

Abstract
Rett syndrome (RTT) is a rare neurodevelopmental disorder affecting almost exclusively females, caused in the overwhelming majority of the cases by loss-of-function mutations in the gene encoding methyl-CpG binding protein 2 (MECP2). High circulating levels of oxidative stress (OS) markers in patients suggest the involvement of OS in the RTT pathogenesis. To investigate the occurrence of oxidative brain damage in Mecp2 mutant mouse models, several OS markers were evaluated in whole brains of Mecp2-null (pre-symptomatic, symptomatic, and rescued) and Mecp2-308 mutated (pre-symptomatic and symptomatic) mice, and compared to those of wild type littermates. Selected OS markers included non-protein-bound iron, isoprostanes (F2-isoprostanes, F4-neuroprostanes, F2-dihomo-isoprostanes) and 4-hydroxy-2-nonenal protein adducts. Our findings indicate that oxidative brain damage 1) occurs in both Mecp2-null (both −/y and stop/y) and Mecp2-308 (both 308/y males and 308/+ females) mouse models of RTT; 2) precedes the onset of symptoms in both Mecp2-null and Mecp2-308 models; and 3) is rescued by Mecp2 brain specific gene reactivation. Our data provide direct evidence of the link between Mecp2 deficiency, oxidative stress and RTT pathology, as demonstrated by the rescue of the brain oxidative homeostasis following brain-specifically Mecp2-reactivated mice. The present study indicates that oxidative brain damage is a previously unrecognized hallmark feature of murine RTT, and suggests that Mecp2 is involved in the protection of the brain from oxidative stress.
Abbreviations: 4-HNE, 4-hydroxy-2-nonenal; 4-HNE PAs, 4-hydroxy-2-nonenal protein adducts; AdA, adrenic acid; ARA, arachidonic acid; ASDs, autism spectrum disorders; AUs, arbitrary units; BDNF, brain-derived neurotrophic factor; CRE, Cre-Recombinase; DHA, docosahexaenoic acid; F2-IsoPs, F2-isoprostanes; F2-dihomo-IsoPs, F2-dihomo-isoprostanes; F4-NeuroPs, F4-neuroprostanes; IsoPs, isoprostanes; 4-HNE PAs, 4-HNE protein adducts; MECP2, methyl-CpG-binding protein 2 — human gene; Mecp2, methyl-CpG-binding protein 2 — mouse gene; MeCP2, methyl-CpG-binding protein 2 — human protein; Mecp2, methyl-CpG-binding protein 2 — mouse protein; Mecp2 −/y, hemizygous null mice; Mecp2 stop/y, Lox/stop pre-symptomatic hemizygous mice; Mecp2 stop/y NestinCre, rescued Lox/stop mice (Mecp2 reactivated in the nervous tissue); Mecp2 308/y, symptomatic Mecp2 308-mutated hemizygous males; Mecp2 308/x, symptomatic Mecp2 308-mutated females; NPBI, non-protein-bound iron; OS, oxidative stress; PSV, Preserved Speech Variant; PUFAs, polyunsaturated fatty acids; ROS, reactive oxygen species; RTT, Rett syndrome; wt, wild type; wt-Cre, wild type expressing Cre recombinase
Keywords: Rett syndrome, Lipid peroxidation, Brain damage, Neurodevelopmental disorder, Murine models, Oxidative stress
Highlights
-
•
Oxidative damage is demonstrated in the brain, and more specifically in the neurons, of Mecp2 mutant mouse models.
-
•
A direct evidence between enhanced oxidative stress and Mecp2 deficiency is provided.
-
•
Oxidative damage precedes the behavioral abnormalities in Mecp2 mutant mice.
-
•
Mecp2 is likely involved in the protection of the brain from oxidative stress.
Introduction
Rett syndrome (RTT, MIM 312750) is a progressive neurodevelopmental disorder affecting almost exclusively the female gender with a frequency of approximately 1:10,000 live births, and is a leading cause of severe intellectual disability and autistic features (Chahrour and Zoghbi, 2007; Weaving et al., 2005). Other features include stereotypic hand movements, communication dysfunction, seizures, postural hypotonia, tremors, autonomic dysfunction, microcephaly and growth failure (Chahrour and Zoghbi, 2007). The classical clinical picture of the disease (Rett, 1966) is characterized by a period of 6 to 18 months of apparently normal neurodevelopment, followed by an early neurological regression, with a progressive loss of acquired cognitive, social, and motor skills in a typical 4-stage neurological regression pattern (Hagberg, 2002; Neul et al., 2010).
RTT is known to be caused in the overwhelming majority of the cases by sporadic de novo loss-of-function mutations in the X-linked methyl-CpG-binding protein 2 (MECP2) gene (Amir et al., 1999) encoding methyl-CpG binding protein 2 (MeCP2), a nuclear protein that binds to methylated CpGs and regulates gene expression (Chahrour et al., 2008; Jones et al., 1998). Different types of mutations within MECP2 are known to cause RTT, including missense, nonsense, deletions and insertions (Bienvenu and Chelly, 2006).
Despite almost two decades of research into the functions and role of MeCP2, surprisingly little is known about the mechanisms leading from MeCP2 deficiency to disease expression, with many questions still unsolved regarding the role of MeCP2 in the brain and, more generally, during development and in physiopathology (Guy et al., 2011; Zachariah et al., 2012).
Over the last decade, several cellular and mouse models have been developed (Bertulat et al., 2012; Calfa et al., 2011; Cheung et al., 2011; Delepine et al., 2013; Yazdani et al., 2012). Recently, primary fibroblasts from RTT patients highlighted a role of MeCP2 in stabilizing microtubule dynamics, explaining in part the observed dendritic abnormalities found in the absence of functional MeCP2 (Delepine et al., 2013).
Mouse models, in which the Mecp2 allele has been modified to prevent production of a fully functional Mecp2 protein, have been established. Mice range from Mecp2-null mutations to specific point mutations mimicking those observed in humans, phenocopying several motor and cognitive features of RTT patients (Chen et al., 2001; Guy et al., 2001; Moretti et al., 2005, 2006; Picker et al., 2006; Santos et al., 2007; Shahbazian et al., 2002). Although mice cannot model all aspects of the human RTT, certainly they recapitulate many features of the disease and are generally accepted as excellent tools to study MeCP2 function (Ricceri et al., 2008). Although no treatments able to fully arrest or rescue the neurological regression are to date available for the human disease, intriguingly, delayed reintroduction of Mecp2 into fully affected Mecp2-null mice is sufficient to rescue RTT-like phenotypes (Guy et al., 2007; Robinson et al., 2012). Restoration of Mecp2 function in astrocytes alone significantly improves the developmental outcome of Mecp2-null mice (Lioy et al., 2011). These findings strongly indicate that the RTT phenotype is reversible upon restoration of Mecp2 function. A recent report on the feasibility of a systemic delivery of Mecp2, rescuing behavioral and cellular deficits in female mouse model of RTT strongly supports this point (Garg et al., 2013). In this scenario, microglia was shown to be a major player in the pathophysiology of RTT, thus suggesting that bone marrow transplantation might offer a feasible therapeutic approach for this disorder (Derecki et al., 2012, 2013).
The occurrence of a redox imbalance in RTT has been previously reported both in patients (De Felice et al., 2009, 2011; Durand et al., 2013; Grillo et al., 2013; Leoncini et al., 2011; Pecorelli et al., 2011; Sierra et al., 2001; Signorini et al., 2011) and in an experimental mouse model (Grosser et al., 2012). However, a clear evidence of oxidative damage in the brain, the key organ in this neurodevelopmental disease, is still lacking to date.
Oxidative stress is a condition in which the free radical insult is predominant on the antioxidant defense, with a consequent oxidative-mediated damage of biomolecules known to be relevant in different pathologies (Halliwell and Gutteridge, 2007). To this regard, in the brain, given its high content in lipids, the lipid peroxidation end-products isoprostanes (IsoPs) have a major pathogenetic relevance. IsoPs are a unique series of prostaglandin-like compounds generated, via a free radical-catalyzed mechanism, from a number of different polyunsaturated fatty acids (PUFAs), including arachidonic acid (ARA), eicosapentaenoic acid (EPA), adrenic acid (AdA), and docosahexaenoic acid (DHA). Plasma F2-IsoPs originating from ARA are considered as an index of generalized lipid peroxidation, whereas the IsoPs originating from DHA are usually termed F4-NeuroPs due to its main localization in the nervous tissue. F2-dihomo-IsoPs, deriving from Ada oxidation, have been characterized as potential markers of free radical damage to the myelin in the human brain (Signorini et al., 2013). All types of IsoPs can been evaluated in their esterified form at the cellular site to supply specific information on the lipid cell oxidation, and IsoPs have been extensively investigated in neurological disease (Durand et al., 2013; Singh et al., 2010). At the same time, redox active iron, such as the non-protein-bound iron (NPBI), is considered a trigger of free radical reaction and the relevance of the iron homeostasis in the brain pathologies is well documented (Rouault, 2013; Schroder et al., 2013).
As for isoprostanes, there are also numerous findings supporting the important presence of 4-hydroxy-2-nonenal (4-HNE) protein adducts in many oxidative stress related neurological diseases. For example, increased 4-HNE levels have been observed in the brain tissue from patients with Alzheimer's disease, Pick's disease, Lewy bodies related diseases, amyotrophic lateral sclerosis, Huntington's disease and Parkinson's disease, indicating therefore, a pathophysiological role of this aldehyde and its ability to form protein adducts in several pathologies (Poli et al., 2008). Moreover, a marked increase of 4-HNE was also detectable in the blood of patients with neurodegenerative and neuropsychiatric diseases (Pecorelli et al., 2013; Poli et al., 2008; Valacchi et al., 2014), confirming that this is a reliable marker of oxidative stress not only at the tissue levels, but also at the systemic level.
