

Abstract
A Bayesian mechanism–based pharmacokinetic/pharmacodynamic model of cytochrome P450 3A4 (CYP3A4) activity was developed based on a clinical study of the effects of ketoconazole and rifampin on midazolam exposure and plasma 4β-hydroxycholesterol (4βHC) concentrations. Simulations from the model demonstrated that the dynamic range of 4βHC as a biomarker of CYP3A4 induction or inhibition was narrower than that of midazolam; an inhibitor that increases midazolam area under the curve by 20-fold may only result in a 20% decrease in 4βHC after 14 days of dosing. Likewise, an inducer that elevates CYP3A4 activity by 1.2-fold would reduce the area under the curve of midazolam by 50% but would only increase 4βHC levels by 20% after 14 days of dosing. Elevation in 4βHC could be reliably detected with a twofold induction in CYP3A4 activity with study sample sizes (N ~ 6–20) typically used in early clinical development. Only a strong CYP3A4 inhibitor (e.g., ketoconazole) could be detected with similar sample sizes.
It is important to understand the potential of a new chemical entity to have significant pharmacokinetic (PK) interactions with commonly administered comedications as early in the exploratory drug development process as possible. This knowledge enables important decisions to be made about either compound selection or revisions to the clinical development program before the investment of significant resources in intermediate- and late-phase clinical trials. PK interactions involving metabolic routes of drug elimination are generally well understood and tend to be the most straightforward to screen for using in vitro and animal models before selection of a compound for exploratory clinical development. Strong inhibitors and inducers of cytochrome P450 3A4 (CYP3A4) are generally excluded by in vitro and in vivo preclinical experiments before selection of a compound to move to first-in-human clinical studies. However, some compounds with the potential for clinically meaningful drug interactions do advance into human clinical studies, and some are eventually approved for use in patients.1
The traditional approach for detecting the potential for CYP3A4-mediated drug interactions for compounds in clinical development is to conduct a clinical PK drug interaction study in human subjects, using a probe substrate of CYP3A4, such as midazolam (MDZ). These studies are typically required as part of the regulatory approval process for drugs in which clinically relevant CYP3A4 induction or inhibition cannot be ruled out based on nonclinical experiments.2 4β-Hydroxycholesterol (4βHC) is an endogenous oxidized metabolite of cholesterol that is specifically produced via metabolism of cholesterol by CYP3A4 in the liver.3 Induction of CYP3A4 causes elevation in plasma concentrations of 4βHC, whereas inhibition of CYP3A4 leads to reduction.4,5 Therefore, 4βHC has the potential to be used in early-phase clinical trials as an endogenous biomarker to determine the potential for a compound to induce or inhibit CYP3A4. 4α-Hydroxycholesterol (4αHC) is a cholesterol-oxidation product that is not produced through metabolism by CYP3A4. It has been proposed as a possible endogenous metabolite that could be used to normalize for differences in general metabolic activity between subjects, thus reducing the impact of intersubject variability on estimation of changes in 4βHC.
Before the routine implementation of 4βHC as a biomarker in clinical development, it is important to understand its dynamic properties in response to well-accepted inducers and inhibitors of CYP3A4. In addition, the inter- and intrasubject variability in the steady-state levels of 4βHC and the response to CYP3A4 modulation in healthy subjects and patients will determine its reliability as a biomarker in clinical studies. Several experimental clinical studies in healthy subjects have already been conducted using strong inhibitors (atazanavir/ritonavir and itraconazole)5,6 or inducers (rifampin, efavirenz, and carbamazepine)7,8,9 of CYP3A4 to evaluate the potential of 4βHC to be used as a clinical biomarker. The results of these studies suggest that because of the relatively long half-life of 4βHC (1–3 weeks)10 and the fact that inducers result in elevation of 4βHC concentrations, it may be a more valuable measure of CYP3A4 induction as opposed to inhibition. Indeed, Yang and Rodrigues11 demonstrated that these results would be expected based on a theoretical model of 4βHC dynamics. However, a model-based characterization of the pharmacodynamics (PD) of 4βHC, and its variability, in humans will provide a thorough understanding of its dynamic properties as a biomarker. A model developed in such a way would permit prospective prediction of the 4βHC biomarker response in clinical studies based on preclinical data. This would enable an early decision to be made regarding whether to collect this biomarker, as well as the timing and frequency of collection.
