

Abstract
In this article we introduce modern statistical machine learning and bioinformatics approaches that have been used in learning statistical relationships from big data in medicine and behavioral science that typically include clinical, genomic (and proteomic) and environmental variables. Every year, data collected from biomedical and behavioral science is getting larger and more complicated. Thus, in medicine, we also need to be aware of this trend and understand the statistical tools that are available to analyze these datasets. Many statistical analyses that are aimed to analyze such big datasets have been introduced recently. However, given many different types of clinical, genomic, and environmental data, it is rather uncommon to see statistical methods that combine knowledge resulting from those different data types. To this extent, we will introduce big data in terms of clinical data, single nucleotide polymorphism and gene expression studies and their interactions with environment. In this article, we will introduce the concept of well-known regression analyses such as linear and logistic regressions that has been widely used in clinical data analyses and modern statistical models such as Bayesian networks that has been introduced to analyze more complicated data. Also we will discuss how to represent the interaction among clinical, genomic, and environmental data in using modern statistical models. We conclude this article with a promising modern statistical method called Bayesian networks that is suitable in analyzing big data sets that consists with different type of large data from clinical, genomic, and environmental data. Such statistical model form big data will provide us with more comprehensive understanding of human physiology and disease.
Keywords: Bayesian analysis, Statistical data interpretation, Systems biology
INTRODUCTION
In medicine and the biomedical sciences, we want to find out how genes interact between themselves and with their environment and how they influence selected traits at any given point in life. For bioinformaticians, biostatisticians, and epidemiologists, the clinical data, gene-gene and gene-environment causal interactions are defined by statistical probabilities. This contrasts strongly against the view physicians and biological scientists take, who think that the mere statistical clinical data, gene-gene and gene-environment interactions aren't as sufficient basis for the actual clinical and biological interactions.
The primary objective of this article is to examine the clinical data, gene-gene and gene-environment interactions, obtained from big data, i.e., large datasets from different types of clinical and genomic data, using statistical and bioinformatics approaches. There has been many in depth articles in analyzing clinical data using traditional statistical analysis methods, i.e., linear or logistic regression [1,2,3,4,5,6]. The gene-gene causal interactions have been modeled using high throughput data from single nucleotide polymorphism (SNP) studies [7,8,9,10] and gene expression studies [11,12,13,14,15]. Recent research in biology shows that the way that genes interact between themselves cannot be described without mentioning the environment in which the interactions are taking place. Moreover, recent studies in the field of epigenetics provide us with possible gene-environment interactions that can potentially alter the genome. The complexity of a statistical model for clinical data, not even including gene-gene and gene-environment causal interactions, is already overwhelming; nevertheless, we need to be aware that additional to the clinical data, gene-gene causal interactions and gene-environment interactions should be also modeled to complete the understanding of the disease progression.
In the following sections, we will look more into different types of clinical and genomic data, i.e., electronic patient data, gene expression data, SNP data, and epigenetic data, and review what bioinformatics and statistical approaches have been used to analyze these data. In conclusion, we will show what traditional statistical methods and recent biostatistical methods can be used in modeling clinical data, gene-gene and gene-environment interactions. In addition, we will present a new promising bioinformatics approach called causal Bayesian networks (BNs), which provides a natural way of describing causal relationships among modeled variables.
CLINICAL DATA
In the past few years, the federal government has spent billions of dollars to improve clinical data analysis through the use electronic patient records. It is believed that the use of electronic records has the capacity of improving the errors that occur in surgery and Emergency Department (ED) visits, hospitalizations, and office visits for patients. In addition, using statistical regression analyses, the use of electronic health records has allowed to better understanding the interconnection among the clinical variables and also allows to better understand the progress, prognosis, and treatment of diseases. Typically, clinical data are analyzed using linear or logistic regressions.