In the present study we investigated the relationship between oxidative damage and phenotypic expression of RTT, by assessing several oxidative stress (OS) markers in whole brain tissues from different Mecp2 mutant experimental models, as well as in a model of brain specific reactivation of Mecp2.
Materials and methods
Breeding
Mecp2 −/y (B6.129P(C) −Mecp2tm1.1Bird/J Jax stock number: 003890), Mecp2-308 (B6.129S-Mecp2tm1Hzo/J Jax stock number: 005439), Mecp2 stop/y (B6.129P2-Mecp2tm2Bird/J Jax stock number: 006849) and NestinCre mice (B6.Cg-Tg(Nest-cre)1Jln/J Jax stock number: 003771) all back crossed to C57BL6/J for at least 12 generations were maintained under standard conditions and in accordance with Home Office regulations and licenses.
Mecp2 mutant hemizygous males and heterozygous females were obtained by mating heterozygous females with wt males. Wild type littermates were used as controls. Mecp2 stop/y NestinCre males were produced by mating heterozygous Mecp2 +/stop females with hemizygous NestinCre males.
The animals were sacrificed and the tissues were recovered and stored at − 80 °C. The national or institutional guidelines were used for the care and use of animals, and approval for the experiments were obtained from the ethical committees of the Italian Ministry of Health, and the UK Home Office.
Genotyping
Genomic DNA was extracted from ear clips or tail tips of pups. The genotype of the mice was determined by polymerase chain reaction using PCR primers and following the conditions described in the web site of the Jackson Laboratories (USA).
Scoring of symptoms
Mice were scored on a weekly basis for a number of symptoms arising from Mecp2 deficiency as previously reported (Guy et al., 2007). Phenotype severity was expressed as aggregate score.
Blood sampling
Blood was collected in heparinized tubes and all manipulations were carried out within 2 h after sample collection. The blood samples were centrifuged at 2400 g for 15 min at 4 °C and plasma was collected. Butylated hydroxytoluene (BHT) (90 μM) was added to platelet poor plasma as an antioxidant. The resulting plasma samples, strictly hemoglobin-free, were stored at − 80 °C until assay. Plasma was used for free F2-IsoPs, F4-NeuroPs, and F2-dihomo-IsoPs determinations.
Brain collection
After transcardial perfusion with saline, brains were removed and bisected on the sagittal plane. Brain hemispheres were immediately frozen in dry ice and stored at − 80 °C until assay. At the time of the assays, brain was homogenized (10% W/V) in phosphate-buffered saline (PBS), pH 7.4. Brain homogenate was used for the determination of total (sum of free and esterified) F2-IsoPs, F4-NeuroPs, and F2-dihomo-IsoPs, as well as for NPBI quantification. Brain tissue lysates were also used for 4-HNE-PA adduct determination.
Indirect ImmunoFluorescence (IIF) analysis
Brains were dissected out, fixed in ethanol (60%), acetic acid (10%), and chloroform (30%), and included in paraffin. Paraffin embedded tissue sections of a thickness of 4 μm were deparaffinized in xylene and rehydrated in graded ethanol solutions (100%, 95%, 80% and 70%) for 5 min each.
Sections were rinsed twice in dH2O for 5 min each.
Briefly, antigen retrieval was obtained by incubation with buffer 10 mM citrate pH 6.0, at a temperature sub-boiling for 20 min. Slides were left to cool for 10 min.
After blocking with PBS containing 5% BSA for 60 min, the sections were incubated with the primary antibody (mouse anti-GFAP clone GA5 Millipore 1:200, mouse anti-βIII tubulin isoform clone TU-20 Millipore 1:50; rabbit anti-8 isoProstaglandin F2 alpha Abcam 1:200), overnight at 4 °C.
Incubation in secondary antibody fluorochrome conjugate (goat anti-rabbit Alexa Fluor 488, goat anti-mouse Alexa Fluor 568) diluted 1:100 in antibody dilution buffer was performed for 1 h at room temperature in the dark.
The nuclei were counterstained by incubating the sections for 10 min with 4′,6-diamidino-2-phenylindole (DAPI). Slides were washed with PBS, and mounted with Antifade. Negative controls were generated by omitting the primary antibody. The fluorescence was observed under a microscope Leica AF CTR6500HS (Microsystems).
Western blot analysis
Protein extracts for western blot analysis were obtained from whole brains. Tissues were collected in ice cold PBS, then homogenized in RIPA buffer (20 mM Tris–Cl pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mM EDTA, 0.1% SDS) with Protease Inhibitor Cocktail by Turrax homogenizer. After 20 min of incubation in ice, the homogenate was centrifuged at maximum speed for 20 min at 4 °C, and the supernatant was stored at − 80 °C. Protein extracts were run on 10% SDS-PAGE gel with 50 μg protein per lane. Western blot assays were performed with 1:2000 dilution of MeCP2 rabbit polyclonal antibody (Sigma-Aldrich, M9317). β-Actin rabbit polyclonal antibody (Sigma-Aldrich, 1:2500 dilution) was used as loading control. Following washes in PBS–Tween and incubation with specific secondary antibody (goat anti-rabbit horseradish peroxidase-conjugated, Santa Cruz Biotechnology Inc., CA, USA) for 1 h at RT, the membranes were incubated with Supersignal West Pico Chemiluminescent Substrate (Pierce Biotechnology, Rockford, USA). Signals were visualized on Amersham Hyperfilm ECL (GE Healthcare Europe GmbH, Milan, Italy).
Isoprostane and F4-neuroprostane determinations
All isoprostane and neuroprostane determinations were carried out by gas chromatography/negative ion chemical ionization tandem mass spectrometry (GC/NICI–MS/MS) analysis after solid phase extraction and derivatization steps.
Solid phase extraction and derivatization procedures
Each plasma sample was spiked with tetradeuterated prostaglandin F2α (PGF2α-d4) (500 pg in 50 ml of ethanol), as an internal standard. After acidification (2 ml of acidified water, pH 3), the extraction and purification procedures were carried out. It consisted of two solid-phase separation steps: an octadecylsilane (C18) cartridge followed by an aminopropyl (NH2) cartridge (Signorini et al., 2003). Each brain homogenate sample was purified as previously reported (Signorini et al., 2009). Briefly, to an aliquot (1 ml) of brain homogenate aqueous KOH (1 mM, 500 μl) was added. After incubation at 45 °C for 45 min, the pH was adjusted to 3 by adding HCl (1 mM, 500 μl). Each sample was spiked with tetradeuterated prostaglandin F2α (PGF2α-d4) (500 pg in 50 μl of ethanol), as an internal standard, and ethyl acetate (10 ml) was added to extract total lipids by vortex-mixing and centrifugation at 1000 g for 5 min at room temperature. The total lipid extract was applied onto an NH2 cartridge and isoprostanes were eluted.
For both plasma and brain eluted samples, the carboxylic group was derivatized as the pentafluorobenzyl ester, whereas the hydroxyl groups were converted to trimethylsilyl ethers (Signorini et al., 2003).
F2-isoprostane GC/NICI–MS/MS
The measured ions were the product ions at m/z 299 and m/z 303 derived from the [M − 181]− precursor ions (m/z 569 and m/z 573) produced from 15-F2t-IsoPs and the tetradeuterated derivative of prostaglandin F2α (PGF2α-d4), respectively (Signorini et al., 2003, 2009).
F4-NeuroPs GC/NICI–MS/MS
Quantification of F4-NeuroPs was performed by gas chromatography/negative ion chemical ionization tandem mass spectrometry (GC/NICI–MS/MS) according to a new method recently setup in our laboratory (Signorini et al., 2003, 2009). The measured ions were the product ions at m/z 323 and m/z 303 derived from the [M − 181]− precursor ions (m/z 593 and m/z 573) produced from oxidized DHA and the tetradeuterated derivative of PGF2α, respectively.
F2-dihomo-IsoPs GC/NICI–MS/MS
For F2-dihomo-IsoPs, the measured ions are the product ions at m/z 327 and m/z 303 derived from the [M − 181]− precursor ions (m/z 597 and m/z 573) produced from the derivatized ent-7(RS)-F2t-dihomo-IsoP and 17-F2t-dihomo-IsoP, and the PGF2α-d4, respectively (De Felice et al., 2011).
Non-protein-bound-iron determination
NPBI is a pro-oxidant factor, associated with hypoxia, hemoglobin oxidation and subsequent heme iron release (Ciccoli et al., 2008). Non-protein-bound-iron was determined as deferoxamine (DFO)–chelatable free iron (DFO–iron complex, ferrioxamine). DFO 25 μM was added to the brain homogenate. The homogenate was ultrafiltered in centrifugal filters with a 30-kDa molecular weight cut-off and the DFO excess removed by silica column chromatography. The DFO–iron complex was determined by high-performance liquid chromatography at the detection wavelength of 229 nm (Signorini et al., 2009).