In this article, we report the development and utility of a Bayesian mechanism–based PK/PD model of 4βHC using data from a clinical study that evaluated 4βHC as a biomarker of both CYP3A4 induction by rifampin and CYP3A4 inhibition by ketoconazole. The objectives for developing this model based on mechanistic information and clinical data are as follows: (i) to provide a tool that permits integration of nonclinical information on the potential of an new molecular entity to have CYP3A4 interaction, so that decisions can be made about how to use 4βHC in early clinical development; (ii) to provide a tool to conduct clinical trial simulations to optimize the design of clinical studies in which 4βHC is used to evaluate CYP3A4 inhibition/induction; and (iii) to evaluate the utility and dynamic range of 4βHC relative to MDZ as a measure of CYP3A4 induction/inhibition by compounds with different induction/inhibition potential.
Results
Development of the PK/PD model for 4βHC
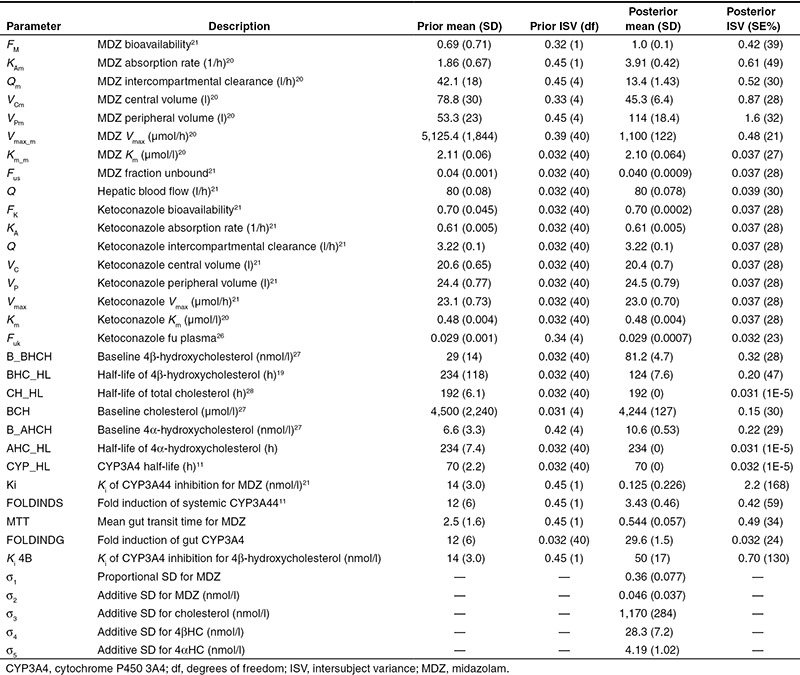
A schematic diagram of the nonlinear mixed effects PK/PD model for CYP3A4-mediated drug interactions is shown in Figure 1. The model describes the PK of MDZ and ketoconazole, the formation of 4βHC and 4αHC from cholesterol (the PD measure), and the induction caused by 600 mg of rifampin. 4αHC is not a substrate for CYP3A4 but may be useful in normalizing for differences in general metabolic rates among individuals when estimating changes in 4βHC. Separate compartments for induction and inhibition of CYP3A4 in the gut vs. the liver were implemented. Bayesian priors were used for all the model parameters. Informative priors were used, when possible, based on the availability of published data. Plasma ketoconazole concentration was not measured in the clinical study; therefore, highly informative priors for ketoconazole PK parameters were used based on a previously published meta-analysis of ketoconazole PK (Table 1). Similarly, the priors for the PK parameters for MDZ and the interaction with ketoconazole were also based on a previously published meta-analysis (Table 1). The objective function and the fixed effects parameters appeared to converge to their optimal stationary values based on examination of the caterpillar plots and evaluation of the Gelman and Rubin interchain diagnostics for the three independent Markov chain Monte Carlo (MCMC) chains (Supplementary Figure S1 and Supplementary Table S1). The prior and posterior means and SDs of the model parameters can be found in Table 1. Separate Ki values were required to describe inhibition of CYP3A4 for MDZ (Ki = 0.125 nmol/l) vs. 4βHC (Ki = 50 nmol/l). Separate values were also required to describe induction of CYP3A4 by rifampin in the gut (29.6-fold) vs. the liver (3.43-fold).
