

Abstract
The Noncanonical NF-κB pathway induces processing of the NF-κB2 precursor protein p100 and, thereby, mediates activation of p52-containing NF-κB complexes. This pathway is crucial for B-cell maturation and humoral immunity, but its role in regulating T-cell function is less clear. Using mutant mice that express a non-processible p100, NF-κB2lym1, we show that the noncanonical NF-κB pathway has a T cell-intrinsic role in regulating the pathogenesis of a T cell-mediated autoimmunity, experimental autoimmune encephalomyelitis (EAE). Although the lym1 mutation does not interfere with naïve T-cell activation, it renders the Th17 cells defective in the production of inflammatory effector molecules, particularly the cytokine GM-CSF. We provide evidence that p52 binds to the promoter of the GM-CSF-encoding gene (Csf2) and cooperates with c-Rel in the transactivation of this target gene. Introduction of exogenous p52 or GM-CSF to the NF-κB2lym1 mutant T cells partially restores their ability to induce EAE. These results suggest that the noncanonical NF-κB pathway mediates induction of EAE by regulating the effector function of inflammatory T cells.
Keywords: NF-κB2, p100 processing, noncanonical NF-κB, p52, NIK, EAE, GM-CSF
Introduction
NF-κB represents a family of structurally related transcription factors, which in mammalian cells includes NF-κB1, NF-κB2, RelA, RelB, and c-Rel (1). The NF-κB proteins function as various homo- and hetero-dimers that transactivate a large array of target genes involved in diverse aspects of immune functions, such as the development, activation, and differentiation of lymphocytes and innate immune cells (2, 3). NF-κB1 and NF-κB2 are translated as precursor proteins, p105 and p100, which generate mature NF-κB subunits, p50 and p52, upon proteasome-mediated processing (4). Under normal conditions, the NF-κB members are sequestered in the cytoplasm by inhibitory proteins, IκBs. The precursor proteins p105 and p100 also function as IκB-like molecules and regulate specific NF-κB members (4).
NF-κB activation is mediated by two major pathways, the canonical and noncanonical pathways, although other atypical pathways also exist (5). The canonical NF-κB relies on the IκB kinase (IKK) complex, composed of two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ (also called NEMO). In response to various immune stimuli, IKK phosphorylates the prototypical NF-κB inhibitor IκBα, causing its degradation and nuclear translocation of p50-containing NF-κB members, particularly the p50/RelA heterodimer (1). On the other hand, the noncanonical NF-κB pathway involves the inducible processing of p100, which requires the NF-κB inducing kinase (NIK) and its downstream kinase IKKα (5). In response to specific signals, NIK/IKKα mediate phosphorylation of two C-terminal residues of p100, Ser-862 and Ser-866, thereby triggering the ubiquitination and processing of p100 (6–8). At least in some cells types, such as B cells, p100 preferentially binds to RelB, and the p100 processing leads to generation of p52 and nuclear translocation of the p52/RelB heterodimer. However, since p52 also dimerize with other NF-κB members, the processing of p100 likely also mediates activation of additional p52-containing NF-κB complexes.
To date, the best-known functions of the noncanonical pathway include B-cell maturation, lymphoid organogenesis, and osteoclast differentiation (9). Nevertheless, emerging evidence suggests a role for this pathway in the regulation of T-cell functions (10–12). In particular, recent work from others and us reveals that mice deficient in NIK are refractory to the induction of a T cell-dependent autoimmunity, experimental autoimmune encephalomyelitis (EAE) (10, 13). However, it remains controversial whether such a function of NIK is T-cell intrinsic or extrinsic (10, 13). Moreover, NIK appears to regulate both noncanonical NF-κB and other signaling pathways, making the results complicated to interpret (10).
Recently, a mutant mouse strain, nfkb2lym1, was identified through chemical mutagenesis screening approach (14). These mice carry a point mutation in the nfkb2 gene, leading to the production of a p100 mutant (lym1) that lacks its C-terminal phosphorylation site and is incapable of undergoing phosphorylation-dependent processing. Both the nfkb2lym1/lym1 homozygous (hereafter called lym1/lym1) and the lym1/+ heterozygous mice display defect in B-cell maturation and osteoclastogenesis (14). The lym1 mice provide a powerful model system for studying the in vivo function of noncanonical NF-κB pathway in different cellular environments. In the present study, we employed the nfkb2lym1 mice to study the role of noncanonical NF-κB pathway in the regulation of T-cell activation and in vivo function. Our study clearly demonstrated a T cell-intrinsic role for this pathway in the regulation of the T cell-dependent autoimmunity EAE. Interestingly, although p100 processing is dispensable for naïve T-cell activation in vitro and T-cell priming in vivo, the lym1 effector T cells are defective in production of the pathogenic cytokine GM-CSF. We provide evidence that noncanonical NF-κB regulates the expression of GM-CSF and several other effector molecules involved in the pathogenesis of EAE.
Materials and Methods
Mice
Nfkb2lym1 mice (14) were provided were provided by Dr. Robyn Starr and The Walter and Eliza Hall Institute of Medical Research. Since both homozygous lym1/lym1 heterozygous lym1/+ mice have a severe defect in p100 processing (14), we used the lym1/+ and age-matched WT controls in the experiments. Mice with genetic ablation of the NIK-encoding gene Map3k14, Map3k14−/− and wild-type control, were provided by Amgen Inc and the Relb−/− mice were from Bristol-Myers Squibb Pharmaceutical Research Institute. Rag1−/− and Tcrb−/−Tcrd−/− mice were from Jackson Laboratory. Mice were maintained in a specific pathogen–free facility of The University of Texas MD Anderson Cancer Center, and all animal experiments were done in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Texas MD Anderson Cancer Center.
Plasmids, antibodies and reagents
pMIGR1-mP52 and pMIGR1-mGM-CSF were generated by inserting mouse p52 (encoding amino acids 1–405 of NF-κB2) and mouse Csf2 cDNAs, respectively, into the EcoRI and BglII sites of pMIGR1-GFP vector. pcDNA-based expression vectors encoding FLAG-tagged mouse p52 (p52-cFlag-pcDNA3), mouse c-Rel (c-Rel-cFlag-pcDNA3), and mouse RelB (RelB-cFlag-pcDNA3) were from Addgene. The Csf2 promoter luciferase plasmid (pGL3-mCsf2), covering the region −225 to +26 of mouse Csf2 promoter (15), was provided by Dr. Takeshi Matsumura (Kumamoto University).
Functional grade anti–mouse (m) CD3ɛ (145-2C11) and anti-mCD28 (37.51) antibodies and blocking antibodies for mIFN-γ (XMG1.2) and mIL-4 (11B11) were from eBioscience. Fluorescence-labeled antibodies for mCD4 (L3T4), mCD8 (53-6.7), mCD3 (145-2C11), CD44 (IM7), CD62L (MEL-14), IL-17A (eBio17B7), GM-CSF (MP1-22E9), and IFN-γ (XMG1.2) were purchased from eBioscience. Antibodies for mouse p50 (C-19), c-Rel (sc-70), RelB (C-19), NIK (H-248), Lamin B (C-20), and Hsp60 (H-1) were from Santa Cruz Biotechnology. An antibody recognizing both p100 and p52 (Anti-p52/p100) was provided by NCI Preclinical Repository.