Linear Regression
Sir Galton first introduced linear regression in the 18th century [16]. Linear regression is a statistical method for modeling the relationship between a dependent variable and one or more explanatory variables. It assumes the outcome can be predicted via weighted sums of input variables. Typically this is the very first model that you will look into before going into more complex models when the outcome variable is continuous. Reed et al. [2] researched the association between implementing a highly available electronic health record (EHR) and ED visits, hospitalizations, and office visits for patients with diabetes mellitus. They applied a linear regression model with fixed effects at the patient level and found that, among patients with diabetes, the use of an EHR was associated with a modest reduction in ED visits and hospitalizations but not on office visit rates. Jaffe et al. [3] measured the annual control rates from the Kaiser Permanente Northern California hypertension registry by accounting for the nonindependence of proportions as a time series, fitting a log-linear regression of the proportion on time, allowing for autocorrelated errors. They found that, among adults with hypertension, implementation of a large-scale hypertension program was associated with a significant increase of hypertension control compare with state and national control rates. Yuasa et al. [6] studied the correlations between the initial tumor size and size reduction rate in patients treated with targeted agents. They used both univariate and multivariate linear regression analyses to discover that only the initial tumor size was associated with the rate of reduction in individual tumors. This could be useful for physicians who treat patients with metastatic renal cell carcinoma.
Logistic Regression
Logistic regression is similar in many aspects to linear regression, they differ in a very critical aspect. Logistic regression assumes outcome can be explained through weighted sum that goes through a special mathematical transformation, called logit. This transformation allows all weighted sum to be mapped into a value in between 0 and 1, which can be interpreted as a probability of a binary outcome. Thus, logistic regression is widely used in outcome variable that has two outcome, e.g., whether you have a disease or not. De Vries et al. [1] researched the relationship between mortality and iatrogenic illnesses that occur outside the surgical room. The researchers implemented a multidisciplinary surgical safety checklist in which six hospitals had to check for medication, operative side, and medication. Logistic regression was performed to assess the relationship between the checklist and mortality. The study showed an association between the comprehensive checklist and a reduction in surgical complication and mortality and hospitals with high standard of care. Shnorhavorian et al. [4] investigated the relationship between maternal risk factors and congenital urinary tract anomalies. The performed a case-control study in which they accessed birth-hospital discharge records from Washington State from 1987-2007, in which cases were children diagnosed with urinary anomalies while controls did not display such urinary tract anomalies. In the analysis, gestational diabetes, preexisting diabetes, and maternal renal disease were all associated with an increased risk of kidney anomalies. Peterson et al. [5] researched in-flight medical emergencies and the outcomes of these events. They characterized the most common medical problems and the type of on board assistance rendered. Through logistic regression, they identified that most in-flight medical emergencies are associated to syncope, respiratory symptoms, and gastrointestinal symptoms.
GENE EXPRESSION DATA
This section is partially adopted and summarized from [17]. Microarray techniques positively impacted the course of molecular biology. Before these techniques existed, there were labor-intensive methods to measure a single gene's expression patterns in cells. Current microarray techniques can measure the expression level of about 10,000 genes at a time. A successful sequencing of the entire genome of yeast Saccharomyces cerevisiae in April 1996 initiated many experimental studies in other forms of yeast [18,19,20]. These studies fit under a new approach in biology that is called systems biology. Systems biology seeks in part to model large networks of cellular function, including the causal pathways that capture how genes regulate each other.
Before describing gene-gene causal interaction models, we first place them in the context of gene clustering methods, which have been very popular the last few years. Indeed, most of the early work on gene expression data analyses used clustering methods. A cluster analysis typically searches for groups of genes that show similarities among different conditions. Other analyses followed using similar cluster analyses applied to microarray data [21,22,23].
Clinical studies also used cluster analysis on microarray data [24,25]. For example, Alizadeh et al. [24] used cluster analysis to find different types of lymphoma among diagnosed patients by comparing the clusters of similarly expressed genes and whether or not they responded to the current therapy. Along with cluster analyses, gene pathway analyses were performed on the gene expression data. Analyses to construct pathways among the genes yield more information than do cluster or classification analyses. Cluster and classification analyses do not necessarily provide causal information, which is at the heart of gene pathway discovery. On the other hand, knowledge of causal pathways can be used to produce a causal clustering of the genes.
In the following subsections, we will briefly review gene-gene causal interaction models. More detail review can be found in Yoo [17].
Boolean Networks
Boolean networks were first introduced by Somogyi and Sniegoski [26] in1996. With its simple representation, Boolean networks were easily implemented as genetic networks. However, since Boolean networks do not explicitly model the uncertainty that the data can have, they cannot model the vague nature of a biological system. Also note that when a Boolean network is created, no arrows are used; thus, there is no sense of direction or causality in the model.