Figure 1.

Diagram of CYP3A4 semi-mechanistic PK/PD model for CYP3A4 interactions. 4αHC, 4α-hydroxycholesterol; 4βHC, 4β-hydroxycholesterol; CKeto, concentration of ketoconazole; CL, clearance from central compartment; CLINT, intrinsic clearance; CMDZ, concentration of MDZ; CYP, fold induction in CYP3A4; FPE, first-pass effect for MDZ; Fu, fraction unbound for MDZ; Fuk, fraction unbound for ketoconazole; INH4βHC, inhibition function for effect of ketoconazole on formation of 4β-hydroxycholesterol; INHMDZ, inhibition function for effect of ketoconazole on midazolam's intrinsic clearance; Kc, central compartment for ketoconazole; Kdeg,4αHC, elimination rate for 4α-hydroxycholesterol; Kdeg,4βHC, elimination rate for 4β-hydroxycholesterol; Ki, inhibition constant for ketoconazole; Km, concentration for half maximal metabolic clearance rate; Kp, peripheral compartment for ketoconazole; Ksyn, synthesis rate for 4β-hydroxycholesterol in the absence of induction or inhibition; Ksyn,4αHC, synthesis rate for 4α-hydroxycholesterol; Ksyn,4βHC, synthesis rate for 4β-hydroxycholesterol; Ksyn,TC, synthesis rate for total cholesterol; KTR, gut transit rate constant; Mc, central compartment for MDZ; Mp, peripheral compartment for MDZ; MTT, mean transit time in gut; N, number of compartments; QH, hepatic blood flow; Rif, rifampin; Vc, volume of central compartment; Vmax, maximum rate of metabolic clearance.
Table 1. Prior and posterior model parameters.
Evaluation of the PK/PD model for 4βHC
The posterior predictions of MDZ plasma concentrations and 4βHC serum concentrations from the model were compared with the observed data, both visually and by comparison of the summary statistical measures. The posterior distribution of predicted MDZ plasma concentrations vs. time compared with the observed data following oral and i.v. MDZ, either alone or in the presence of ketoconazole or rifampin, are shown in Figure 2a–f. The model is able to capture the time course of MDZ concentrations and the effects of CYP3A4 inhibition by ketoconazole and induction by rifampin. Comparison of the summary statistics for the posterior model predictions of MDZ exposures to the observed statistics reveals some minor discrepancies in the model predictions (Table 2). There was a slight overprediction of the area under the curve (AUC) for oral MDZ in the presence of ketoconazole (~20%) and a slight overprediction of the AUC of oral MDZ in the presence of rifampin (~29%). However, the observed oral and i.v. MDZ AUC ratios following ketoconazole or rifampin administration are within the credible intervals of the posterior predictions.
Figure 2.