Flow cytometry analysis and cell sorting
Single-cell suspensions of splenocytes were subjected to flow cytometry and cell sorting as previously described (16) using a FACSAria (BD Biosciences). For intracellular cytokine staining (ICS) assays, T cells were isolated from the spleen or central nervous system (CNS) (brain and spinal cord) of immunized mice or from in vitro cultures were stimulated for 4 hours with PMA (50 ng/mL) and ionomycin (500 ng/mL) in the presence of monensin (10 μg/mL). The stimulated cells were fixed in 2% paraformaldehyde and permeablized in 0.5% saponin and then subjected to cytokine staining and flow cytometry analyses.
Mixed Bone-Marrow Chimera
Rag1−/− mice were subjected to lethal-dose (992 rads) irradiation and, 1 day later, were adoptively transferred with bone-marrow cells harvested from the tibiae and femurs of the indicated mice. For mixed bone marrow transfers, an equal number (6.4 × 106) of bone marrow cells from the Rag1−/− and the WT or lym1/+ mice were mixed and injected intravenously into the irradiated Rag1−/− mice. After 6 weeks, the resulting chimeras were subjected to experimental analyses. Where indicated, the T cell deficient Tcrb−/−Tcrd−/− mice were used as recipient for adoptive transfer of mixed bone-marrow cells from lym1/+ and Tcrb−/−Tcrd−/− mice in a 1:4 ratio.
Induction and evaluation of EAE
The encephalitogenic peptide (residues 35–55, Met-Glu-Val-Gly-Trp-Tyr-Arg-Ser-Pro-Phe-Ser-Arg-Val-Val-His-Leu-Tyr-Arg-Asn-Gly-Lys) of myelin oligodendrocyte glycoprotein (MOG) was purchased from Genemed Synthesis Inc. (San Francisco, CA). To induce acute EAE, mice were injected s.c. (in the back region) with 200 μg of the MOG35–55 peptide in CFA containing 5 mg/mL heat-killed Mycobacterium tuberculosis (H37Ra strain; BD Diagnostics, Franklin Lakes, NJ). On the day of immunization and 48 hours later, the mice were also injected i.v. with Pertussis toxin (200 ng/mouse; List Biological Laboratories, Campbell, CA) in PBS. Mice were examined daily for EAE disease symptoms, which were scored using a standard method: 0, no clinical signs; 1, limp tail; 2, paraparesis (weakness, incomplete paralysis of 1 or 2 hind limbs); 3, paraplegia (complete paralysis of 2 hind limbs); 4, paraplegia with fore limb weakness or paralysis; and 5, moribund state or death.
Retroviral infection of CD4 T cells
Total CD4 T cells were activated with plate-bound anti-CD3 (1 μg/mL) plus anti-CD28 (1 μg/mL) in 24-well plates for 24 hours and then infected with retroviruses carrying the pMIGR1 empty vector, pMIGR1-mP52, or pMIGR1-mGM-CSF. The GFP+ transduced cells were sorted to perform adoptive transfer and EAE induction.
Analysis of in vivo T-cell differentiation and CNS infiltration
At the indicated time points after MOG35–55 immunization, the mice were sacrificed for splenocyte preparation and CNS infiltration analysis. CD4+ T cells were isolated from the splenocytes using magnetic beads (Invitrogen) and subjected to ICS as described above. For the preparation of CNS lymphocytes, brains and spinal cords were excised and dissociated for 1 hour at 37°C by digestion with collagenase IV (0.5 mg/mL; Invitrogen) and DNase I (10 μg /mL; Roche, Indianapolis, IN) in RPMI medium. Dispersed cells were passed through a 40-μm nylon mesh and collected by centrifugation. The cells were then resuspended in RPMI medium, layered onto a Percoll density gradient (Biochrom, Berlin, Germany), and centrifuged for 30 minutes (625 g, 22°C). CNS lymphocytes were isolated by collection of the interphase fraction between 30% and 70% Percoll. After intensive washing in Hanks balanced-salt solution, cells were analyzed by flow cytometry.
T cell proliferation and in vitro differentiation
CD4+ T cells were purified from splenocytes with anti-CD4-conjugated magnetic beads (Invitrogen). For ELISA and proliferation assays, naïve CD4 T cells were stimulated in replicate wells of 96-well plates (1 × 105 cells per well). Culture supernatants were analyzed by ELISA (eBioScience), and the cells were pulse-labeled for 6 h with [3H] thymidine for proliferation assays.
For in vitro CD4+ T-cell differentiation assays, naïve CD4+ T cells (CD4+CD25−CD44loCD62Lhi) were sorted from splenic CD4+ T cells, prepared using a CD4 T-cell Isolation Kit (Miltenyi Biotec, Auburn, CA), and stimulated with plate-bound anti-CD3 and anti-CD28 under Th0 (5 μg/mL anti–IL-4, 5μg/mL anti–IFN-γ), Th1 (10 ng/mL IL-12, 5μg/mL anti–IL-4), or Th17 (20 ng/mL IL-6, 5 ng/mL TGF-β, 5 μg/mL anti–IL-4, 5 μg/mL anti–IFN-γ) conditions. After the indicated times, the cells were subjected to ICS to quantify the production of their signature cytokines. For pathogenic Th17 differentiation, naïve CD4+ T cells (CD4+CD25−CD44loCD62Lhi) were firstly induced with plate-bound anti-CD3 and anti-CD28 under Th17 (20 ng/mL IL-6, 2.5 ng/mL TGF-β, 5 μg/mL anti–IL-4, 5μg/mL anti–IFN-γ). Differentiated Th17 cells were then allowed to ‘rest’ for 2 d in the presence of IL-2 (2 ng/ml), then were washed and repeated for a second stimulation of 72 h with anti-CD3 and anti-CD28 in the presence of TGF-β (2ng/ml) plus IL-6 (10ng/ml), IL-23 (40 ng/ml) or IL-1b (10ng/ml). After each stimulation period, supernatants were used for ELISA.
Real-time quantitative RT-PCR (qRT-PCR)
RNA was extracted with TRIzol reagent (Sigma) for qRT-PCR analyses using the SYBR regent (Bio-Rad). The expression of individual genes was calculated by a standard curve method and was normalized to the expression of Actb. The gene-specific primer sets used (all for mouse genes) were as in Table 1.
Table 1.