4-HNE protein adducts
4-HNE PAs are markers of protein oxidation due to aldehyde binding from lipid peroxidation sources (Signorini et al., 2013). Brain 4-HNE protein adducts were determined by western blot technique. Brain tissue proteins (30 μg protein, as determined by using Bio-Rad protein assay; BioRad, Hercules, CA, USA) were resolved on 4–20% SDS-PAGE gels (Lonza Group Ltd., Switzerland) and transferred onto a hybond ECL nitrocellulose membrane (GE Healthcare Europe GmbH, Milan, Italy). After blocking in 3% non-fat milk (Bio-Rad, Hercules, CA, USA), the membranes were incubated overnight at 4 °C with goat polyclonal anti 4-HNE adduct antibody (cod. AB5605; Millipore Corporation, Billerica, MA, USA). Following washes in TBS–Tween and incubation with specific secondary antibody (mouse anti-goat horseradish peroxidase-conjugated, Santa Cruz Biotechnology Inc., CA, USA) for 1 h at RT, the membranes were incubated with ECL reagents (Bio-Rad, Hercules, CA, USA) for 1 min. The bands were visualized by autoradiography. Quantification of the relevant bands was performed by digitally scanning the Amersham Hyperfilm ECL (GE Healthcare Europe GmbH, Milan, Italy) and measuring immunoblotting image densities with ImageJ software.
Statistical analysis
Results were expressed as medians with inter-quartile ranges, or means ± SD. Differences between groups were evaluated by the non-parametric Mann–Whitney rank sum test, Wilcoxon rank test, or Kruskal–Wallis test analysis of variance (ANOVA), as appropriate. Associations between variables were tested by univariate regression analysis. Multiple of medians (MoMs) for the brain OS markers were used to account for the possible effect for potential sources of variation including inter- and intra-group differences in strain, age, diet or breeding. The MedCalc ver. 12.0 statistical software package (MedCalc. Software, Mariakerke, Belgium) was used for data analysis. A two-tailed P < 0.05 was considered to indicate statistical significance.
Results
The elevated concentrations of F2-isoprostanes (F2-IsoPs) in plasma of symptomatic Mecp2 −/y (median age 9 weeks) and hemizygous Mecp2 308/y mutated (median age 32 weeks) mice compared with wild type (wt) indicate the presence of a systemic OS status in the symptomatic phase of the disease (Figs. 1A–B), thus suggesting that these strains constitute reliable RTT animal models to further investigate the link between OS and Mecp2 deficiency.
Fig. 1.

Oxidative stress in plasma of two murine models of Rett syndrome. Plasma levels of F2-IsoPs are significantly increased in symptomatic Mecp2 −/y mice (N = 16, median age, M ± SD, age 9 ± 1.25 weeks) vs. matched wt littermates (N = 15, median age 9 ± 1.2 weeks) (A) and in symptomatic Mecp2 308/y (N = 9, median age 32 ± 11 weeks) vs. matched wt littermates (N = 10, median age 32 ± 11 weeks) (B). Data are expressed as medians (columns) and semi-interquartile range (bars). *P < 0.05; **P < 0.01.
In order to evaluate whether the oxidative damage observed in this peripheral body fluid is actually associated with oxidative damage in the brain, likely the main target organ of RTT given the major neurological dysfunctions in the patients, the following OS markers, in additions to F2-IsoPs were evaluated in whole brain from Mecp2 −/y 7 to 9 weeks symptomatic null mice (median age 9 weeks; mean aggregate score, M ± SD, 4.5 ± 0.43), and compared to age-matched wt littermates: non-protein-bound iron (NPBI), F2-dihomo-isoprostanes (F2-dihomo-IsoPs), F4-neuroprostanes (F4-NeuroPs) and 4-hydroxy-2-nonenal protein adducts (4-HNE PAs). The severity of the Mecp2-null phenotype was quantified using a simple phenotypic scoring method (Guy et al., 2007), which assesses a number of RTT like features seen in Mecp2 mutant mice. Significantly elevated NPBI, F2-IsoP, and F4-NeuroP levels were evident in the brain of symptomatic null mice as compared to wt, thus demonstrating the occurrence of brain oxidative damage in the symptomatic phase of the disease (Figs. 2A–C).
Fig. 2.

Evidence of oxidative brain damage in symptomatic Mecp2 −/y mice (N = 16, median age 9 weeks). Significant increased levels of NPBI (A), F2-IsoPs (B), and F4-NeuroPs (C) vs. matched wt littermates (N = 15, mean age 9 ± 1.2 weeks) are observed in whole brain tissue, whereas no significant changes in F2-dihomo-IsoPs (D), and 4-HNE PAs (E) are detected. Data are expressed as medians (columns) and semi-interquartile range (bars). *P < 0.05; **P < 0.01. N.S.: no significant differences (P > 0.05).
These data indicate that the oxidative damage is mainly the consequence of the peroxidation of arachidonic acid (ARA) and docosahexaenoic acid (DHA), i.e., fatty acid precursors of F2-IsoPs and F4-NeuroPs, respectively, as triggered by NPBI as pro-oxidant factor. On the other hand, no significant changes for F2-dihomo-IsoPs or 4-HNE PAs were detectable in this model at this disease stage (Figs. 2D–E; Supplementary Fig. 1A).
These data indicate that the oxidative damage is mainly the consequence of the peroxidation of arachidonic acid (ARA) and docosahexaenoic acid (DHA), i.e., fatty acid precursors of F2-IsoPs and F4-NeuroPs, respectively, as triggered by NPBI as pro-oxidant factor. On the other hand, no significant changes for F2-dihomo-IsoPs or 4-HNE PAs were detectable in this model at this disease stage (Figs. 2D–E; Supplementary Fig. 1A).
To better evaluate the cellular origin of the OS alteration, an immunohistochemical analysis with a specific F2-IsoP antibody was performed. The assay revealed a strong increase in F2-IsoPs in βIII tubulin positive cells (neurons, Fig. 3A) but not in glial fibrillary acidic protein (GFAP) positive cells (astroglia, Fig. 3B) of Mecp2 −/y mice compared to wt, thus indicating the presence of an oxidative damage in neuronal more than in astroglial cells.
Fig. 3.
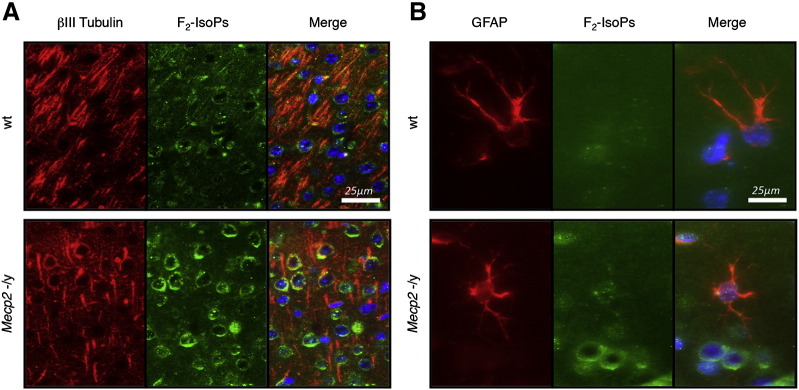
Double immunofluorescence in the brains of symptomatic Mecp2 −/y and matched wt littermates at 9 weeks, for F2-IsoPs (green)/βIII tubulin (red) (A) and for F2-IsoPs (green)/GFAP (red) (B). In the merge image the nuclei were identified by counterstaining with the nuclear marker DAPI (blue).
Significant inverse relationships of F4-NeuroPs with brain weight and body weight (Figs. 4A–B) were evidenced, suggesting an involvement of the DHA-derived peroxidation products in the pathogenesis of microcephaly and somatic growth deficiency in the Mecp2 −/y mouse model of RTT.
Fig. 4.

Inverse linear relationship of brain F4-NeuroPs vs. brain weight in symptomatic Mecp2 −/y mice (A) and of brain F4-NeuroPs vs. body weight in symptomatic Mecp2 −/y mice (B).
In order to evaluate the timing of the oxidative brain damage, we subsequently tested the same OS markers in whole brains from Mecp2 −/y 5 weeks pre-symptomatic null mice (median age 5 weeks; mean aggregate score 0.25 ± 0.25). As with the symptomatic null animals, brains of pre-symptomatic null mice also showed significantly increased NPBI, F2-IsoP, and F4-NeuroP tissue levels compared with wt, thus indicating that the oxidative brain damage, unlike other epiphenomena of the disease, precedes the onset of overt behavioral abnormalities (Figs. 5A–C). On the contrary, as observed in symptomatic mice, no statistical differences for F2-dihomo-IsoPs or 4-HNE PAs were observed (Figs. 5D–E; Supplementary Fig. 1B).
Fig. 5.

Evidence of oxidative brain damage in pre-symptomatic Mecp2 −/y mice vs. matched wt littermates with significant increase of NPBI (A), F2-IsoP (B), and F4-NeuroP (C) levels (N = 13, median age 5 weeks). No significant changes for F2-dihomo-IsoPs (D), and 4-HNE PAs (E) are observed. Data are expressed as medians (columns) and semi-interquartile range (bars). *P < 0.05; **P < 0.01. N.S.: no significant differences (P > 0.05).
In order to evaluate the timing of the oxidative brain damage, we subsequently tested the same OS markers in whole brains from Mecp2 −/y 5 weeks pre-symptomatic null mice (median age 5 weeks; mean aggregate score 0.25 ± 0.25). As with the symptomatic null animals, brains of pre-symptomatic null mice also showed significantly increased NPBI, F2-IsoP, and F4-NeuroP tissue levels compared with wt, thus indicating that the oxidative brain damage, unlike other epiphenomena of the disease, precedes the onset of overt behavioral abnormalities (Figs. 5A–C). On the contrary, as observed in symptomatic mice, no statistical differences for F2-dihomo-IsoPs or 4-HNE PAs were observed (Figs. 5D–E; Supplementary Fig. 1B).