Continuous Models
In mathematics, using differential equations to model a biological system has a long history [27,28,29]. Chen et al. [30] modeled a simplified dynamic system of gene regulation (with feedback on transcription). Differential equations can model biological dynamics better than Boolean networks, but the computational cost of using differential equations is high, and often many of the parameters are required in order to use differential equation modeling are not available. Since most of the dynamics of the actual genetic pathways appear to be non-linear, a linear model seems to work on only limited dynamics of the genetic pathway.
Bayesian Networks
The BN model has been widely used to learn predictive models from data. BNs can model causality based on either the researcher's knowledge, data or both. It is also used in many medical related domains because of its ability to perform inferences easily [31,32,33]. One practical limitation of BNs is that inference within them is not practically feasible with large a number (>50) of modeled variables [34], which is a frequent limitation of many reasoning methodologies; in response, researchers have developed different methodologies to address the issue.
A causal BN (or causal network for short) is a BN in which each arrow is interpreted as a direct causal influence between a parent variable and the variable to which it is directly related to, which is called the child variable [35]. Fig. 1 illustrates the structure of a hypothetical causal BN structure containing five variables that represent genes.
Fig. 1.

A causal Bayesian network that represents a hypothetical gene-regulation pathway.
The causal network structure in Fig. 1 indicates, for example, that the Gene1 can regulate (causally influence) the expression level of the Gene3, which in turn can regulate the expression level of the Gene5. The causal Markov condition gives the conditional independence relationships specified by a causal BN:
A variable is independent of its nondescendants given that its parents occur (i.e., its direct causes).
Murphy and Mian [36] showed that the Boolean network model [26], the linear model [37], and the non-linear weighted model [38] are all special cases of dynamic BNs (DBNs). A DBN incorporates time in BNs (which is then usually called a temporal BN).
Mixture Models and Other Models
McAdams and Shapiro [39] modeled the E. coli λ phage lysis-lysogeny genetic switch using a mixture of Boolean networks and continuous input-output relations. Yuh et al. [40] was able to model a single gene within the sea urchin embryo with a similar hybrid model. Matsuno et al. [41] used a Petri net that models continuous variables and analyzes the genetic switch mechanism of λ phage. Goss and Peccoud [42] used stochastic Petri nets to model the stabilizing effect of proteinson the genetic network regulating plasmid replication.
There are many different kinds of statistical classification methods. A method commonly used for statistical classification is k-Nearest Neighbor (kNN), which classifies a new case by calculating the minimum distance between the new case and a set of training cases. kNN has been used in areas such as radiology and immunology. Variations of kNN have recently been used in classifying and clustering genes from large gene expression datasets [18,21,22,23].
Petri nets are a formal graphical language appropriate for modeling systems where concurrency occurs. Petri nets were used in guidelines for patient care flow [43]. It has also been used in modeling mechanisms in a cell [41,42,44].
Genetic programming uses the three basic mechanisms that drive natural evolution-reproduction, mutation, and selection-in its search for a model that best fits the training data. Evolutionary methods allow a program to evolve, giving it great freedom to search through a large space of possible models. Koza et al. [45] has used genetic programming to learn gene networks from simulated data that was generated by a computer model of the cell, called E cell [46].
SINGLE NUCLEOTIDE POLYMORPHISMS
Recent genome-wide association studies have discovered significant associations between complex diseases and SNPs. A SNP is a DNA sequence variation resulting from an alteration of a single nucleotide in the genome. It differs from a mutation in that the variation must occur within at least 1% of the population. SNPs are the most common genetic variations and thus are the most thoroughly investigated. It is believed that SNP-SNP interactions, not the individual SNPs themselves, play an important role in the development of complex diseases. Multiple models have been employed in SNP-SNP analysis, most notably logistic regression, combinatorial methods, support vector machines (SVMs), and logic regression.
Logistic regression, a fairly traditional model used for SNP analysis, is capable of linking SNPs to disease outcome using a function called logit. SNP-SNP interactions can be considered by including interaction terms in the model. This of course can result in a large number of variables. When stratification is present within the data, the conditional logistic regression (CLR) method can be used. By stratifying the data, the CLR method is able to adjust for the matching of the the variables with each other [47].