Gene specific primers used for QPCR assays
| Primer name | Forward primer | Reverse primer |
|---|---|---|
| GM-CSF | TCGAGCAGGGTCTACGGGGC | TCCGTTTCCGGAGTTGGGGG |
| IL-23R | GCCAAGAAGAC CATTCCCGA | TCAGTGCTACAATCTTC |
| IL-1b | AAGCCTCGTGCTGTCGGACC | TGAGGCCCAAGGCCACAGGT |
| CD80 | ACGTGTCAGAGGACTTCACCTGGG | GCGCCGAATCCTGCCCCAAA |
| CD86 | AGCACCCACGATGGACCCCA | GCACGGCAGATATGCAGTCCCA |
| TGFb3 | ATGATCCAGGGACTGGCGGAGC | ACCCGGAACTCTGCCCGGA |
| IL17a | CTCCAGAAGGCCCTCAGACTACC | AGCTTTCCCTCCGCATTGACACAG |
| Rorc | CAAGTCATCTGGGATCCACTAC | TGCAGGAGTAGGCCACATTACA |
| Actin | CGTGAAAAGATGACCCAGATCA | CACAGCCTGGATGGCTACGT |
ChIP assays
ChIP assays were performed as previously described (17). In brief, CD4+ T cells (1 × 107), freshly isolated from the spleen of MOG35–55 immunized mice (7 days), were resuspended in 9 ml RPMI supplemented with 2% fetal bovine serum in a 15 ml conical tube. After adding 0.25 ml of 37% formaldehyde to the tubes, the samples were rocked for 15 min at room temperature, followed by adding glycine (1.25M) to a final concentration of 0.125M. Cells were pelleted for 3 min at 3000 rpm at 4ºC and washed twice with 10 ml cold PBS containing 1 mM PMSF and 1μg/μl pepstatin A. Cell pellets were resuspended in 600 μl RIPA buffer containing 0.8M NaCl, incubated for 10 min on ice, and then sonicated to shear the DNA to 300–1000 base pairs. The chromatin DNA was added to rabbit Ig-loaded beads and rocked 1 hr at 4 °C for pre-clearance. Pre-cleared chromatin DNA was mixed with salmon sperm DNA (Stratagene, #201190-81) (final concentration 1.5 μg/μl), BSA (final concentration 0.5μg/μl), and subjected to IP using antibodies for RelA RelB, c-Rel, p50, p52 (all from NCI Preclinical Repository) and normal Rabbit IgG (SC-2027, Santa Cruz). After purification, the precipitated DNA was analyzed by quantitative PCR using the Csf2 primers listed in Table 2.
Table 2.
GM-CSF PCR primer used for ChIP assays
| Primer name | Forward primer | Reverse primer |
|---|---|---|
| GM-CSF | CCTGACAACCTGGGGGAAGGCT | CCAAGGCCGGGTGACAGTGATG |
Luciferase reporter assay
HEK293T cells were transfected with pGL3-Csf2 together with a control renillia luciferase reporter. Cells were also transfected with additional cDNA expression vectors. After 48h, cells were lysed and followed by a ducal luciferase assay (promega). Specific luciferase activity was normalized to the activity of renilla luciferase in each sample (internal control).
Statistical analysis
Two-tailed unpaired T test statistical analysis was performed using the Prism software. P values less than 0.05 were considered significant, and the level of significance was indicated as *P<0.05, **P<0.01, ***P<0.001.
For EAE clinical scores, differences between groups were evaluated by two-way ANOVA with Bonferroni’s post-test.
Results
p100 processing is largely dispensable for TCR/CD28-stimulated canonical NF-κB activation and naïve T-cell activation
To understand the role of noncanonical NF-κB in the regulation of T-cell activation, we employed the nfkb2lym1 mice. As indicated above, the nfkb2lym1 mice express a mutant form of p100 that lacks its C-terminal phosphorylation site and is unable to respond to processing-stimulating signals (14). We used the heterozygous mice, since both the heterozygous and homozygous mice were shown to have strong phenotype in B cells and osteoclasts (14) and since the generation of homozygous mice had very low efficiency (data not shown). Stimulation of WT T cells with anti-CD3 plus anti-CD28 led to the potent induction of p100 expression and its conversion to p52 (Fig. 1A). Although the T-cell activation also induced the expression of the p100-lym1, this p100 mutant was unable to be processed to generate p52 (Fig. 1A). This finding suggests that the T-cell activation signals stimulate the processing of p100, which is blocked by the lym1 mutation. Consistent with these results, p52 was abundantly expressed in the nucleus of WT T cells following prolonged stimulation, but little p52 could be detected in the lym1/+ T cells (Fig. 1A). Furthermore, although the lym1 mutation did not affect TCR/CD28-stimulated expression of RelB, it attenuated the nuclear translocation of RelB. In contrast, the p100 lym1 mutation did not appreciably affect the nuclear induction of canonical NF-κB members, c-Rel, RelA, or p50 (Fig. 1A). This latter finding was also in agreement with the normal induction of p100 and RelB expression, a molecular event known to be dependent on canonical NF-κB (18–20). Quantification of the protein bands from three independent experiments revealed significant defects of the lym1/+ T cells in the processing of p100 to p52 and in the nuclear expression of p52 and RelB (Supplementary Fig. 1). We also repeated these studies using T cells isolated from Map3k14−/− and control mice. As seen with the lym1/+ T cells, the Map3k14−/− T cells had a defect in the generation and nuclear translocation of p52 as well as reduction in the nuclear translocation of RelB (Fig. 1B). Furthermore, stimulation of WT T cells with anti-CD3 plus anti-CD28 led to the accumulation of NIK (Fig. 1C), a central step in noncanonical NF-κB signaling (5). These results suggest that T-cell activation is associated with the induction of p100 processing that is dependent on its C-terminal phosphorylation site and the upstream kinase NIK.
FIGURE 1.
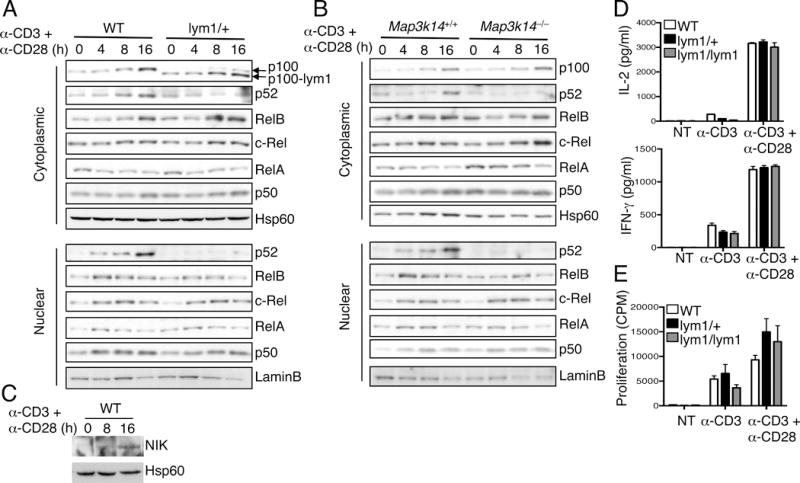
TCR/CD28-stimulated p100 processing is dispensable for naïve T-cell activation. (A and B) CD4+ T cells from WT and lym1/+ mice (A) or Map3k14+/+ and Map3k14−/− mice (B) were stimulated with plate-bound anti-CD3 plus anti-CD28 for the indicated time periods. Cytoplasmic and nuclear extracts were subjected to IB analysis of the indicated NF-κB proteins and loading controls Hsp60 and Lamin B. (C) IB analysis of NIK and Hsp60 in whole-cell lysates of WT T cells stimulated as indicated. (D and E) ELISA of IL-2 and IFN-γ (D) and proliferation assays (E) using CD4+ T cells purified from the spleen of young (6 wk, n=4) WT, lym1/+, and lym1/lym1 mice and stimulated for 48 h with plate-bound anti-CD3 or plus anti-CD28. Data are representative of three or more independent experiments.