These data were confirmed by OS marker analysis in an independent strain, in which the endogenous Mecp2 allele is silenced by a targeted stop cassette (Mecp2 stop/y) (Guy et al., 2007). Mecp2 stop/y mice are phenotypically equivalent to Mecp2 −/y animals and the observed residual expression of Mecp2 of around 2.5% compared with wt levels is not correlated with the severity of symptom progression (Robinson et al., 2012). Altered concentrations of NPBI, F2-IsoPs, and F4-NeuroPs are significantly detected in the brain at the pre-symptomatic stage (median age 5 weeks; mean aggregate score 0.25 ± 0.42) (Supplementary Figs. 2A–C), whereas no statistical differences were detectable regarding F2-dihomo-IsoPs or 4-HNE PAs (Supplementary Figs. 2D–E).
These data were confirmed by OS marker analysis in an independent strain, in which the endogenous Mecp2 allele is silenced by a targeted stop cassette (Mecp2 stop/y) (Guy et al., 2007). Mecp2 stop/y mice are phenotypically equivalent to Mecp2 −/y animals and the observed residual expression of Mecp2 of around 2.5% compared with wt levels is not correlated with the severity of symptom progression (Robinson et al., 2012). Altered concentrations of NPBI, F2-IsoPs, and F4-NeuroPs are significantly detected in the brain at the pre-symptomatic stage (median age 5 weeks; mean aggregate score 0.25 ± 0.42) (Supplementary Figs. 2A–C), whereas no statistical differences were detectable regarding F2-dihomo-IsoPs or 4-HNE PAs (Supplementary Figs. 2D–E).
We then evaluated OS alterations in a different RTT mouse model (Shahbazian et al., 2002), with a truncating mutation (Mecp2-308). In this specific RTT model, males show a much milder phenotype than human males with RTT-causing mutations, and heterozygous females also display a milder phenotype than that of RTT girls. These mutant mice live longer and are therefore easier to study as compared to the Mecp2-null models.
Therefore, OS markers were tested in the brain tissue of symptomatic Mecp2 308/y and Mecp2 308/x mice. Mecp2 308/y (median age 32 weeks) showed significant increase in NPBI, F2-IsoPs, F4-NeuroPs, and 4-HNE PAs as compared to wt mice (Figs. 6A–C, E; Supplementary Fig. 1C), thus confirming the relationship between the symptomatic phase of the disease and the fatty acid peroxidation leading to lipid and protein damage. On the other hand, no statistical difference for F2-dihomo-IsoPs was detectable (Fig. 6D).
Fig. 6.

Evidence of oxidative brain damage in symptomatic hemizygous male Mecp2-308 mutated mice vs. matched wt littermates (N = 9, median age 32 weeks) showing significant increase of the assessed OS markers (A–C, E), with the single exception of F2-dihomo-IsoPs (D). Data are expressed as medians (columns) and semi-interquartile range (bars). *P < 0.05; **P < 0.01. N.S.: no significant differences (P > 0.05).
Therefore, OS markers were tested in the brain tissue of symptomatic Mecp2 308/y and Mecp2 308/x mice. Mecp2 308/y (median age 32 weeks) showed significant increase in NPBI, F2-IsoPs, F4-NeuroPs, and 4-HNE PAs as compared to wt mice (Figs. 6A–C, E; Supplementary Fig. 1C), thus confirming the relationship between the symptomatic phase of the disease and the fatty acid peroxidation leading to lipid and protein damage. On the other hand, no statistical difference for F2-dihomo-IsoPs was detectable (Fig. 6D).
Interestingly, heterozygous 308 mutated females (Mecp2 308/x), which exhibit a milder form of the disease with a delayed onset of the behavioral manifestations (median age 54 weeks), showed biochemical signs of oxidative brain damage limited to F2-IsoPs, and F4-NeuroPs (Figs. 7B–C), whereas no statistical differences were observed for NPBI, F2-dihomo-IsoPs, or 4-HNE PAs (Figs. 7A, D–E; Supplementary Fig. 1D).
Fig. 7.

Evidence of oxidative brain damage in symptomatic heterozygous female Mecp2-308 mutated mice vs. matched wt littermates (N = 5, median age 54 weeks) showing significant increase of F2-IsoPs (B), and F4-NeuroPs (C). No significant changes in NPBI (A), F2-dihomo-IsoPs (D), and 4-HNE PAs (E) are observed. Data are expressed as medians (columns) and semi-interquartile range (bars). *P < 0.01. N.S.: no significant differences (P > 0.05).
Interestingly, heterozygous 308 mutated females (Mecp2 308/x), which exhibit a milder form of the disease with a delayed onset of the behavioral manifestations (median age 54 weeks), showed biochemical signs of oxidative brain damage limited to F2-IsoPs, and F4-NeuroPs (Figs. 7B–C), whereas no statistical differences were observed for NPBI, F2-dihomo-IsoPs, or 4-HNE PAs (Figs. 7A, D–E; Supplementary Fig. 1D).
Likewise, brain oxidative damage precedes the symptomatic phase also in the Mecp2 308/x mice, given that presymptomatic animals (median age 22 weeks) show a significant increase in NPBI, F2-IsoPs, and F4-NeuroPs (Figs. 8A–C). In contrast, no significant differences for F2-dihomo-IsoPs, or 4-HNE PAs were observed between mutant mice and their wt counterparts (Figs. 8D–E).
Fig. 8.

Evidence of oxidative brain damage in pre-symptomatic heterozygous female Mecp2-308 mutated mice showing significant increase of NPBI (A), F2-IsoPs (B), and F4-NeuroPs (C) vs. matched wt littermates (N = 3, median age 22 weeks). No significant changes in F2-dihomo-IsoPs (D), and 4-HNE PAs (E) are observed. Data are expressed as medians (columns) and semi-interquartile range (bars). *P = 0.0339. N.S.: no significant differences (P > 0.05).
In order to compare the entity of the different oxidative events in the different Mecp2 mutant mouse models, the levels of each marker were expressed as a function of the median levels in the age-matched wt controls (multiple of medians of the wt, MoMs) (Supplementary Figs. 3A–E). Besides the need to level off the methodological variability, MoMs were used to account for potential confounders including inter- and intra-group differences in strain, age, diet or breeding. Normalized F2-IsoP levels were found to be significantly lower in the symptomatic Mecp2 308/x and in the presymptomatic Mecp2 −/y-null mice, thus indicating that brain ARA peroxidation is relatively lower in these models. Symptomatic Mecp2 308/y and Mecp2 stop/y show relatively higher brain levels of 4-HNE PAs, thus indicating that the oxidative protein damage consequent to aldehyde binding is increased in this murine models of the disease.
In order to compare the entity of the different oxidative events in the different Mecp2 mutant mouse models, the levels of each marker were expressed as a function of the median levels in the age-matched wt controls (multiple of medians of the wt, MoMs) (Supplementary Figs. 3A–E). Besides the need to level off the methodological variability, MoMs were used to account for potential confounders including inter- and intra-group differences in strain, age, diet or breeding. Normalized F2-IsoP levels were found to be significantly lower in the symptomatic Mecp2 308/x and in the presymptomatic Mecp2 −/y-null mice, thus indicating that brain ARA peroxidation is relatively lower in these models. Symptomatic Mecp2 308/y and Mecp2 stop/y show relatively higher brain levels of 4-HNE PAs, thus indicating that the oxidative protein damage consequent to aldehyde binding is increased in this murine models of the disease.
When comparing the differences between relative OS marker levels in the different mouse models, a trend just at the borders of the statistical significance was observed for F4-NeuroPs (ANOVA, P = 0.0587), whereas no significant differences were detectable for NPBI and F2-dihomo-IsoPs (Kruskal Wallis ANOVA, P = 0.2325 and, P = 0.1380, respectively).
No significant relationships between the entity of the brain oxidative damage (as expressed as MoMs for the age-matched wt control population) and the clinical phenotype severity, as expressed as aggregate score (Guy et al., 2007), were observed (r ≤ 0.3499; P ≤ 0.2010, data not shown).
In order to further test a potential cause–effect relationship between oxidative brain damage and Mecp2 loss-of-function, brain levels of OS markers were evaluated in brain specific Mecp2 rescued mice. Specifically, the endogenous Mecp2 allele silenced by a targeted stop cassette (Mecp2 stop/y) was activated specifically in the brain during embryogenesis by expressing the Cre recombinase under the control of Nestin promoter (Mecp2 stop/y NestinCre mice) (Tronche et al., 1999). As expected (Robinson et al., 2012), a variable residual expression of the Mecp2 protein was observed in the brain tissues of the symptomatic Mecp2 stop/y (mean age 17 weeks; median aggregate score 6.5 ± 0.7), whereas a normal or near to normal Mecp2 expression was detectable in the brain of rescued Mecp2 stop/y NestinCre animals (mean age 17 weeks; median aggregate score 0) (Fig. 9A).
Fig. 9.

Rescue of oxidative brain damage in Mecp2 stop/y NestinCre mice. Western blot analysis of Mecp2 protein in the brains of wt, wt-Cre, Mecp2 stop/y, and Mecp2 stop/y NestinCre mice. β-Actin was used as loading control (A). Analysis of NPBI, F2-IsoPs, F4-NeuroPs, F2-dihomo-IsoPs and 4-HNE PAs in the brains of wt-Cre (N = 5, median age 17 weeks), Mecp2 stop/y (N = 2, median age 17 weeks), and Mecp2 stop/y NestinCre (N = 6, median age 17 weeks) mice (B). OS markers are expressed as medians (columns) and semi-interquartile range (bars). ANOVA: Kruskal–Wallis analysis of variance. *P < 0.05. N.S.: no statistically significant differences (P > 0.05).