A widely used combinatorial method for SNP analysis is multifactor dimensionality reduction (MDR). MDR attempts to combine two or more attributes, in this case SNPs, into a single attribute to improve disease prediction. The combination of SNPs is a great predictor of a disease because it minimizes error. A number of MDR variations have been proposed, including pair-wise MDR, which addresses the problem of MDR's inability to classify empty cells [9] and robust MDR which makes use of the Fisher exact test [10].
Goodman [48] developed an approach similar to MDR, known as polymorphism interaction analysis (PIA) to explore SNP interactions and colon cancer risk. Like MDR, PIA examines all possible SNP combinations to find the interaction that best predicts the risk of disease. They differ in that PIA uses two unique scoring functions, the Gini index and the percentage wrong (i.e., the percentage of misclassified subjects), to find the interactions most likely associated with disease risk. In addition, PIA makes use of ten-fold cross validation and, excludes SNPs or SNP combinations that have a ratio greater than 1.2 from the analysis [48].
SVMs have also been recently used in SNP-SNP analyses. SVMs are a collection of supervised learning methods used for both classification and regression. Whereas many classifiers aim to minimize prediction error, SVMs are trained to maximize accuracy. Observations are represented as points in space while a hyperplane is constructed and treated as the decision boundary between the outcome categories. The prediction accuracy is maximized by finding the hyperplane that has the greatest distance to the nearest training data points [8].
Chen et al. [8] proposed the following four search algorithms to detect interaction among SNPs: recursive feature addition SVM (SVM-RFA), recursive feature elimination SVM (SVM-RFE), SVM with local search (SVM-local), and SVM with genetic algorithm (SVM-GA). RFA/E discovers the optimal subset of SNP combinations by ranking the subsets according to a ranking criterion. SNP combination(s) are added/eliminated at each iteration using the correlation coefficients as the ranking criteria [8].
The SVM-local algorithm is similar to most local searches in that a random subset of SNP interactions is initially generated. A search is then conducted through the initial subset's neighbors in an attempt to find a "better" subset. If one is found, the "better" subset is accepted. This continues until a subset is selected in which no "better" subset exists. In order for a given subset of SNP interactions to have a neighbor(s), they both must differ by a single element [8].
Unlike SVM-local, SVM-GA is a stochastic search that is based upon natural selection and genetics. The search begins by generating a random set of SNP combinations, called the population. Genetic operations, crossovers, and mutations are performed on randomly selected chromosomes (individual SNP combinations within the population) to yield the next generation. An evolution process, called selection, is then performed on both generations to improve the chromosomes. New generations are created and the above is repeated until the chromosomes in the population converge. The final chromosome is considered the best subset of SNP interactions [8].
In logic regression, the interactions among SNPS are represented in logic trees and logic expressions. Both make use of the logic operators "or" and "and", the latter signifying an interaction. Traditional logic regression uses the Monte Carlo Markov Chain (MCMC) method to find the collection of best logic regression models. From that collection, the SNP combinations occurring most frequently are identified and assumed to be important interactions. The importance of interactions is quantified by the proportion of models in which the SNP combinations appear. Interactions that are only significant in small subgroups of the population thus have the potential to be overlooked. The LogicFS [7] approach to logic regression uses sampling to address this issue. Another advantage of the LogicFS approach is that, unlike MCMC logic regression, it uses two unique measures that allow for the comparison of very distinct interactions. Logic regression is considered more practical than other methods used in SNP-SNP analysis because it does not require interaction terms to be included in the model as inputs.
Logistic regression, MDR, SVMs, and logic regression are all methods that are capable of identifying important SNP-SNP interactions. Algorithms that use different search mechanisms, different ranking/importance criterion, and/or that are geared toward specific situations have been proposed. Despite these advantages, the literature seems to lack studies that seek out causal discovery among SNPs. Like the other models, BNs are able to identify important associations among SNPs. It is being proposed that BNs are also capable of extracting causal information from those SNP-SNP and SNP-disease associations.
EPIGENETIC REGULATION OF THE GENOME
Epigenetics modify genomes functions without altering the DNA sequence. Thus, the epigenetic modifications change the transcriptions of genes.
DNA methylation, which involves the addition of a methyl group onto cytosines in the DNA, was thought to be active only during embryonic development. However, recent studies show that DNA methylation occurs in even fully differentiated cells [49]. This shows biological examples of gene-environmental interactions. Such interactions need to be considered in modeling gene expression. The gene-environment interactions also arise from gene transcription.