The lym1/+ mice provide an excellent model for studying the role of noncanonical NF-κB signaling in T-cell activation. Taking advantage of this new model system, we examined the effect of the lym1 mutation on naïve T-cell activation. Compared with the WT T cells, the lym1/+ and lym1/lym1 T cells had a moderate defect in the production of cytokines, IL-2 and IFN-γ, when stimulated with anti-CD3 in the absence of anti-CD28 (Fig. 1D). However, these mutant T cells were competent in both cytokine production and proliferation when activated in the presence of the costimulatory stimuli anti-CD28 (Fig. 1D and E). Thus, the defect in p100 processing does not affect the initial activation of naïve T cells.
Noncanonical NF-κB signaling mediates EAE pathogenesis
To further examine the role of noncanonical NF-κB signaling in the regulation of T-cell function, we employed a T cell-dependent autoimmunity model, EAE. Immunization of WT mice with MOG35–55, along with an injection of pertussis toxin, led to the induction of EAE clinical scores (Fig. 2A). Remarkably, the lym1/+ mice were completely refractory to EAE induction. This phenotype was associated with a significant reduction in the number of CD4+ T cells and CD11b+ monocytes infiltrating to the CNS (Fig. 2B). Furthermore, the CNS of the lym1/+ mice had a profoundly lower number and frequency of the Th1 and Th17 inflammatory T cells (Fig. 2B). Analysis of the effector T cells in the spleen of the EAE-induced mice revealed a significant reduction in the frequency of Th17, although not Th1 cells (Fig. 2C and 2D). Since the in vivo differentiation of T cells involves both T cells and supporting cells, such as dendritic cells, we performed in vitro T-cell differentiation experiments to further assess the T cell-intrinsic role of the noncanonical NF-κB signaling in regulating T-cell differentiation. Interestingly, the naïve CD4+ T cells from the lym1/+ mice did not show any significant defect in Th17 cell differentiation in vitro, although there was a moderate reduction in the Th1 cells (Fig. 2E and 2F). These mutant T cells were also normal in the expression the Th17-specific transcription factor gene Rorc as well as the Th17-signature cytokine gene Il17a (Fig. 2G). Collectively, these results suggest that the defect in noncanonical NF-κB signaling renders the lym1/+ mice refractory to EAE induction, coupled with reduced immune cell infiltration to the CNS.
FIGURE 2.
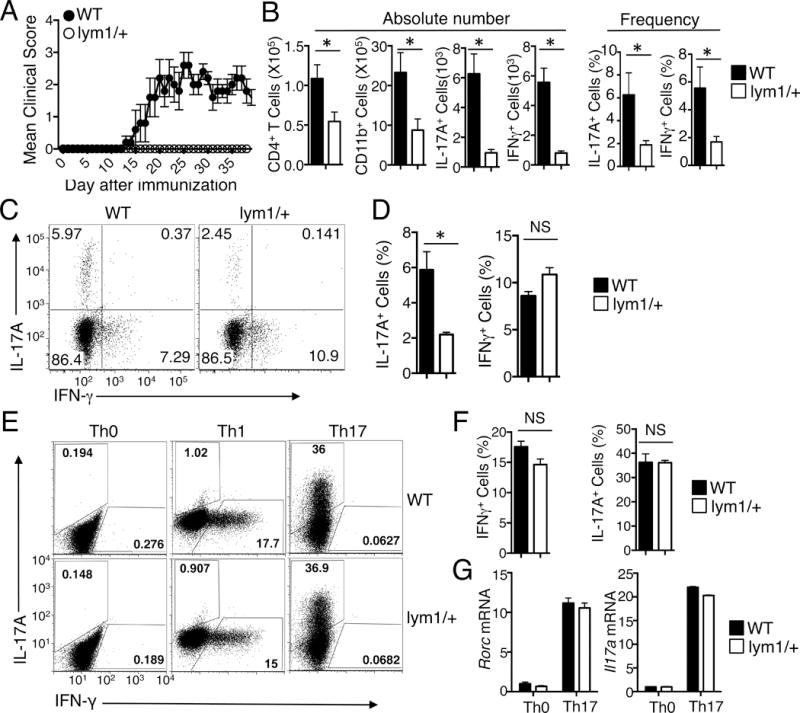
The processing of p100 is essential for EAE induction. (A) Age- and sex-matched WT and lym1/+ mice (5 WT and 4 lym1/+) were immunized with MOG35–55 peptide on day 0 and day 8 and monitored daily for EAE disease symptoms. (B) Flow cytometric analyses of the number or frequency of CD4+ T cells, CD11b+ cells, IL-17A+T cells, and IFN-γ+ T cells in the CNS of WT and lym1/+ mice at 14 days after EAE induction. Data are presented as mean ± SD values of 5 WT and 5 lym1/+ mice. (C and D) Splenic CD4+ T cells from mice of (B) were stimulated for 4 hours with PMA plus ionomycin and subjected to ICS and flow cytometry to determine the frequency of Th17 and Th1 cells based on their production of IL-17A and IFN-γ, respectively. Data are presented as a representative flow cytometry plot (C) and mean ± SD values (D). (E–G) Naïve CD4+ T cells isolated from WT and lym1/+ mice were stimulated for 84 hours with plate-bound anti-CD3 and anti-CD28 under Th0, Th1, or Th17 conditions followed by flow cytometry to measure the frequency of IFN-γ-producing Th1 cells and IL-17-producing Th17 cells (E and F) or by qRT-PCR to measure Rorc and Il17a mRNA expression level relative to the internal control Actb (G). Data are presented as a representative flow cytometry graph (E) and mean ± s.d. values of triplicate samples (F and G). Data are representative of three independent experiments.