Symptomatic Mecp2 stop/y mice, like the Mecp2 −/y mice, showed oxidative damage in the brain, with NPBI, F2-IsoP, and F4-NeuroP levels being significantly elevated as compared to those of age-matched wt expressing Cre recombinase (wt-Cre) littermates, whereas no statistical differences were observed for F2-dihomo-IsoPs and 4-HNE PAs (Fig. 9B and Supplementary Fig. 3). On the other hand, rescued stop/y mice showed levels of brain OS comparable to those of age-matched wt littermates (Fig. 9B; Supplementary Fig. 1E), thus indicating a full rescue of the brain OS damage following brain specific Mecp2 gene reactivation, and demonstrating that the altered redox homeostasis at the brain level in this RTT murine model can be fully reversed following restoration of the Mecp2 function.
Symptomatic Mecp2 stop/y mice, like the Mecp2 −/y mice, showed oxidative damage in the brain, with NPBI, F2-IsoP, and F4-NeuroP levels being significantly elevated as compared to those of age-matched wt expressing Cre recombinase (wt-Cre) littermates, whereas no statistical differences were observed for F2-dihomo-IsoPs and 4-HNE PAs (Fig. 9B and Supplementary Fig. 3). On the other hand, rescued stop/y mice showed levels of brain OS comparable to those of age-matched wt littermates (Fig. 9B; Supplementary Fig. 1E), thus indicating a full rescue of the brain OS damage following brain specific Mecp2 gene reactivation, and demonstrating that the altered redox homeostasis at the brain level in this RTT murine model can be fully reversed following restoration of the Mecp2 function.
Discussion
OS has been widely implicated in several pathological conditions including neurological disease (Ferguson, 2010; Halliwell and Gutteridge, 2007; Praticò, 2010). Lipid peroxidation, a critical component of OS, is a process well known to induce oxidative damage to key cellular components, implicated in several diseases. In particular, free radicals and specifically reactive oxygen species (ROS) are able to attack polyunsaturated fatty acids (PUFAs) of cell membranes, thus generating the prostaglandin-like end-products IsoPs, along with a family of α,β-unsaturated reactive aldehydes, such as 4-HNE.
Isoprostanes are considered as the gold standard for the OS in vivo evaluation (Galano et al., 2013; Signorini et al., 2013). Specifically, F2-IsoPs are the oxidation end-products of ARA, a polyunsaturated fatty acid, abundant in both brain gray and white matter, F4-NeuroPs are the end-products of DHA, abundant in neuronal membranes, whereas F2-dihomo-IsoPs are known to derive from oxidation of AdA (De Felice et al., 2011), a fatty acid abundant in white matter, specifically myelin and can be considered a marker of white matter oxidative damage (Supplementary Fig. 4).
Isoprostanes are considered as the gold standard for the OS in vivo evaluation (Galano et al., 2013; Signorini et al., 2013). Specifically, F2-IsoPs are the oxidation end-products of ARA, a polyunsaturated fatty acid, abundant in both brain gray and white matter, F4-NeuroPs are the end-products of DHA, abundant in neuronal membranes, whereas F2-dihomo-IsoPs are known to derive from oxidation of AdA (De Felice et al., 2011), a fatty acid abundant in white matter, specifically myelin and can be considered a marker of white matter oxidative damage (Supplementary Fig. 4).
Our data, obtained in established mouse models of Rett syndrome, appear to be in line with the emerging view that a lipid abnormality may be key to the pathogenesis of the Rett syndrome (Buchovecky et al., 2013; De Felice et al., 2013; Nagy and Ackerman, 2013; Sticozzi et al., 2013). Of course, it should always be kept in mind that experimental models for a disease unavoidably carry intrinsic limitations related to inter-species differences with the mimicked human pathology. To this regard, a puzzling discrepancy with the behavior of the OS markers in blood samples from RTT patients is represented by the lack of changes in F2-dihomo-IsoP levels in the tested RTT mouse models (data not shown), which is in good agreement with the presence of increased level of F2-IsoPs in neurons but not in astroglia (Fig. 3), but is in contrast with the marked increase in F2-dihomo-IsoPs previously documented in plasma samples from patients at an early stage of the disease (De Felice et al., 2011).
Nonetheless, the data presented here point out to several interesting considerations:
-
i)
alterations of the redox balance have been confirmed in murine models of RTT. More importantly, imbalances of OS “gold standard” markers are well evident especially in neurons;
-
ii)
our findings indicate that an OS-driven brain damage occurs in two different mouse models of RTT: the Mecp2-null and Mecp2-308 animals. Thus, our findings further strengthen the above reported observations, having extended our investigation on OS markers to murine RTT models in which Mecp2 is hypofunctional, rather than limiting our studies to Mecp2-null mice in which the Mecp2 protein is totally absent (Katz et al., 2012);
-
iii)
brain oxidative damage precedes the clinical manifestations by several weeks in Mecp2 −/y, stop/y and Mecp2-308/x models, where we detected a significant brain redox alteration prior to symptoms onset. These data are consistent with a close relationship between Mecp2 deficiency and development of RTT, and indicate the existence of a phase of the disease in which biochemical signs of enhanced OS are present in the brain, well before the clinical signs of the pathology, although some clinical evidence suggests that the disease could start at birth or even prenatally (Leonard and Bower, 1998). Notably, prior experimental data obtained with a mouse model carrying Mecp2 T158A mutation suggest that the underlying deficits in neural activity precede the establishment of behavioral symptoms (Goffin et al., 2012). Furthermore, in vitro electrophysiological studies showed reduced cortical excitability in Mecp2 −/y mice even at 2–3 weeks of age, that is well before the onset of neurological symptoms (Dani et al., 2005);
-
iv)
in a translational perspective, these findings would strongly suggest that neurology of RTT girls may be abnormal long before the onset of clinical signs, in line with several clinical (Burford et al., 2003; Einspieler et al., 2005a,b; Marschik et al., 2011; Temudo et al., 2007) and preclinical evidence (De Filippis et al., 2010; Picker et al., 2006);
-
v)
the correction of Mecp2 deficient genotype in the rescued Mecp2 stop/y NestinCre animals, re-establish the correct level of IsoPs. With this experiment we can affirm that the OS imbalance is a reversible phenomenon, which may be corrected by the re-introduction of a functional MeCP2. Moreover, the re-expression of Mecp2 in a Nestin-driven manner strongly suggests that the brain OS imbalance is due to a neural specific impairment of Mecp2 function, although the underlying molecular mechanism is still obscure.
In fact, the occurrence of alterations in OS brain markers, here evidenced when the Mecp2 gene is knocked out/silenced or mutated, does not necessarily mean that redox control could be a new, direct, function for the Mecp2 protein; our data do only provide clear evidence that Mecp2 deficiency is associated with a brain redox abnormality, thus indicating that oxidative brain damage is a previously unrecognized hallmark feature of murine RTT, and suggesting that Mecp2 is likely involved in the protection of the brain from OS.
Given that a loss of Mecp2 likely leads to the dysregulation of thousands of genes (Chahrour et al., 2008), with all the complex downstream consequences of this, it is not possible, to date, to relate any specific phenotypic features to the increased OS marker levels in the brain and/or plasma of the affected animals. At the same time, it is undeniable that RTT patients and Mecp2 mutant animal models are facing remarkable breathing challenges, exemplified by recurrent apneas and breath-holds (De Felice et al., 2010; Ramirez et al., 2013), which ultimately lead to a clinical phenotype defined not only by complex genetic causes (Grillo et al., 2013), but also by a series of interacting mechanisms involving a variety of compensatory, synaptic and neuromodulatory alterations, as well as disturbed homeostasis and OS (Grosser et al., 2012; Ramirez et al., 2013). Since several receptors and ion-channels are known to be redox-modulated (Poli et al., 2008; Sticozzi et al., 2013), it is possible that the mitochondrial (Grosser et al., 2012) and redox changes (De Felice et al., 2009; Grosser et al., 2012) evidenced in patients and animal models could contribute to the hyperexcitability and diminished synaptic plasticity in MeCP2 deficiency.
The key role of OS mechanisms in determining some of the characteristic neurological features in RTT appears to be also confirmed by the recent report on reduction in neuronal hyperexcitability, improvement in synaptic short-term plasticity, and restoration of synaptic long-term potentiation in a Mecp2 null mouse model of the disease following the incubation of hippocampal slices with a free radical scavenger vitamin E derivative compound (Janc and Muller, 2014).
It is important to underline that biochemical signs of brain oxidative damage predate the onset of symptoms, including the respiratory features, in the examined mutant mice. Although human and experimental evidence indicate that Obstructive Sleep Apnea Hypopnea Syndrome and intermittent hypoxia can be associated with enhanced OS, conflicting reports exist (De Felice et al., in press and references therein). However, the relationships between apneas/upper airways obstruction/intermittent hypoxia and OS status in RTT patients appear to be limited to the generation of a pro-oxidant status, as indicated by a reported link between intraerythrocyte-NPBI, but not F2-IsoPs, and apneas (De Felice et al., in press). Therefore, it becomes clear that mechanisms other than apneas/intermittent hypoxia should be the major sources of enhanced OS in human RTT and, by inference, mouse models of the disease.
Taken as a whole, the oxidative hypothesis of RTT (De Felice et al., 2012b) would seem to explain several intriguing features of the human disease. For instance, the risk of OS-driven brain damage may represent one of the possible reasons why MeCP2 activity must be finely tuned and tightly regulated, including embryonal microRNA control (Han et al., 2013). In addition, the occurrence of biochemical signs of oxidative brain damage preceding the neurological symptoms may explain the inconsistency of the apparently normal developmental phase (i.e., latency period) before clinical onset in the RTT patients. Finally, the previously unrecognized key role of Mecp2 in the regulation of redox homeostasis could explain the potential reversibility of the disease following functional restoration of the Mecp2 protein.