Fu et al. [50] developed Bayesian inference methods for epigenetic data to study the transmission of DNA methylation patterns over cell divisions. Genome-wide methylation data were analyzed using the genome-wide statistical significance calculation for increased variability [51] and Bayesian hierarchical model [52]. A beta-mixture model was used in analyzing genome-wide methylation patterns of colon cancer.
CONCLUSIONS
We have reviewed bioinformatics and statistical methods for clinical data, gene-gene and gene-environment causal interactions using big data, typically from different sources; i.e. genomic and clinical data. Traditionally in statistics, modeling clinical data and even complex gene-gene and gene-environment interactions are given in a linear equation among modeled variables [53]. However, note that there are pros and cons of the traditional statistical approach. Moreover, modeling causality is not a straight forward extension.
Recently, there have been many or statistical methods that have been used in order to study complex gene-gene and gene-environment interactions. These bioinformatics methods were presented in the previous sections. Here we present causal BNs as a method that can model complex clinical, gene-gene and gene-environment interactions using big data, from different types of genomic and clinical data.
Given the emergence of datasets in medicine and biology with large number of variables, BNs have been successful in developing efficient algorithms that are able to handle very large datasets and develop high quality predictive models from genomic and clinical data [12]. A BN is a directed acyclic graph in which each node represents a variable and each arc represents a relationship. In BNs, each arc is interpreted as a direct influence between a parent node (variable) and a child node.
BNs are also built based on the causal Markov conditions [35]. This can be understood with the following hypothetical example BN:
In Fig. 2, either Rain occurs or you turn or the Sprinkler, both of which can make your lawn wet. Also, if your lawn gets enough water, it gets green (My Lawn Green). Of course, your neighbor, who does not have a sprinkler, can get his lawn wet when it rains. In the above network, we can identify the following three sub networks:
Fig. 2.
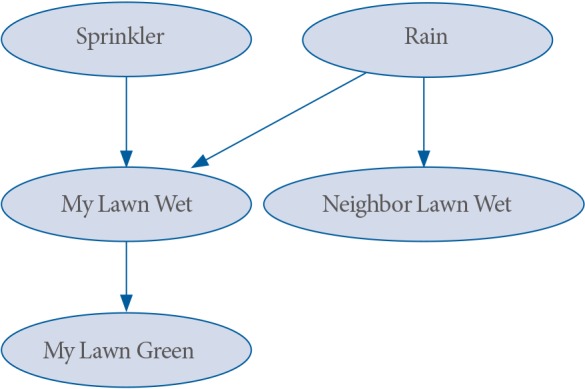
A simple example Bayesian network.
In Fig. 3A, which are called converging arcs, if you know your lawn is wet and you know it didn't rain then there is a high chance that your sprinkler is on (Sprinkler). In other words, if nodes A and B converge into node C, then A and B becomes dependent given that C occurs. Also note that in Fig. 3B, called diverging arcs, if it rains, your lawn and your neighbor lawn get wet. If you know it rained (Rain), knowing your lawn is wet does not tell you about your neighbor's lawn being wet, in other words, if variable C diverges into variables A and B, then A and B becomes independent given that C occurs. In Fig. 3C, called serial arcs, if it rains (Rain), then my lawn gets wet, and eventually, your lawn gets green. In this case, if you know your lawn is wet, then knowing whether it rained or not will not tell you much more about your lawn getting green, in other words, if the serial arcs goes from a variable A to a variable B to a variable C, then A and C are independent given that B occurs.
Fig. 3.
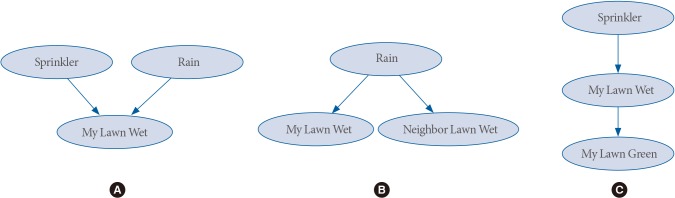
Sub networks from Fig. 2. (A) Converging arcs, (B) diverging arcs, and (C) serial arcs.