T-cell intrinsic function of p100 processing, but not RelB expression, in the regulation of EAE pathogenesis
One caveat of the EAE experiment described above is that it could not tell whether noncanonical NF-κB has a T-cell intrinsic role in EAE regulation, since the lym1 mutation occurs in all cell types. Moreover, the lym1 mutation is known to perturb the development of T cells likely due to the function of noncanonical NF-κB in thymic epithelial cells (14). To overcome this problem, we performed mixed bone marrow transfer studies by mixing the lym1/+ and Rag1−/− bone marrows and transferring them into the Rag1−/− recipient mice. This strategy could eliminate the contribution of noncanonical NF-κB functions in stromal cells and dendritic cells. Under these conditions, the lym1 mutation did not alter the number of different stages of thymocytes (Supplementary Fig. 2A) or the peripheral T cells (Supplementary Fig. 2B), although it reduced the number of mature follicular B cells and marginal zone B cells in the spleen (Supplementary Fig. 2C). Moreover, the the lym1/+ mixed chimeric mice displayed a delayed onset and reduced severity of the EAE clinical symptoms compared to the WT mixed bone marrow chimeric mice, suggesting a lymphocyte-intrinsic role for noncanonical NF-κB in regulating EAE pathogenesis (Fig. 3A). Interestingly, despite their severe defect in EAE induction, the lym1/+ chimeric mice did not have a significant reduction in the frequency or number of the inflammatory Th1 and Th17 cells in the spleen (Fig. 3B), which was in agreement with the normal differentiation of lym1/+ CD4+ T cells in vitro (Fig. 2E and 2F).
FIGURE 3.
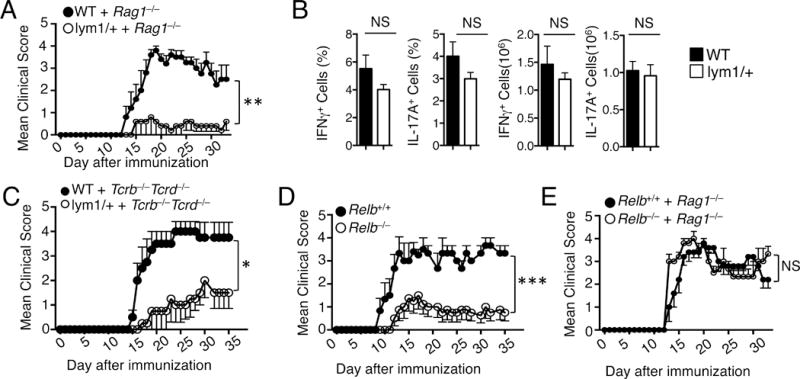
T cell-intrinsic function of p100 processing, but not RelB expression, in EAE pathogenesis. (A) Lethally irradiated Rag1−/− mice were reconstituted with mixed bone marrow cells of Rag1−/− and WT or lym1/+ mice (1:1). 6 weeks after reconstitution, bone marrow chimeric mice were immunized with MOG35–55 peptide to induce EAE and monitored daily for EAE disease symptoms (5 WT, 4 lym1/+). (B) Splenic CD4+ T cells from mice of (A) were stimulated for 4 hours with PMA plus ionomycin and subjected to ICS and flow cytometry to determine the frequency of Th17 and Th1 cells based on their production of IL-17A and IFN-γ, respectively. (C) Lethally irradiated Tcrb−/−Tcrd−/− mice were reconstituted with mixed bone marrow cells of Tcrb−/−Tcrd−/− and WT or lym1/+ mice (4:1). 6 weeks after reconstitution, bone marrow chimeric mice were immunized with MOG35–55 peptide to induce EAE and monitored daily for EAE disease symptoms (5 WT, 5 lym1/+). (D) Age- and sex-matched Relb+/+ and Relb−/− mice (5 WT and 5 Relb−/−) were immunized with MOG35–55 peptide on day 0 and day 8 and monitored daily for EAE disease symptoms. (E) Lethally irradiated Rag1−/− mice were reconstituted with mixed bone marrow of Rag1−/− and Relb+/+ or Relb−/− mice (1:1) to make bone marrow chimera. 6 weeks after reconstitution, bone marrow chimeric mice were immunized with MOG35–55 peptide to induce EAE and monitored daily for EAE disease symptoms (5 Relb+/+ and 5 Relb−/−). Data are representative of three or more independent experiments.
To specifically examine the role of noncanonical NF-κB in T cells, we performed mixed bone marrow transfer studies by mixing lym1/+ and Tcrb−/−Tcrd−/− bone marrows and transferring them into the T cell deficient Tcrb−/−Tcrd−/− recipient mice. Under these conditions, the lym1 mutation would be limited to the T cells in the chimeric mice. The chimeric mice had normal thymocyte and peripheral T-cell numbers, suggesting normal development and homeostasis (Supplementary Fig. 2D, 2E). However, the Tcrb−/−Tcrd−/− chimeric mice receiving the lym1/+ and Tcrb−/−Tcrd−/− bone marrows were still refractory to EAE induction, thus confirming a T-cell intrinsic role of p100 processing in EAE regulation (Fig. 3C).
RelB is generally considered as a noncanonical NF-κB member based on the dependence of its activation on p100 processing in certain cell types, such as dendritic cells. However, the activation of RelB is not always identical to noncanonical NF-κB signaling. As shown in Fig. 1A, the activation of RelB in T cells was only partially dependent on p100 processing. We thus examined the role of RelB in EAE pathogenesis under both direct immunization and bone marrow adoptive transfer conditions. Like the lym1/+ mice, the Relb−/− mice were refractory to EAE induction (Fig. 3D). Interestingly, however, the role of RelB in EAE regulation was not lymphocyte-intrinsic, as revealed by mixed bone marrow chimera studies. Chimeric mice reconstituted with mixed bone marrows from the Relb−/− and Rag1−/− mice did not show any EAE-resistance phenotype (Fig. 3E). These results uncovered a major difference between RelB expression and p100 processing.
NF-κB2 p52 regulates Th17 effector genes and is required for EAE pathogenesis
The finding that p100 processing was largely dispensable for the generation of Th17 cells raised the question of whether the noncanonical NF-κB pathway regulates the expression of inflammatory effector molecules of Th17 cells. We prepared T cells from the spleen of EAE-induced WT and lym1/+ mice and examined the gene expression by qRT-PCR. Interestingly, the lym1/+ T cells had a profound reduction in the expression of GM-CSF (Fig. 4A), a cytokine known to be crucial for the encephalitogenicity of Th17 cells (21, 22). These mutant T cells also had reduced expression of IL-23R and CD80, known to regulate the terminal differentiation and effector function of Th17 cells (23, 24). On the other hand, the lym1/+ and WT control T cells had comparable levels of expression of several other genes, including those encoding IL-1R, CD86, and TGF-β3 (Fig. 4A). Consistent with these findings, flow cytometric analyses also revealed reduced frequency of GM-CSF-producing CD4+ T cells in the CNS of EAE-induced lym1/+ and NIK-deficient mice as compared their WT controls (Fig. 4B and Supplementary Fig. 3).
FIGURE 4.