Evaluation of posterior and prospective predictive performance of the PK/PD model. Observed and posterior predicted concentrations for MDZ and 4βHC for: (a) 2 mg oral MDZ; (b) 2 mg oral MDZ with ketoconazole; (c) 2 mg oral MDZ with rifampin; (d) 0.8 mg i.v. MDZ; (e) 0.8 mg i.v. MDZ with ketoconazole; (f) 0.8 mg i.v. MDZ with rifampin; (g) 4βHC after 14 days of 400 mg q.d. ketoconazole; (h) 4βHC after 14 days of 600 mg q.d. rifampin; (i) observed and prospectively predicted MDZ ratio for 13 compounds; and (j) observed and prospectively predicted 4βHC vs. time following carbamazepine administration. Plots a through h show the observed concentrations vs. time, the median and 90% intervals for the posterior predictions, and the 90% credible intervals. Plot i shows the observed vs. predicted with the line of unity (solid line) and the lines representing twofold prediction error (dotted lines). Plot j shows the pooled 4βHC observations from Wide et al., as well as the median and 95% interval of the population predictions. 4βHC, 4β-hydroxycholesterol; CYP3A4, cytochrome P450 3A4; MDZ, midazolam; PD, pharmacodynamics; PK, pharmacokinetics.
Table 2. Observed and posterior predicted midazolam exposures and 4β-hydroxycholesterol.
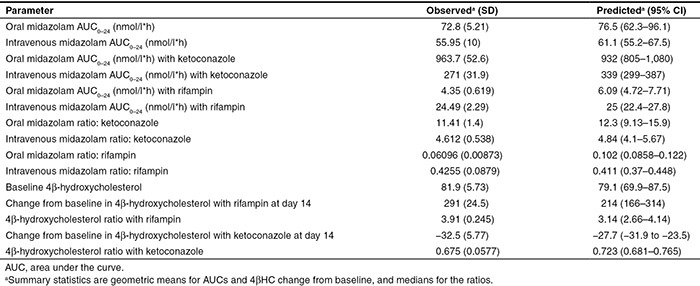
The posterior predictions of 4βHC concentration vs. time compared with the observed data are shown in Figure 2g,h. The predictions of 4βHC levels following inhibition by ketoconazole or induction by rifampin appear to be adequate. Table 2 provides the summary statistics of predicted and observed means for 4βHC serum concentrations at baseline and on day 14.
To evaluate the prospective predictive performance of the model, predictions were generated for 13 known inhibitors and inducers of CYP3A4 that were not included in the analysis dataset. Figure 2i shows the observed vs. predicted MDZ AUC ratio for these compounds. The data and references used in the prospective predictions can be found in Supplementary Table S2. Figure 2i shows the predicted and observed time course for the change from baseline in 4βHC following administration of carbamazepine (CYP3A4 inducer) for 24 weeks. The observed 4βHC vs. time data for carbamazepine treatment come from a clinical study by Wide et al.9 in pediatric epilepsy patients.
Evaluation of dynamic range of 4βHC vs. MDZ
Simulations using the model were conducted to characterize the dynamic range of 4βHC as a biomarker of CYP3A4 induction and inhibition in comparison to MDZ. Figure 3a shows the percent increase in 4βHC compared with the fold decrease in MDZ AUC in the presence of inducers with different CYP3A4 induction potentials after 14 days of treatment. 4βHC does not appear to be as sensitive as MDZ for inducers; for an inducer with 1.05-fold CYP3A4 induction potential (equivalent to a 105% increase in enzyme activity), the AUC of MDZ would be expected to be reduced by 20%, whereas the AUC of 4βHC would be expected to increase by only 4%. To see an increase in 4βHC of 20%, CYP3A4 induction would need to be 1.2-fold (equivalent to a 120% increase in enzyme activity), sufficient to reduce MDZ AUC by 50%. Figure 3b,c show the fold increase in MDZ AUC vs. the percent decrease in 4βHC in the presence of CYP3A4 inhibitors, with different Ki values and a half-life of 2 h, administered as 400 mg q.d. or 200 mg b.i.d. The difference in dynamic range between 4βHC and MDZ is more pronounced for inhibitors than that for inducers. Inhibitors that reduce 4βHC by 20% would be expected to increase MDZ AUC by 20- to 26-fold, depending on the dosing regimen.
Figure 3.