These three sub networks (converging, diverging, and serial) provide ways to express causal interactions in intuitive ways. The fact that causal BNs can provide a myriad of combinations with the statistical analysis of collected data, makes an excellent bioinformatics statistical tool in modeling complex clinical parameters, gene-gene, and gene-environment interactions from different types of genomic and clinical data.
Footnotes
No potential conflict of interest relevant to this article was reported.
References
- 1.de Vries EN, Prins HA, Crolla RM, den Outer AJ, van Andel G, van Helden SH, et al. Effect of a comprehensive surgical safety system on patient outcomes. N Engl J Med. 2010;363:1928–1937. doi: 10.1056/NEJMsa0911535. [DOI] [PubMed] [Google Scholar]
- 2.Reed M, Huang J, Brand R, Graetz I, Neugebauer R, Fireman B, et al. Implementation of an outpatient electronic health record and emergency department visits, hospitalizations, and office visits among patients with diabetes. JAMA. 2013;310:1060–1065. doi: 10.1001/jama.2013.276733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaffe MG, Lee GA, Young JD, Sidney S, Go AS. Improved blood pressure control associated with a large-scale hypertension program. JAMA. 2013;310:699–705. doi: 10.1001/jama.2013.108769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shnorhavorian M, Bittner R, Wright JL, Schwartz SM. Maternal risk factors for congenital urinary anomalies: results of a population-based case-control study. Urology. 2011;78:1156–1161. doi: 10.1016/j.urology.2011.04.022. [DOI] [PubMed] [Google Scholar]
- 5.Peterson DC, Martin-Gill C, Guyette FX, Tobias AZ, McCarthy CE, Harrington ST, et al. Outcomes of medical emergencies on commercial airline flights. N Engl J Med. 2013;368:2075–2083. doi: 10.1056/NEJMoa1212052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yuasa T, Urakami S, Yamamoto S, Yonese J, Nakano K, Kodaira M, et al. Tumor size is a potential predictor of response to tyrosine kinase inhibitors in renal cell cancer. Urology. 2011;77:831–835. doi: 10.1016/j.urology.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Schwender H, Ickstadt K. Identification of SNP interactions using logic regression. Biostatistics. 2008;9:187–198. doi: 10.1093/biostatistics/kxm024. [DOI] [PubMed] [Google Scholar]
- 8.Chen SH, Sun J, Dimitrov L, Turner AR, Adams TS, Meyers DA, et al. A support vector machine approach for detecting gene-gene interaction. Genet Epidemiol. 2008;32:152–167. doi: 10.1002/gepi.20272. [DOI] [PubMed] [Google Scholar]
- 9.He H, Oetting WS, Brott MJ, Basu S. Pair-wise multifactor dimensionality reduction method to detect gene-gene interactions in a case-control study. Hum Hered. 2010;69:60–70. doi: 10.1159/000243155. [DOI] [PubMed] [Google Scholar]
- 10.Gui J, Andrew AS, Andrews P, Nelson HM, Kelsey KT, Karagas MR, et al. A robust multifactor dimensionality reduction method for detecting gene-gene interactions with application to the genetic analysis of bladder cancer susceptibility. Ann Hum Genet. 2011;75:20–28. doi: 10.1111/j.1469-1809.2010.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman N, Linial M, Nachman I, Pe'er D. Using Bayesian networks to analyze expression data. J Comput Biol. 2000;7:601–620. doi: 10.1089/106652700750050961. [DOI] [PubMed] [Google Scholar]
- 12.Yoo C, Cooper GF. An evaluation of a system that recommends microarray experiments to perform to discover gene-regulation pathways. Artif Intell Med. 2004;31:169–182. doi: 10.1016/j.artmed.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 13.Lin Y, Lin S, Watson M, Trinkaus KM, Kuo S, Naughton MJ, et al. A gene expression signature that predicts the therapeutic response of the basal-like breast cancer to neoadjuvant chemotherapy. Breast Cancer Res Treat. 2010;123:691–699. doi: 10.1007/s10549-009-0664-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedraza V, Gomez-Capilla JA, Escaramis G, Gomez C, Torne P, Rivera JM, et al. Gene expression signatures in breast cancer distinguish phenotype characteristics, histologic subtypes, and tumor invasiveness. Cancer. 2010;116:486–496. doi: 10.1002/cncr.24805. [DOI] [PubMed] [Google Scholar]