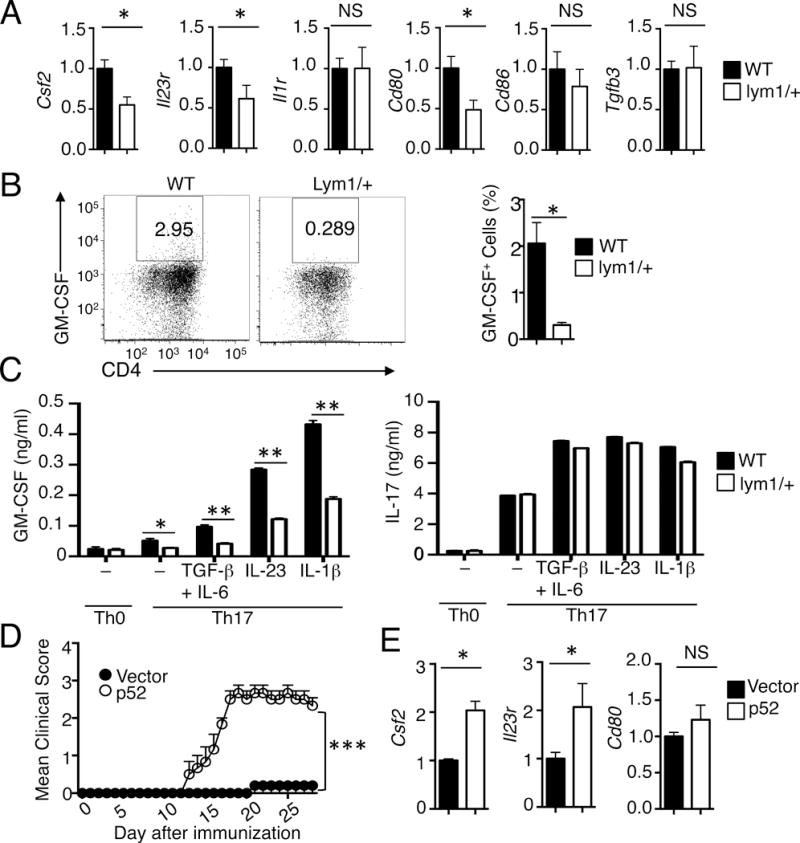
Regulation of GM-CSF induction and encephalitogenicity of T cells. (A) qRT-PCR analysis of relative mRNA levels (normalized to the control Actb) of the indicated genes in CD4+ T cells freshly isolated from WT and lym1/+ mice immunized with MOG35–55 for 7 days. (B) CNS CD4+ T cells from EAE-induced WT and lym1/+ mice (day 14 after immunization) were stimulated for 4 hours with PMA plus ionomycin and subjected to ICS and flow cytometry to determine the frequency of GM-CSF producing cells. Data are presented as a representative flow cytometry graph (left) and mean value (right). (C) Naive CD4+ T cells isolated from WT and lym1/+ mice were stimulated for 72 hours with plate-bound anti-CD3 and anti-CD28 under Th0 or Th17 conditions. Differentiated Th17 cells were rested for 2 days, followed by the second stimulation of 72 h with anti-CD3 and anti-CD28 in the absence (−) or presence of the indicated cytokines. ELISAs were performed for GM-CSF and IL-17 produced. (D) lym1/+ CD4+ T cells were infected with vector or p52. After 48h, cells were adoptively transferred into Rag1−/− mice (n=4). The recipient mice were immunized for EAE induction. (E) qRT-PCR analysis of the indicated genes in CD4+ T cells isolated from mice of (D). Data are representative of two or more independent experiments.
Since GM-CSF is a crucial factor mediating the inflammatory function of Th17 cells in the CNS, we further examined the role of p100 processing in the regulation of this cytokine production during in vitro T-cell differentiation. Consistent with previous studies, the expression of GM-CSF was strongly induced in the WT CD4+ T cells under Th17 differentiation conditions, particularly in the presence of IL-23 and IL-1β (Fig. 4C). Importantly, the induction of GM-CSF was profoundly reduced in the lym1/+ T cells under different Th17 differentiation conditions (Fig. 4C). This result suggested that the noncanonical NF-κB signaling pathway might promote GM-CSF induction by mediating the TCR/CD28 signal. Parallel analyses revealed that the lym1 mutation did not affect the induction of IL-17 expression (Fig. 4C).
The studies presented above suggested a crucial role for p100 processing in the regulation of Th17 effector gene induction and EAE pathogenesis. Since the major NF-κB member affected by the blockade of p100 processing was p52 (Fig. 1A), we sought to examine whether exogenous p52 could rescue the function of the lym1/+ T cells in effector gene expression and EAE induction. We reconstituted the lym1/+ T cells with a GFP-expressing retroviral vector encoding p52. Following enriching the infected cells by flow cytometric sorting, we adoptively transferred the T cells into Rag1−/− mice. Introduction of endogenous p52, but not the empty vector, into the lym1/+ T cells profoundly restored their EAE-inducing function, coupled with increased expression of Csf2 and Il23r genes (Fig. 4D and 4E). The exogenous p52 did not promote the expression of CD80, suggesting the involvement of additional factors or a higher threshold of p52 requirement in the induction of this gene. Together, these data suggest that the noncanonical NF-κB member p52 is crucial for the induction of T-cell effector molecules, particularly GM-CSF, and the pathogenic function of T cells in EAE induction.
p52 cooperates with c-Rel in the regulation of Csf2 promoter activity
We next examined the functional significance and the underlying mechanism of p52-mediated Csf2 gene expression. To examine whether the reduced GM-CSF expression in lym1/+ T cells contributes to the EAE-refractory phenotype of the lym1/+ mice, we reconstituted the lym1/+ CD4+ T cells with the a GFP-expressing retroviral vector or the same vector encoding GM-CSF and then adoptively transferred the cells into Rag1−/− mice. We also used the vector-transduced wild-type T cells as control in this experiment. As expected, the recipient mice of the vector-transduced lym1/+ T cells had delayed onset and profoundly reduced severity of EAE than those transferred with the wildtype T cells (Fig. 5A). Interestingly, however, the recipient mice of the GM-CSF-transduced lym1/+ T cells developed severe EAE clinical symptoms. Of note, the expression of GM-CSF in lym1/+ T cells failed to rescue the delayed disease onset but almost completely restored the disease severity at later time points. These findings suggest that GM-CSF is an important, but not the only, EAE-regulatory factor that is defective in the lym1/+ T cells, consistent with the finding that p100 processing regulates the expression of additional genes during EAE induction (Fig. 4A).
FIGURE 5.