Comparison of dynamic range of 4βHC vs. MDZ after 14 days of treatment with inducers or inhibitors of CYP3A4. (a) Fold change in 4βHC vs. MDZ AUC ratio for different levels of CYP3A4 induction; blue dotted lines show an inducer with 1.05-fold induction potential (0.8 MDZ AUC ratio and 4% increase in 4βHC). Green dotted lines show an inducer with 1.2-fold induction potential (0.5 MDZ AUC ratio and 20% increase in 4βHC); black dotted lines show an inducer with 4.6-fold induction potential (0.1 MDZ AUC ratio and 260% increase in 4βHC). (b) Fold change in 4βHC vs. MDZ AUC ratio for inhibitors with different CYP3A4 Ki values and a 2-h half-life administered at 400 mg q.d. Red dotted lines show an inhibitor with an [I]/KiCmin of 0.003 (20-fold increase in MDZ AUC ratio and 20% decrease in 4βHC). (c) Fold change in 4βHC vs. MDZ AUC ratio for inhibitors with different CYP3A4 Ki values and a 2-h half-life administered at 200 mg b.i.d. Red dotted lines show an inhibitor with an [I]/KiCmin of 0.0012 (26-fold increase in MDZ AUC ratio and 20% decrease in 4βHC). 4βHC, 4β-hydroxycholesterol; AUC, area under the curve; CYP3A4, cytochrome P450 3A4; MDZ, midazolam.
Clinical trial simulations of CYP3A4 induction and inhibition
To characterize the potential utility of 4βHC as a biomarker of CYP3A4 induction in clinical trials, simulations were conducted using compounds with different CYP3A4 induction potentials. Figure 4a shows the change in 4βHC vs. time for four CYP3A4 inducers with different CYP3A4 induction potentials, following 14 days of treatment. Induction potential is defined as the fold increase in CYP3A4 by day 14. Induction potentials of 1.5-, 2-, 3-, and 4.6-fold would be expected to result in changes in 4βHC of 40, 75, 150, and 244%, respectively, on day 14. Simulation of clinical trials with different sample sizes show the relationship between sample size and the probability to detect a significant difference in 4βHC, relative to placebo-treated subjects, for compounds with different induction potentials (Figure 4b). A significant difference was defined as lack of overlap between the 95% confidence intervals of the treated group relative to placebo, with the assumption of equal sample sizes between the groups. There is a relatively linear relationship between induction potential and the sample size to achieve 80% probability (Figure 4c).
Figure 4.

Clinical trial simulation of 4βHC following 14 days of once-daily treatment with potential inducers of liver CYP3A4. (a) Mean levels of 4βHC vs. time. (b) Probability of detecting a difference relative to placebo vs. sample size on day 14. (c) Fold induction in CYP3A4 vs. the sample size to achieve 80% probability. 4βHC, 4β-hydroxycholesterol; CYP3A4, cytochrome P450 3A4.
The change in 4βHC as a result of liver CYP3A4 inhibition in the model is driven by unbound concentrations of inhibitor in the systemic circulation as a function of time and therefore depends on the Ki for the enzyme, the PK properties, and the dosing regimen of the compound. Figure 5a shows the change in 4βHC vs. time for five CYP3A4 inhibitors with different PK properties (defined primarily by half-life), Ki values, and dosing regimens, following 14 days of treatment. Simulation of clinical trials with different sample sizes show the relationship between sample size and probability to detect a significant difference in 4βHC relative to placebo-treated subjects for CYP3A4 inhibitors with different properties (Figure 5b). Because several compound-specific factors influence the change in 4βHC on day 14, an examination of different compound properties was conducted to determine the measure that would best correlate with the effect on 4βHC. The concentration of inhibitor divided by the Ki (I/Ki) appears to be the most useful parameter for predicting the change in 4βHC at the end of treatment. Both I/Ki for average plasma concentration at steady state (Cavg) and steady-state Cmin show similarly strong correlations with the change in 4βHC (data not shown). Figure 5c shows the relationship between I/Ki for Cmin at steady state vs. the sample size to achieve 80% probability to detect a significant difference in 4βHC vs. placebo treatment. In contrast to the inducers, there is greater variability in this relationship because of the several factors involved in producing changes in 4βHC (i.e., CYP3A4 Ki, PK properties, and dosing regimen). It is important to determine that the type 1 error rate (i.e., false-positive rate) is consistent with expectations when interpreting the results of clinical trial simulations. Clinical trial simulations using the PK/PD model and assuming no induction/inhibition of CYP3A4 (n = 8 each in placebo and treated) determined the type 1 error to be ~5%.