- 15.Yoo C, Brilzb EM, Wilcox M, Pershousec MA, Putnam EA. Gene pathways discovery in asbestos-related diseases using local causal discovery algorithm. Commun Stat Simul Comput. 2012;41:1840–1859. [Google Scholar]
- 16.Stanton JM. Galton, Pearson, and the Peas: a brief history of linear regression for statistics instructors. J Stat Educ. 2001;9(3) [Google Scholar]
- 17.Yoo C. Discovering gene-gene and gene-environment causal interactions using Bioinformatics approaches. In: Roy D, Dorak MT, editors. Environmental factors, genes, and the development of human cancers. New York: Springer; 2010. pp. 115–138. [Google Scholar]
- 18.Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, et al. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ideker T, Thorsson V, Ranish JA, Christmas R, Buhler J, Eng JK, et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- 20.Smith EN, Kruglyak L. Gene-environment interaction in yeast gene expression. PLoS Biol. 2008;6:e83. doi: 10.1371/journal.pbio.0060083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michaels GS, Carr DB, Askenazi M, Fuhrman S, Wen X, Somogyi R. Cluster analysis and data visualization of large-scale gene expression data. Pac Symp Biocomput. 1998:42–53. [PubMed] [Google Scholar]
- 22.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 23.Herwig R, Poustka AJ, Muller C, Bull C, Lehrach H, O'Brien J. Large-scale clustering of cDNA-fingerprinting data. Genome Res. 1999;9:1093–1105. doi: 10.1101/gr.9.11.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 25.Getz G, Levine E, Domany E. Coupled two-way clustering analysis of gene microarray data. Proc Natl Acad Sci U S A. 2000;97:12079–12084. doi: 10.1073/pnas.210134797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Somogyi R, Sniegoski CA. Modeling the complexity of genetic networks: understanding multigenetic and pleiotropic regulation. Complexity. 1996;1:45–63. [Google Scholar]
- 27.Goodwin BC. Oscillatory behavior in enzymatic control processes. Adv Enzyme Regul. 1965;3:425–438. doi: 10.1016/0065-2571(65)90067-1. [DOI] [PubMed] [Google Scholar]
- 28.Griffith JS. Mathematics of cellular control processes. I. Negative feedback to one gene. J Theor Biol. 1968;20:202–208. doi: 10.1016/0022-5193(68)90189-6. [DOI] [PubMed] [Google Scholar]
- 29.Griffith JS. Mathematics of cellular control processes. II. Positive feedback to one gene. J Theor Biol. 1968;20:209–216. doi: 10.1016/0022-5193(68)90190-2. [DOI] [PubMed] [Google Scholar]
- 30.Chen T, He HL, Church GM. Modeling gene expression with differential equations. Pac Symp Biocomput. 1999:29–40. [PubMed] [Google Scholar]
- 31.Citro G, Banks G, Cooper G. INKBLOT: a neurological diagnostic decision support system integrating causal and anatomical knowledge. Artif Intell Med. 1997;10:257–267. doi: 10.1016/s0933-3657(97)00395-3. [DOI] [PubMed] [Google Scholar]
- 32.Chevrolat JP, Golmard JL, Ammar S, Jouvent R, Boisvieux JF. Modelling behavioral syndromes using Bayesian networks. Artif Intell Med. 1998;14:259–277. doi: 10.1016/s0933-3657(98)00037-2. [DOI] [PubMed] [Google Scholar]
- 33.Lucas PJ, de Bruijn NC, Schurink K, Hoepelman A. A probabilistic and decision-theoretic approach to the management of infectious disease at the ICU. Artif Intell Med. 2000;19:251–279. doi: 10.1016/s0933-3657(00)00048-8. [DOI] [PubMed] [Google Scholar]
- 34.Cooper GF. Probabilistic inference using belief networks is NP-hard. Stanford, CA: Stanford University; 1987. [Google Scholar]
- 35.Pearl J. Probabilistic reasoning in intelligent systems: networks of plausible inference. San Mateo, CA: Morgan Kaufmann; 1988. [Google Scholar]