Taken together, our data suggest the existence of a window in which an early OS-modulating therapy could reduce/limit phenotype severity. As there are no currently proven effective pharmacological therapies for human RTT that can either halt progression or reverse the neurological and cognitive abnormalities, although many strategies are ongoing (Panayotis et al., 2011) our findings could pave the way for an early OS-modulating intervention during the preclinical window in RTT. This concept is supported by a previous pilot study in RTT patients at an early stage of the disease using ω-3 PUFAs (De Felice et al., 2012a).
The following are the supplementary data related to this article.
Evidence of oxidative brain damage, i.e. 4-HNE PAs, in Mecp2 mutant mouse models. Panels show western blot from 6 representative samples (3 Mecp2 mutant mice vs. 3 matched wt littermates) for each animal model. Brain 4-HNE PAs in symptomatic Mecp2 −/y mice (median age 9 weeks) (A); brain 4-HNE PAs in pre-symptomatic Mecp2 −/y mice (median age 5 weeks) (B); brain 4-HNE PAs in symptomatic hemizygous male Mecp2-308 mutated mice (median age 32 weeks) (C); brain 4-HNE PAs in symptomatic heterozygous female Mecp2-308 mutated mice (median age 54 weeks) (D); brain 4-HNE PAs in Mecp2 stop/y NestinCre and Mecp2 stop/y mice (median age 17 weeks) (E).
Evidence of oxidative brain damage in pre-symptomatic Mecp2 stop/y mice at 5 weeks of age vs. matched wt littermates (N = 7, mean age 5 weeks), with significant increase of NPBI (A), F2-IsoPs (B), and F4-NeuroPs (C). No significant changes for F2-dihomo-IsoPs (D), and 4-HNE PAs (E) are observed. Data are expressed as medians (columns) and semi-interquartile range (bars). *P < 0.05; **P < 0.01; ***P < 0.001. N.S.: no statistically significant differences (P > 0.05).
Differences in the entity of oxidative stress in the brain as a function of different murine models of Rett syndrome. F2-IsoPs (B) are significantly increased in the group formed by pre-symptomatic Mecp2 stop/y 5 weeks, symptomatic Mecp2 −/y 7–9 weeks, symptomatic Mecp2 stop/y 17 weeks, and symptomatic hemizygous male Mecp2 308/y 32 weeks as compared to pre-symptomatic Mecp2 −/y 5 weeks, symptomatic heterozygous female 308/x 54 weeks and pre-symptomatic female 308/x 22 weeks (Kruskal–Wallis ANOVA, P = 0.0025). 4-HNE PAs (E) are significantly higher in the symptomatic Mecp2 stop/y 17 weeks and symptomatic hemizygous male Mecp2 308/y 32 weeks as compared to other murine models of RTT (P = 0.0276). A trend just above the statistical significance threshold was observed for F4-NeuroPs (C) (P = 0.0587), whereas no significant differences for NPBI (A) (P = 0.2325) and F2-dihomo-IsoPs (D) (P = 0.1380) were detectable.
Data are expressed as medians (columns) and semi-interquartile range (bars) of multiple of medians (MoMs) for the corresponding wt littermate groups in order to account for potential confounder variables including age, strain, breeding, or diet. *P < 0.05.
Biosynthesis of isoprostanes and neuroprostanes. The metabolism and structural rearrangement of the main fatty acid precursors leading to oxidized products, including some of the isoprostane families examined in the present study, under the action of reactive oxygen species (ROS) are indicated.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.nbd.2014.04.006.
Author contributions
Concept: CDF, CS, SL, MDE, SF, and JH.
Experimental design: CDF, CS, SL, MDE, FDR, SF, and JH.
Mecp2-null mice breeding and sample collection: JG, SF, FDR, FS, MDE, FM, and MM.
Mecp2-308 truncated mice breeding and sample collection: LR, BDF, and GL.
Sample (brain and blood) preparation: JG, CS, SL, AP, FS, FM, and MM.
Isoprostane and neuroprostane assays: CS and LC.
NPBI assays: SL and LC.
4-HNE-PA assays: AP, LC, and GV.
Isoprostane synthesis: TD, CO, JMG, AG, and VBP.
Double fluorescence immunostaining: GB, AP, CS, and GV.
Data analysis: CDF, CS, SL, FDR, and BDF.
Data interpretation: All the Authors.
Manuscript drafting: all the authors.
Approval: all the authors.
Conflict of interest
The authors have declared that no conflict of interest exists.
Acknowledgments
The present research project has been funded by the Tuscany Region (Bando Salute 2009 project no. TR142, Italy); and Italian Association for Rett Syndrome (AIR; call 2011). It was also funded by the UE Initial Training Network project no. 238242 “DISCHROM”, by the EPIGENOMICS flagship project EPIGEN, MIUR-CNR to MDE, and by the IRE-IFO (RF 2008) “MECP2 phosphorylation and related kinase in Rett syndrome” to GL.
We sincerely thank the Round Table Club 41 and the Kiwanis Club of Siena for donations and continued support.
We heartily thank professional singer Matteo Setti (www.matteosetti.com) for his many charity concerts and continued interest in the scientific aspects of our research in Rett syndrome.
We are very grateful for a generous anonymous donation used to purchase some of the experimental mice evaluated in this study and to Dr. Pierluigi Tosi, Dr. Silvia Briani and Dr. Roberta Croci from the Administrative Direction of the Azienda Ospedaliera Senese for continued support to our studies and prior purchasing of the gas spectrometry instrumentation.
This research is dedicated to all the Rett girls and their families who represented the true inspiration for our research.
Contributor Information
Claudio De Felice, Email: geniente@gmail.com.
Maurizio D'Esposito, Email: maurizio.desposito@igb.cnr.it.
References
- Amir R.E., Van den Veyver I.B., Wan M., Tran C.Q., Francke U., Zoghbi H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Bertulat B., De Bonis M.L., Della Ragione F., Lehmkuhl A., Milden M., Storm C., Jost K.L., Scala S., Hendrich B., D'Esposito M., Cardoso M.C. MeCP2 dependent heterochromatin reorganization during neural differentiation of a novel Mecp2-deficient embryonic stem cell reporter line. PLoS One. 2012;7:e47848. doi: 10.1371/journal.pone.0047848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu T., Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat. Rev. Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Buchovecky C.M., Turley S.D., Brown H.M., Kyle S.M., McDonald J.G., Liu B., Pieper A.A., Huang W., Katz D.M., Russell D.W., Shendure J., Justice M.J. A suppressor screen in Mecp2 mutant mice implicates cholesterol metabolism in Rett syndrome. Nat. Genet. 2013;45:1013–1020. doi: 10.1038/ng.2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burford B., Kerr A.M., Macleod H.A. Nurse recognition of early deviation in development in home videos of infants with Rett disorder. J. Intellect. Disabil. Res. 2003;47:588–596. doi: 10.1046/j.1365-2788.2003.00476.x. [DOI] [PubMed] [Google Scholar]
- Calfa G., Percy A.K., Pozzo-Miller L. Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp. Biol. Med. (Maywood) 2011;236:3–19. doi: 10.1258/ebm.2010.010261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M., Zoghbi H.Y. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M., Jung S.Y., Shaw C., Zhou X., Wong S.T., Qin J., Zoghbi H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R.Z., Akbarian S., Tudor M., Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- Cheung A.Y., Horvath L.M., Grafodatskaya D., Pasceri P., Weksberg R., Hotta A., Carrel L., Ellis J. Isolation of MECP2-null Rett syndrome patient hiPS cells and isogenic controls through X-chromosome inactivation. Hum. Mol. Genet. 2011;20:2103–2115. doi: 10.1093/hmg/ddr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccoli L., Leoncini S., Signorini C., Comporti M. Iron and erythrocytes: physiological and pathophysiological aspects. In: Valacchi G., Davis P., editors. Oxidant in Biology: A Question of Balance. Springer; 2008. pp. 167–181. [Google Scholar]
- Dani V.S., Chang Q., Maffei A., Turrigiano G.G., Jaenisch R., Nelson S.B. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U. S. A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice C., Ciccoli L., Leoncini S., Signorini C., Rossi M., Vannuccini L., Guazzi G., Latini G., Comporti M., Valacchi G., Hayek J. Systemic oxidative stress in classic Rett syndrome. Free Radic. Biol. Med. 2009;47:440–448. doi: 10.1016/j.freeradbiomed.2009.05.016. [DOI] [PubMed] [Google Scholar]
- De Felice C., Guazzi G., Rossi M., Ciccoli L., Signorini C., Leoncini S., Tonni G., Latini G., Valacchi G., Hayek J. Unrecognized lung disease in classic Rett syndrome: a physiologic and high-resolution CT imaging study. Chest. 2010;138:386–392. doi: 10.1378/chest.09-3021. [DOI] [PubMed] [Google Scholar]
