MF wrote the first draft of the manuscript. MF and Shilu Mathew contributed to the writing of the manuscript. SM, SM, and MF jointly developed the structure and arguments for the paper. MF, Shilu Mathew, GA, NB, SJ, IQ, MQ and MI made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.
REFERENCES
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Book World Cancer Report 2008 IARC PUBLICATIONS Edited by Peter Boyle and Bernard Levin. 2008. http://www.iarc.fr/en/publications/pdfs-online/wcr/2008/IARC. IARC World Cancer Report.
- 3.Seeff LB. Natural history of chronic hepatitis C. Hepatology. 2002;36(1):35–46. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- 4.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–63. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 5.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436(7053):933–8. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 6.Penin F, Dubuisson J, Rey FA, Moradpour D, Pawlotsky JM. Structural biology of hepatitis C virus. Hepatology. 2004;39(1):5–19. doi: 10.1002/hep.20032. [DOI] [PubMed] [Google Scholar]
- 7.Mousseau G, Kota S, Takahashi V, Frick DN, Strosberg AD. Dimerization-driven interaction of hepatitis C virus core protein with NS3 helicase. J Gen Virol. 2011;92(1):101–11. doi: 10.1099/vir.0.023325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boulant S, Montserret R, Hope RG, Ratinier M, Targett-Adams P. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J Biol Chem. 2006;281:22236–47. doi: 10.1074/jbc.M601031200. [DOI] [PubMed] [Google Scholar]
- 9.Blair WS, Pickford C, Irving SL, et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010;6:e1001220. doi: 10.1371/journal.ppat.1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McHutchison JG, Gordon SC, Schiff ER, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N Engl J Med. 1998;339:1485–92. doi: 10.1056/NEJM199811193392101. [DOI] [PubMed] [Google Scholar]
- 11.Di Bisceglie AM Hoofnagle JH. Optimal therapy of hepatitis C. Hepatology. 2002;36:121–7. doi: 10.1053/jhep.2002.36228. [DOI] [PubMed] [Google Scholar]
- 12.Hofmann WP, Zeuzem S. A new standard of care for the treatment of chronic HCV infection. Nat Rev Gastroenterol Hepatol. 2011;8:257–64. doi: 10.1038/nrgastro.2011.49. [DOI] [PubMed] [Google Scholar]
- 13.Calland N, Dubuisson J, Rouille Y, Seron K. Hepatitis C virus and natural compounds: a new antiviral approach? Viruses. 2012;4(10):2197–217. doi: 10.3390/v4102197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee KH. Research and future trends in the pharmaceutical development of medicinal herbs from Chinese medicine. Public Health Nutr. 2000;3(4):515–22. doi: 10.1017/s1368980000000604. [DOI] [PubMed] [Google Scholar]
- 15.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–35. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Panigrahi AK, Roca J, Acharya SK, Jameel S, Panda SK. Genotype determination of hepatitis C virus from northern India: identification of a new subtype. J Med Virol. 1996;48(2):191–8. doi: 10.1002/(SICI)1096-9071(199602)48:2<191::AID-JMV12>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 17. Available at http://www.clcbio.com/products/clc-drug-discovery-workbench/#features.
- 18. Available at http://www.phylogeny.fr.
- 19.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dereeper A, Audic S, Claverie JM, Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010;10(8):1471–2148. doi: 10.1186/1471-2148-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dereeper A, Blanc G, Audic S, et al. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;1(36):465–9. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guindon S, Gascuel O. A simple, fast and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 23.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 24.Benigni R, Bossa C, Netzeva T, Rodomonte A, Tsakovska I. Mechanistic QSAR of aromatic amines: new models for discriminating between homocyclic mutagens and nonmutagens, and validation of models for carcinogens. Environ Mol Mutagen. 2007;48(9):754–71. doi: 10.1002/em.20355. [DOI] [PubMed] [Google Scholar]
- 25.Fjodorova N, Vracko M, Novič M, Roncaglioni A, Benfenati E. New public QSAR model for carcinogenicity. Chem Cent J. 2010;4:S3. doi: 10.1186/1752-153X-4-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Available at http://www.vega-qsar.eu/download.html.
- 27.Polyak SJ, Morishima C, Shuhart MC, Wang CC, Liu Y, Lee DY. Inhibition of T-cell inflammatory cytokines, hepatocyte NF-kappaB signaling, and HCV infection by standardized Silymarin. Gastroenterology. 2007;132(5):1925–36. doi: 10.1053/j.gastro.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 28.Polyak SJ, Morishima C, Lohmann V, et al. Identification of hepatoprotective flavonolignans from silymarin. Proc Natl Acad Sci USA. 2010;107(13):5995–9. doi: 10.1073/pnas.0914009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagoner J, Morishima C, Graf TN, et al. Differential in vitro effects of intravenous versus oral formulations of silibinin on the HCV life cycle and inflammation. PLoS One. 2011;6(1):e16464. doi: 10.1371/journal.pone.0016464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calland N, Albecka A, Belouzard S, et al. Epigallocatechin-3-gallate is a new inhibitor of hepatitis C entry. Hepatology. 2012;55(3):720–9. doi: 10.1002/hep.24803. [DOI] [PubMed] [Google Scholar]
- 31.Chen C, Qiu H, Gong J, et al. Epigallocatechin-3-gallate inhibits the replication cycle of hepatitis C virus. Arch Virol. 2012;157(7):1301–12. doi: 10.1007/s00705-012-1304-0. [DOI] [PubMed] [Google Scholar]
- 32.Ciesek S, Von HT, Colpitts CC, et al. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology. 2011;54(6):1947–55. doi: 10.1002/hep.24610. [DOI] [PubMed] [Google Scholar]
- 33.Haid S, Novodomská A, Gentzsch J, et al. A plant-derived flavonoid inhibits entry of all HCV genotypes into human hepatocytes. Gastroenterology. 2012;143(1):213–22. doi: 10.1053/j.gastro.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 34.Goldwasser J, Cohen PY, Lin W, et al. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J Hepatol. 2011;55:963–71. doi: 10.1016/j.jhep.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nahmias Y, Goldwasser J, Casali M, et al. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology. 2008;47(5):1437–45. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagoner J, Negash A, Kane OJ, et al. Multiple effects of silymarin on the hepatitis C virus lifecycle. Hepatology. 2010;6(51):1912–21. doi: 10.1002/hep.23587. [DOI] [PMC free article] [PubMed] [Google Scholar]