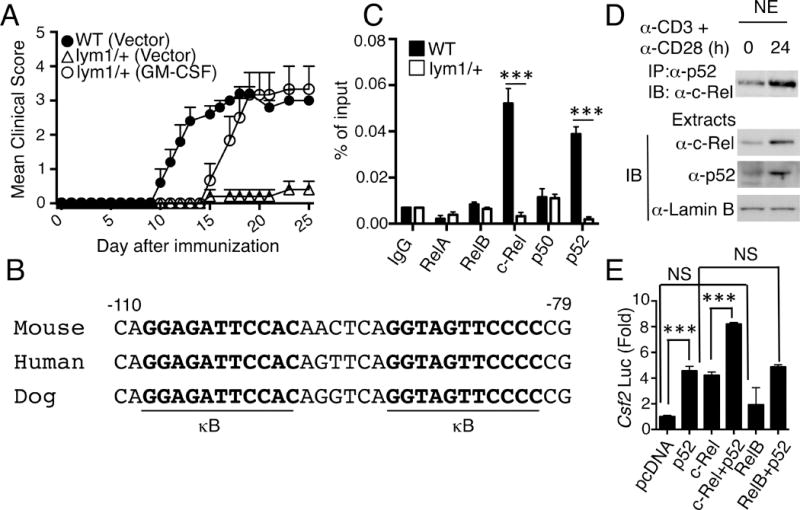
Synergistic induction of Csf2 gene expression by p52 and c-Rel. (A) WT or lym1/+ CD4+ T cells were infected with a retroviral vector or the same vector encoding GM-CSF. 48 h after infection, the cells were adoptively transferred into Rag1−/− mice, and the recipient mice were immunized for EAE induction (n=5). (B) Sequence alignment of mouse, human, and dog Csf2 promoters, showing two conserved NF-κB binding sites (κB). (C) CD4+ T cells were isolated from WT and lym1/+mice immunized with Mog35–55 for 7 days, followed by Csf2 promoter chip assay with different antibodies as indicated. (D) WT T cells were stimulated as indicated, and nuclear extracts were subjected to p52/c-Rel co-IP assay (upper) or direct IB (lower). (E) 293T cells were transfected with Csf2 luciferase reporter, along with the indicated expression vectors. 48h after transfection, luciferase activities were measured and presented as fold relative to the activity of cells transfected with an empty pcDNA vector. Results are expressed as mean±s.d. of three independent experiments.
The promoter region of Csf2 contains an NF-κB binding element, κB, located at positions −91 to −82 (25). Adjacent to this previously identified κB site, there was another κB-like sequence (nucleotide −108 to −98), and these κB elements are highly conserved between mouse, human, and dog (Fig. 5B). By performing ChIP assays, we found that this promoter region was strongly bound by p52 as well as by c-Rel, although it was only weekly or barely bound by the other NF-κB members (Fig. 5C). Although the activation of c-Rel was independent of the noncanonical NF-κB pathway, this canonical NF-κB member physically associated with p52 in the nucleus of activated T cells (Fig. 5D). We then carried out luciferase reporter assays to further assess the function of p52 and c-Rel in the regulation of Csf2 promoter activity. Both p52 and c-Rel were able to induce the luciferase expression from the Csf2 promoter, whereas RelB displayed a much weaker activity (Fig. 5E). Moreover, although RelB has been viewed as the partner of p52, RelB failed to promote p52-mediated Csf2 promoter activation (Fig. 5E). Instead, c-Rel strongly cooperated with p52 in this specific function. Therefore, p52 appeared to regulate the expression of Csf2 gene together with c-Rel.
Discussion
Noncanonical NF-κB is known to regulate B-cell development and activation, although its role in T cells remains poorly understood and somewhat controversial. The major limitation in addressing this issue has been the lack of an appropriate genetic system. RelB-deficient mice have been employed for the study of noncanonical NF-κB function, but the activation of RelB is not always a consequence of noncanonical NF-κB signaling. In the present study, we employed a mutant mouse strain carrying a mutation in the nfkb2 gene, which express a p100 mutant (lym1) with defective in phosphorylation and processing (14). We found that p100 processing is induced along with T-cell activation, which is essential for the activation of p52 but only partially required for the activation of RelB. By performing mixed bone marrow adoptive transfer studies, we demonstrated that the p100 processing, but not RelB expression, had a T cell-intrinsic role in the regulation of EAE induction. Our data suggest that p52 regulates the expression of specific Th17 effector molecules, particularly GM-CSF, which in turn is important for EAE pathogenesis.
We have previously shown that the noncanonical NF-κB activating kinase NIK is crucial for EAE regulation (10). However, it was unclear whether NIK functions via the noncanonical NF-κB pathway, since the NIK-deficient T cells have a defect in the activation of STAT3 (10). Indeed, our present study suggests that the function of NIK in EAE regulation might be partially mediated through the STAT3 signaling axis, since NIK knockout, but not lym1 mutation, had a T cell-intrinsic effect in Th17 cell differentiation. Nevertheless, our data suggest that NIK also plays a role in the regulation of TCR/CD28-stimulated p100 processing, suggesting its involvement in noncanonical NF-κB signaling in T cells. A prior study suggests that transgenic overexpression of NIK in dendritic cells rescues the defect of the Map3k14−/− mice in EAE induction (13). While this work suggests a role for NIK to function in DCs, it does not exclude the T cell-intrinsic function of NIK in this autoimmune response. The extremely high level of NIK transgene may cause abnormal function of DCs in T-cell activation. More importantly, a low level of NIK is also expressed in other cells, including T cells, in the DC-specific NIK transgenic mice (13), further complicating the phenotype. As discussed above, the conclusion based on the use of Relb−/− mice should be taken cautiously, since RelB activation does not always represent noncanonical NF-κB signaling. Using both p100 processing-defective mice (lym1 mice) and p52-mediated functional rescue approaches, our present study clearly demonstrated the T cell-intrinsic role for noncanonical NF-κB signaling in the regulation of EAE pathogenesis. Our work is also in agreement with the finding that NIK has a T cell-intrinsic role in the regulation of graft-versus-host disease (11).
Previous work suggests that overexpression of p100 in Jurkat T cells inhibits TCR-mediated IL-2 induction (26). Interestingly, under the endogenous conditions, the blockade of p100 processing in the lym1/+ or lym1/lym1 T cells did not appreciably affect the IL-2 induction or proliferation of naïve T cells stimulated by anti-CD3 plus anti-CD28. On the other hand, we did detect a moderate reduction in these cellular events when the T cells were stimulated with anti-CD3 alone. It is likely that under co-stimulation conditions, the processing of p100 is not required for initial activation of naïve T cells. Our data suggest that the noncanonical NF-κB pathway is also dispensable for the differentiation of naïve CD4 T cells to Th1 or Th17 cells. Nevertheless, this signaling pathway is essential for the pathological function of T cells in mediating EAE. This T cell-intrinsic function of the noncanonical NF-κB pathway is at least partially mediated through induction of GM-CSF, a T-cell effector cytokine mediating induction of EAE (21, 22, 27). We found that the lym1/+ T cells are defective in GM-CSF induction in Th17 cells. Furthermore, introduction of exogenous p52 largely rescued GM-CSF induction and EAE pathogenic action of T cells. Interestingly, p52 cooperates with c-Rel in the induction of Csf2 promoter. This finding supports the previous report that c-Rel regulates Csf2 gene expression (28) and further revealed an interesting crosstalk between the noncanonical NF-κB p52 and the canonical NF-κB c-Rel in this gene induction event. As such, our current study provides new insight into the signaling mechanism that controls the effector function of Th17 cells.