- De Felice C., Signorini C., Durand T., Oger C., Guy A., Bultel-Ponce V., Galano J.M., Ciccoli L., Leoncini S., D'Esposito M., Filosa S., Pecorelli A., Valacchi G., Hayek J. F2-dihomo-isoprostanes as potential early biomarkers of lipid oxidative damage in Rett syndrome. J. Lipid Res. 2011;52:2287–2297. doi: 10.1194/jlr.P017798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice C., Signorini C., Durand T., Ciccoli L., Leoncini S., D'Esposito M., Filosa S., Oger C., Guy A., Bultel-Ponce V., Galano J.M., Pecorelli A., De Felice L., Valacchi G., Hayek J. Partial rescue of Rett syndrome by omega-3 polyunsaturated fatty acids (PUFAs) oil. Genes Nutr. 2012;7:447–458. doi: 10.1007/s12263-012-0285-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice C., Signorini C., Leoncini S., Pecorelli A., Durand T., Valacchi G., Ciccoli L., Hayek J. The role of oxidative stress in Rett syndrome: an overview. Ann. N. Y. Acad. Sci. 2012;1259:121–135. doi: 10.1111/j.1749-6632.2012.06611.x. [DOI] [PubMed] [Google Scholar]
- De Felice C., Signorini C., Leoncini S., Pecorelli A., Durand T., Galano J.M., Bultel-Poncé V., Guy A., Oger C., Zollo G., Valacchi G., Ciccoli L., Hayek J. Fatty acids and autism spectrum disorders: the Rett syndrome conundrum. Food Nutr. Sci. 2013:71–75. [Google Scholar]
- De Felice C., Rossi M., Leoncini S., Chisci G., Signorini C., Lonetti G., Vannuccini L., Spina D., Ginori A., Iacona I., Cortelazzo A., Pecorelli A., Valacchi G., Ciccoli L., Pizzorusso T., Hayek J. Inflammatory lung disease in Rett syndrome. Mediat. Inflamm. 2014;2014:560120. doi: 10.1155/2014/560120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis B., Ricceri L., Laviola G. Early postnatal behavioral changes in the Mecp2-308 truncation mouse model of Rett syndrome. Genes Brain Behav. 2010;9:213–223. doi: 10.1111/j.1601-183X.2009.00551.x. [DOI] [PubMed] [Google Scholar]
- Delepine C., Nectoux J., Bahi-Buisson N., Chelly J., Bienvenu T. MeCP2 deficiency is associated with impaired microtubule stability. FEBS Lett. 2013;587:245–253. doi: 10.1016/j.febslet.2012.11.033. [DOI] [PubMed] [Google Scholar]
- Derecki N.C., Cronk J.C., Lu Z., Xu E., Abbott S.B., Guyenet P.G., Kipnis J. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–109. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derecki N.C., Cronk J.C., Kipnis J. The role of microglia in brain maintenance: implications for Rett syndrome. Trends Immunol. 2013;34:144–150. doi: 10.1016/j.it.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand T., De Felice C., Signorini C., Oger C., Bultel-Ponce V., Guy A., Galano J.M., Leoncini S., Ciccoli L., Pecorelli A., Valacchi G., Hayek J. F(2)-dihomo-isoprostanes and brain white matter damage in stage 1 Rett syndrome. Biochimie. 2013;95:86–90. doi: 10.1016/j.biochi.2012.09.017. [DOI] [PubMed] [Google Scholar]
- Einspieler C., Kerr A.M., Prechtl H.F. Abnormal general movements in girls with Rett disorder: the first four months of life. Brain Dev. 2005;27(Suppl. 1):S8–S13. doi: 10.1016/j.braindev.2005.03.014. [DOI] [PubMed] [Google Scholar]
- Einspieler C., Kerr A.M., Prechtl H.F. Is the early development of girls with Rett disorder really normal? Pediatr. Res. 2005;57:696–700. doi: 10.1203/01.PDR.0000155945.94249.0A. [DOI] [PubMed] [Google Scholar]
- Ferguson L.R. Chronic inflammation and mutagenesis. Mutat. Res. 2010;690:3–11. doi: 10.1016/j.mrfmmm.2010.03.007. [DOI] [PubMed] [Google Scholar]
- Galano J.M., Mas E., Barden A., Mori T.A., Signorini C., De Felice C., Barrett A., Opere C., Pinot E., Schwedhelm E., Benndorf R., Roy J., Le Guennec J.Y., Oger C., Durand T. Isoprostanes and neuroprostanes: total synthesis, biological activity and biomarkers of oxidative stress in humans. Prostaglandins Other Lipid Mediat. 2013;107:95–102. doi: 10.1016/j.prostaglandins.2013.04.003. [DOI] [PubMed] [Google Scholar]
- Garg S.K., Lioy D.T., Cheval H., McGann J.C., Bissonnette J.M., Murtha M.J., Foust K.D., Kaspar B.K., Bird A., Mandel G. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J. Neurosci. 2013;33:13612–13620. doi: 10.1523/JNEUROSCI.1854-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffin D., Allen M., Zhang L., Amorim M., Wang I.T., Reyes A.R., Mercado-Berton A., Ong C., Cohen S., Hu L., Blendy J.A., Carlson G.C., Siegel S.J., Greenberg M.E., Zhou Z. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat. Neurosci. 2012;15:274–283. doi: 10.1038/nn.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillo E., Lo Rizzo C., Bianciardi L., Bizzarri V., Baldassarri M., Spiga O., Furini S., De Felice C., Signorini C., Leoncini S., Pecorelli A., Ciccoli L., Mencarelli M.A., Hayek J., Meloni I., Ariani F., Mari F., Renieri A. Revealing the complexity of a monogenic disease: Rett syndrome exome sequencing. PLoS One. 2013;8:e56599. doi: 10.1371/journal.pone.0056599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosser E., Hirt U., Janc O.A., Menzfeld C., Fischer M., Kempkes B., Vogelgesang S., Manzke T.U., Opitz L., Salinas-Riester G., Muller M. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol. Dis. 2012;48:102–114. doi: 10.1016/j.nbd.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Guy J., Hendrich B., Holmes M., Martin J.E., Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J., Gan J., Selfridge J., Cobb S., Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J., Cheval H., Selfridge J., Bird A. The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- Hagberg B. Clinical manifestations and stages of Rett syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:61–65. doi: 10.1002/mrdd.10020. [DOI] [PubMed] [Google Scholar]
- Halliwell B., Gutteridge J. Fourth edition. Oxford University Press; 2007. Free Radicals in Biology and Medicine. [Google Scholar]
- Han K., Gennarino V.A., Lee Y., Pang K., Hashimoto-Torii K., Choufani S., Raju C.S., Oldham M.C., Weksberg R., Rakic P., Liu Z., Zoghbi H.Y. Human-specific regulation of MeCP2 levels in fetal brains by microRNA miR-483-5p. Genes Dev. 2013;27:485–490. doi: 10.1101/gad.207456.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janc O.A., Muller M. The free radical scavenger Trolox dampens neuronal hyperexcitability, reinstates synaptic plasticity, and improves hypoxia tolerance in a mouse model of Rett syndrome. Front. Cell. Neurosci. 2014;8:56. doi: 10.3389/fncel.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.L., Veenstra G.J., Wade P.A., Vermaak D., Kass S.U., Landsberger N., Strouboulis J., Wolffe A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Katz D.M., Berger-Sweeney J.E., Eubanks J.H., Justice M.J., Neul J.L., Pozzo-Miller L., Blue M.E., Christian D., Crawley J.N., Giustetto M., Guy J., Howell C.J., Kron M., Nelson S.B., Samaco R.C., Schaevitz L.R., St Hillaire-Clarke C., Young J.L., Zoghbi H.Y., Mamounas L.A. Preclinical research in Rett syndrome: setting the foundation for translational success. Dis. Model Mech. 2012;5:733–745. doi: 10.1242/dmm.011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard H., Bower C. Is the girl with Rett syndrome normal at birth? Dev. Med. Child Neurol. 1998;40:115–121. [PubMed] [Google Scholar]
- Leoncini S., De Felice C., Signorini C., Pecorelli A., Durand T., Valacchi G., Ciccoli L., Hayek J. Oxidative stress in Rett syndrome: natural history, genotype, and variants. Redox Rep. 2011;16:145–153. doi: 10.1179/1351000211Y.0000000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy D.T., Garg S.K., Monaghan C.E., Raber J., Foust K.D., Kaspar B.K., Hirrlinger P.G., Kirchhoff F., Bissonnette J.M., Ballas N., Mandel G. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschik P.B., Lanator I., Freilinger M., Prechtl H.F.R., Einspieler C. Early signs and later neurophysiological correlates of Rett syndrome. Klin. Neurophysiol. 2011;42:22–26. [Google Scholar]
- Moretti P., Bouwknecht J.A., Teague R., Paylor R., Zoghbi H.Y. Abnormalities of social interactions and home-cage behavior in a mouse model of Rett syndrome. Hum. Mol. Genet. 2005;14:205–220. doi: 10.1093/hmg/ddi016. [DOI] [PubMed] [Google Scholar]
- Moretti P., Levenson J.M., Battaglia F., Atkinson R., Teague R., Antalffy B., Armstrong D., Arancio O., Sweatt J.D., Zoghbi H.Y. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J. Neurosci. 2006;26:319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G., Ackerman S.L. Cholesterol metabolism and Rett syndrome pathogenesis. Nat. Genet. 2013;45:965–967. doi: 10.1038/ng.2738. [DOI] [PubMed] [Google Scholar]
- Neul J.L., Kaufmann W.E., Glaze D.G., Christodoulou J., Clarke A.J., Bahi-Buisson N., Leonard H., Bailey M.E., Schanen N.C., Zappella M., Renieri A., Huppke P., Percy A.K. Rett syndrome: revised diagnostic criteria and nomenclature. Ann. Neurol. 2010;68:944–950. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panayotis N., Pratte M., Borges-Correia A., Ghata A., Villard L., Roux J.C. Morphological and functional alterations in the substantia nigra pars compacta of the Mecp2-null mouse. Neurobiol. Dis. 2011;41:385–397. doi: 10.1016/j.nbd.2010.10.006. [DOI] [PubMed] [Google Scholar]