- 36.Murphy K, Mian S. Modelling gene expression data using dynamic bayesian networks. Technical report. Berkeley, CA: Computer Science Division, University of California; Life Sciences Division, Lawrence Berkeley National Laboratory; 1999. [Google Scholar]
- 37.D'haeseleer P, Wen X, Fuhrman S, Somogyi R. Linear modeling of mRNA expression levels during CNS development and injury. Pac Symp Biocomput. 1999:41–52. doi: 10.1142/9789814447300_0005. [DOI] [PubMed] [Google Scholar]
- 38.Weaver DC, Workman CT, Stormo GD. Modeling regulatory networks with weight matrices. Pac Symp Biocomput. 1999:112–123. doi: 10.1142/9789814447300_0011. [DOI] [PubMed] [Google Scholar]
- 39.McAdams HH, Shapiro L. Circuit simulation of genetic networks. Science. 1995;269:650–656. doi: 10.1126/science.7624793. [DOI] [PubMed] [Google Scholar]
- 40.Yuh CH, Bolouri H, Davidson EH. Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science. 1998;279:1896–1902. doi: 10.1126/science.279.5358.1896. [DOI] [PubMed] [Google Scholar]
- 41.Matsuno H, Doi A, Nagasaki M, Miyano S. Hybrid Petri net representation of gene regulatory network. Pac Symp Biocomput. 2000:341–352. doi: 10.1142/9789814447331_0032. [DOI] [PubMed] [Google Scholar]
- 42.Goss PJ, Peccoud J. Analysis of the stabilizing effect of Rom on the genetic network controlling ColE1 plasmid replication. Pac Symp Biocomput. 1999:65–76. doi: 10.1142/9789814447300_0007. [DOI] [PubMed] [Google Scholar]
- 43.Quaglini S, Stefanelli M, Lanzola G, Caporusso V, Panzarasa S. Flexible guideline-based patient careflow systems. Artif Intell Med. 2001;22:65–80. doi: 10.1016/s0933-3657(00)00100-7. [DOI] [PubMed] [Google Scholar]
- 44.Boucher A, Doisy A, Ronot X, Garbay C. A society of goal-oriented agents for the analysis of living cells. Artif Intell Med. 1998;14:183–199. doi: 10.1016/s0933-3657(98)00022-0. [DOI] [PubMed] [Google Scholar]
- 45.Koza JR, Mydlowec W, Lanza G, Yu J, Keane MA. Reverse engineering of metabolic pathways from observed data using genetic programming. Pac Symp Biocomput. 2001:434–445. doi: 10.1142/9789814447362_0043. [DOI] [PubMed] [Google Scholar]
- 46.Tomita M, Hashimoto K, Takahashi K, Shimizu T, Matsuzaki Y, Miyoshi F, et al. E-CELL: Software Environment for Whole Cell Simulation. Genome Inform Ser Workshop Genome Inform. 1997;8:147–155. [PubMed] [Google Scholar]
- 47.Heidema AG, Boer JM, Nagelkerke N, Mariman EC, van der A DL, Feskens EJ. The challenge for genetic epidemiologists: how to analyze large numbers of SNPs in relation to complex diseases. BMC Genet. 2006;7:23. doi: 10.1186/1471-2156-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goodman SN. Probability at the bedside: the knowing of chances or the chances of knowing? Ann Intern Med. 1999;130:604–606. doi: 10.7326/0003-4819-130-7-199904060-00022. [DOI] [PubMed] [Google Scholar]
- 49.Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 50.Fu AQ, Genereux DP, Stoger R, Laird CD, Stephens M. Statistical inference of transmission fidelity of DNA methylation patterns over somatic cell divisions in mammals. Ann Appl Stat. 2010;4:871–892. doi: 10.1214/09-AOAS297SUPPA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaffe AE, Feinberg AP, Irizarry RA, Leek JT. Significance analysis and statistical dissection of variably methylated regions. Biostatistics. 2012;13:166–178. doi: 10.1093/biostatistics/kxr013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu G, Yi N, Absher D, Zhi D. Statistical quantification of methylation levels by next-generation sequencing. PLoS One. 2011;6:e21034. doi: 10.1371/journal.pone.0021034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kraft P, Hunter DJ. The challenge of assessing complex gene-environment and gene-gene interactions. In: Khoury MJ, Bedrosian SR, Gwinn M, editors. Human genome epidemiology: building the evidence for using genetic information to improve health and prevent disease. 2nd ed. New York: Oxford University Press; 2010. pp. 165–187. [Google Scholar]