Supplementary Material
Acknowledgments
We thank Amgen Inc., Bristol-Myers Squibb, Dr. Robyn Starr and The Walter and Eliza Hall Institute of Medical Research for mutant mice, NCI Preclinical Repository for antibodies. We also thank the personnel from the flow cytometry, DNA analysis, and animal facilities at The MD Anderson Cancer Center.
This work was supported by grants from the National Institutes of Health (AI057555, AI064639, GM84459, and AI104519) and partially supported by a Sister Institution Network Fund and a seed fund from the Center for Inflammation and Cancer at the MD Anderson Cancer Center. This study also used the NIH/NCI-supported resources under award number P30CA016672 at The MD Anderson Cancer Center.
Abbreviations used in this article
- EAE
experimental autoimmune encephalomyelitis
- IKK
IκB kinase
- NIK
NF-κB inducing kinase
- CNS
central nervous system
- IB
immunoblot
- ICS
intracellular cytokine staining
- ChIP
chromotin immunoprecipitation
- MOG
myelin oligodendrocyte glycoprotein
- qRT-PCR
real-time quantitative RT-PCR
References
- 1.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 2012;26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 3.Oh H, Ghosh S. NF-kappaB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev. 2013;252:41–51. doi: 10.1111/imr.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J. 2004;382:393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun SC. The noncanonical NF-kappaB pathway. Immunol Rev. 2012;246:125–140. doi: 10.1111/j.1600-065X.2011.01088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao G, Harhaj EW, Sun SC. NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- 7.Senftleben U, et al. Activation of IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- 8.Liang C, Zhang M, Sun SC. beta-TrCP binding and processing of NF-kappaB2/p100 involve its phosphorylation at serines 866 and 870. Cell Signal. 2006;18:1309–1317. doi: 10.1016/j.cellsig.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72:1161–1179. doi: 10.1016/j.bcp.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 10.Jin W, Zhou XF, Yu J, Cheng X, Sun SC. Regulation of Th17 cell differentiation and EAE induction by the MAP3K NIK. Blood. 2009;113:6603–6610. doi: 10.1182/blood-2008-12-192914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray SE, Polesso F, Rowe AM, Basak S, Koguchi Y, Toren KG, Hoffmann A, Parker DC. NF-kappaB-inducing kinase plays an essential T cell-intrinsic role in graft-versus-host disease and lethal autoimmunity in mice. J Clin Invest. 2011;121:4775–4786. doi: 10.1172/JCI44943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rowe AM, Murray SE, Raue HP, Koguchi Y, Slifka MK, Parker DC. A Cell-Intrinsic Requirement for NF-kappaB-Inducing Kinase in CD4 and CD8 T Cell Memory. J Immunol. 2013;191:3663–3672. doi: 10.4049/jimmunol.1301328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofmann J, Mair F, Greter M, Schmidt-Supprian M, Becher B. NIK signaling in dendritic cells but not in T cells is required for the development of effector T cells and cell-mediated immune responses. J Exp Med. 2011;208:1917–1929. doi: 10.1084/jem.20110128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tucker E, et al. A novel mutation in the Nfkb2 gene generates an NF-kappa B2 “super repressor”. J Immunol. 2007;179:7514–7522. doi: 10.4049/jimmunol.179.11.7514. [DOI] [PubMed] [Google Scholar]
- 15.Matsumura T, Sakai M, Matsuda K, Furukawa N, Kaneko K, Shichiri M. Cis-acting DNA elements of mouse granulocyte/macrophage colony-stimulating factor gene responsive to oxidized low density lipoprotein. J Biol Chem. 1999;274:37665–37672. doi: 10.1074/jbc.274.53.37665. [DOI] [PubMed] [Google Scholar]
- 16.Reiley WW, Zhang M, Jin W, Losiewicz M, Donohue KB, Norbury CC, Sun SC. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol. 2006;7:411–417. doi: 10.1038/ni1315. [DOI] [PubMed] [Google Scholar]
- 17.Ji Y, Little AJ, Banerjee JK, Hao B, Oltz EM, Krangel MS, Schatz DG. Promoters, enhancers, and transcription target RAG1 binding during V(D)J recombination. J Exp Med. 2010;207:2809–2816. doi: 10.1084/jem.20101136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liptay S, Schmid RM, Nabel EG, Nabel GJ. Transcriptional regulation of NF-kappa B2: evidence for kappa B-mediated positive and negative autoregulation. Mol Cell Biol. 1994;14:7693–7703. doi: 10.1128/mcb.14.12.7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun S-C, Ganchi PA, Beraud C, Ballard DW, Greene WC. Autoregulation of the NF-κB transactivator Rel A (p65) by multiple cytoplasmic inhibitors containing ankyrin motifs. Proc Natl Acad Sci USA. 1994;91:1346–1350. doi: 10.1073/pnas.91.4.1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bren GD, Solan NJ, Miyoshi H, Pennington KN, Pobst LJ, Paya CV. Transcription of the RelB gene is regulated by NF-kappaB. Oncogene. 2001;20:7722–7733. doi: 10.1038/sj.onc.1204868. [DOI] [PubMed] [Google Scholar]
- 21.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 23.Podojil JR, Kohm AP, Miller SD. CD4+ T cell expressed CD80 regulates central nervous system effector function and survival during experimental autoimmune encephalomyelitis. J Immunol. 2006;177:2948–2958. doi: 10.4049/jimmunol.177.5.2948. [DOI] [PubMed] [Google Scholar]
- 24.McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schreck R, Baeuerle PA. NF-kappa B as inducible transcriptional activator of the granulocyte-macrophage colony-stimulating factor gene. Mol Cell Biol. 1990;10:1281–1286. doi: 10.1128/mcb.10.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Legarda-Addison D, Ting AT. Negative Regulation of TCR Signaling by NF-{kappa}B2/p100. J Immunol. 2007;178:7767–7778. doi: 10.4049/jimmunol.178.12.7767. [DOI] [PubMed] [Google Scholar]
- 27.Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:39–48. doi: 10.4049/jimmunol.178.1.39. [DOI] [PubMed] [Google Scholar]
- 28.Bunting K, Rao S, Hardy K, Woltring D, Denyer GS, Wang J, Gerondakis S, Shannon MF. Genome-wide analysis of gene expression in T cells to identify targets of the NF-kappa B transcription factor c-Rel. J Immunol. 2007;178:7097–7109. doi: 10.4049/jimmunol.178.11.7097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.