- Pecorelli A., Ciccoli L., Signorini C., Leoncini S., Giardini A., D'Esposito M., Filosa S., Hayek J., De Felice C., Valacchi G. Increased levels of 4HNE-protein plasma adducts in Rett syndrome. Clin. Biochem. 2011;44:368–371. doi: 10.1016/j.clinbiochem.2011.01.007. [DOI] [PubMed] [Google Scholar]
- Pecorelli A., Leoncini S., De Felice C., Signorini C., Cerrone C., Valacchi G., Ciccoli L., Hayek J. Non-protein-bound iron and 4-hydroxynonenal protein adducts in classic autism. Brain Dev. 2013;35:146–154. doi: 10.1016/j.braindev.2012.03.011. [DOI] [PubMed] [Google Scholar]
- Picker J.D., Yang R., Ricceri L., Berger-Sweeney J. An altered neonatal behavioral phenotype in Mecp2 mutant mice. Neuroreport. 2006;17:541–544. doi: 10.1097/01.wnr.0000208995.38695.2f. [DOI] [PubMed] [Google Scholar]
- Poli G., Schaur R.J., Siems W.G., Leonarduzzi G. 4-Hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- Praticò D. The neurobiology of isoprostanes and Alzheimer's disease. Biochim. Biophys. Acta. 2010;1801:930–933. doi: 10.1016/j.bbalip.2010.01.009. [DOI] [PubMed] [Google Scholar]
- Ramirez J.M., Ward C.S., Neul J.L. Breathing challenges in Rett syndrome: lessons learned from humans and animal models. Respir. Physiol. Neurobiol. 2013;189:280–287. doi: 10.1016/j.resp.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 1966;116:723–726. [PubMed] [Google Scholar]
- Ricceri L., De Filippis B., Laviola G. Mouse models of Rett syndrome: from behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav. Pharmacol. 2008;19:501–517. doi: 10.1097/FBP.0b013e32830c3645. [DOI] [PubMed] [Google Scholar]
- Robinson L., Guy J., McKay L., Brockett E., Spike R.C., Selfridge J., De Sousa D., Merusi C., Riedel G., Bird A., Cobb S.R. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain. 2012;135:2699–2710. doi: 10.1093/brain/aws096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault T.A. Iron metabolism in the CNS: implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013;14:551–564. doi: 10.1038/nrn3453. [DOI] [PubMed] [Google Scholar]
- Santos M., Silva-Fernandes A., Oliveira P., Sousa N., Maciel P. Evidence for abnormal early development in a mouse model of Rett syndrome. Genes Brain Behav. 2007;6:277–286. doi: 10.1111/j.1601-183X.2006.00258.x. [DOI] [PubMed] [Google Scholar]
- Schroder N., Figueiredo L.S., de Lima M.N. Role of brain iron accumulation in cognitive dysfunction: evidence from animal models and human studies. J. Alzheimer's Dis. 2013;34:797–812. doi: 10.3233/JAD-121996. [DOI] [PubMed] [Google Scholar]
- Shahbazian M., Young J., Yuva-Paylor L., Spencer C., Antalffy B., Noebels J., Armstrong D., Paylor R., Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- Sierra C., Vilaseca M.A., Brandi N., Artuch R., Mira A., Nieto M., Pineda M. Oxidative stress in Rett syndrome. Brain Dev. 2001;23(Suppl. 1):S236–S239. doi: 10.1016/s0387-7604(01)00369-2. [DOI] [PubMed] [Google Scholar]
- Signorini C., Comporti M., Giorgi G. Ion trap tandem mass spectrometric determination of F2-isoprostanes. J. Mass Spectrom. 2003;38:1067–1074. doi: 10.1002/jms.520. [DOI] [PubMed] [Google Scholar]
- Signorini C., Ciccoli L., Leoncini S., Carloni S., Perrone S., Comporti M., Balduini W., Buonocore G. Free iron, total F-isoprostanes and total F-neuroprostanes in a model of neonatal hypoxic–ischemic encephalopathy: neuroprotective effect of melatonin. J. Pineal Res. 2009;46:148–154. doi: 10.1111/j.1600-079X.2008.00639.x. [DOI] [PubMed] [Google Scholar]
- Signorini C., De Felice C., Leoncini S., Giardini A., D'Esposito M., Filosa S., Della Ragione F., Rossi M., Pecorelli A., Valacchi G., Ciccoli L., Hayek J. F(4)-neuroprostanes mediate neurological severity in Rett syndrome. Clin. Chim. Acta. 2011;412:1399–1406. doi: 10.1016/j.cca.2011.04.016. [DOI] [PubMed] [Google Scholar]
- Signorini C., De Felice C., Durand T., Oger C., Galano J.M., Leoncini S., Pecorelli A., Valacchi G., Ciccoli L., Hayek J. Isoprostanes and 4-hydroxy-2-nonenal: markers or mediators of disease? Focus on Rett syndrome as a model of autism spectrum disorder. Oxid. Med. Cell. Longev. 2013;2013:343824. doi: 10.1155/2013/343824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M., Dang T.N., Arseneault M., Ramassamy C. Role of by-products of lipid oxidation in Alzheimer's disease brain: a focus on acrolein. J. Alzheimer's Dis. 2010;21:741–756. doi: 10.3233/JAD-2010-100405. [DOI] [PubMed] [Google Scholar]
- Sticozzi C., Belmonte G., Pecorelli A., Cervellati F., Leoncini S., Signorini C., Ciccoli L., De Felice C., Hayek J., Valacchi G. Scavenger receptor B1 post-translational modifications in Rett syndrome. FEBS Lett. 2013;587:2199–2204. doi: 10.1016/j.febslet.2013.05.042. [DOI] [PubMed] [Google Scholar]
- Temudo T., Maciel P., Sequeiros J. Abnormal movements in Rett syndrome are present before the regression period: a case study. Mov. Disord. 2007;22:2284–2287. doi: 10.1002/mds.21744. [DOI] [PubMed] [Google Scholar]
- Tronche F., Kellendonk C., Kretz O., Gass P., Anlag K., Orban P.C., Bock R., Klein R., Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Valacchi G., Pecorelli A., Signorini C., Leoncini S., Ciccoli L., De Felice C., Hayek J. 4HNE protein adducts in autistic spectrum disorders: Rett syndrome and autism. In: Patel V.B., P.V.R., Martin C.R., editors. Comprehensive Guide to Autism. Springer; New York: 2014. pp. 2667–2688. [Google Scholar]
- Weaving L.S., Ellaway C.J., Gecz J., Christodoulou J. Rett syndrome: clinical review and genetic update. J. Med. Genet. 2005;42:1–7. doi: 10.1136/jmg.2004.027730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdani M., Deogracias R., Guy J., Poot R.A., Bird A., Barde Y.A. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem Cells. 2012;30:2128–2139. doi: 10.1002/stem.1180. [DOI] [PubMed] [Google Scholar]
- Zachariah R.M., Olson C.O., Ezeonwuka C., Rastegar M. Novel MeCP2 isoform-specific antibody reveals the endogenous MeCP2E1 expression in murine brain, primary neurons and astrocytes. PLoS One. 2012;7:e49763. doi: 10.1371/journal.pone.0049763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Evidence of oxidative brain damage, i.e. 4-HNE PAs, in Mecp2 mutant mouse models. Panels show western blot from 6 representative samples (3 Mecp2 mutant mice vs. 3 matched wt littermates) for each animal model. Brain 4-HNE PAs in symptomatic Mecp2 −/y mice (median age 9 weeks) (A); brain 4-HNE PAs in pre-symptomatic Mecp2 −/y mice (median age 5 weeks) (B); brain 4-HNE PAs in symptomatic hemizygous male Mecp2-308 mutated mice (median age 32 weeks) (C); brain 4-HNE PAs in symptomatic heterozygous female Mecp2-308 mutated mice (median age 54 weeks) (D); brain 4-HNE PAs in Mecp2 stop/y NestinCre and Mecp2 stop/y mice (median age 17 weeks) (E).
Evidence of oxidative brain damage in pre-symptomatic Mecp2 stop/y mice at 5 weeks of age vs. matched wt littermates (N = 7, mean age 5 weeks), with significant increase of NPBI (A), F2-IsoPs (B), and F4-NeuroPs (C). No significant changes for F2-dihomo-IsoPs (D), and 4-HNE PAs (E) are observed. Data are expressed as medians (columns) and semi-interquartile range (bars). *P < 0.05; **P < 0.01; ***P < 0.001. N.S.: no statistically significant differences (P > 0.05).
Differences in the entity of oxidative stress in the brain as a function of different murine models of Rett syndrome. F2-IsoPs (B) are significantly increased in the group formed by pre-symptomatic Mecp2 stop/y 5 weeks, symptomatic Mecp2 −/y 7–9 weeks, symptomatic Mecp2 stop/y 17 weeks, and symptomatic hemizygous male Mecp2 308/y 32 weeks as compared to pre-symptomatic Mecp2 −/y 5 weeks, symptomatic heterozygous female 308/x 54 weeks and pre-symptomatic female 308/x 22 weeks (Kruskal–Wallis ANOVA, P = 0.0025). 4-HNE PAs (E) are significantly higher in the symptomatic Mecp2 stop/y 17 weeks and symptomatic hemizygous male Mecp2 308/y 32 weeks as compared to other murine models of RTT (P = 0.0276). A trend just above the statistical significance threshold was observed for F4-NeuroPs (C) (P = 0.0587), whereas no significant differences for NPBI (A) (P = 0.2325) and F2-dihomo-IsoPs (D) (P = 0.1380) were detectable.
Data are expressed as medians (columns) and semi-interquartile range (bars) of multiple of medians (MoMs) for the corresponding wt littermate groups in order to account for potential confounder variables including age, strain, breeding, or diet. *P < 0.05.
Biosynthesis of isoprostanes and neuroprostanes. The metabolism and structural rearrangement of the main fatty acid precursors leading to oxidized products, including some of the isoprostane families examined in the present study, under the action of reactive oxygen species (ROS) are indicated.