Figure 5.

Clinical trial simulation of 4βHC following 14 days of once-daily treatment with potential inhibitors of CYP3A4. (a) Mean levels of 4βHC vs. time. (b) Probability of detecting a difference relative to placebo vs. sample size on day 14. (c) I/Ki at Cmin vs. sample size for 80% probability. (d) Summary table of relevant parameters for CYP3A4 inhibitors. Drugs B and C have PK properties and CYP3A4 Ki similar to ketoconazole. 4βHC, 4β-hydroxycholesterol; CYP3A4, cytochrome P450 3A4.
Discussion
We have reported the development of a nonlinear mixed effects PK/PD model to evaluate the utility of 4βHC as a biomarker of induction and inhibition of CYP3A4 in clinical studies. A Bayesian approach was utilized for estimation of the model parameters, both to leverage prior knowledge in estimation of parameters based on the current clinical trial dataset and to compensate for data that was not collected in the study. The Bayesian approach is useful in facilitating the development of mechanism-based models, in which many parameters are often not identifiable based on data typically collected in clinical trials. The Bayesian approach is more commonly applied in toxicology research, in which the algorithms and software available to implement such approaches have been widely adopted.12,13 However, examples of more clinically oriented PK/PD models developed using Bayesian approaches have also been reported.14,15,16 MCMC Bayesian algorithms offer distinct advantages over more commonly used deterministic estimation algorithms. It does not suffer from the notorious numerical instability that often occurs with linear approximation algorithms (e.g., FO and FOCE) and complex highly dimensional PK/PD models.17 In addition, formal utilization of prior knowledge of PK/PD relationships using MCMC Bayesian algorithms allows leveraging of the wealth of prior knowledge for model development. Thus, a model developed using this approach represents not only knowledge gained from the current analysis dataset but also potentially all prior knowledge.
To be able to accurately estimate oral and i.v. MDZ exposures, separate compartments were needed to represent CYP3A4 interaction in the liver vs. the gut. Interestingly, the fold induction of CYP3A4 estimated for rifampin in the gut was much greater than that in the liver. This is consistent with reported effect of rifampin on verapamil, where oral clearance is elevated 32-fold and systemic clearance only increases 1.3-fold.18 To accurately estimate the effect of ketoconazole on both MDZ and 4βHC, separate parameters for CYP3A4 Ki were necessary, with the Ki for 4βHC estimated to be 400-fold higher than that of MDZ. This finding could be explained by the potential of 4βHC to be generated through metabolism by other metabolic enzymes besides CYP3A4. This is consistent with the observation in a clinical study that propiverine treatment produces an increase in MDZ exposure, suggesting CYP3A4 inhibition, and a significant increase in 4βHC, suggesting induction of an alternative pathway (non-CYP3A4/5) for formation of 4βHC.19 The half-life of 4βHC was estimated to be 10 days, only slightly less than the half-life of 17 days commonly described in the literature.3,10 However, the model-based estimation of the 4βHC half-life is likely to be reliable for healthy subjects given the robustness of model-based approaches for estimating physiological rate constants compared with the noncompartmental approaches typically used in analysis of clinical study data. There were differences in the MDZ PK parameters between the posterior mean estimates and the priors, based on the meta-analyses by Yu et al.20 and Li et al.21, particularly for Vmax and bioavailability. The meta-analyses by Yu et al.20 and Li et al.21 were conducted using aggregate level (i.e., summary level) data, whereas the current analysis is conducted using individual-level data, and this may explain some of this difference. In addition, differences between the population sample used in the current analysis compared with the meta-analyses by Yu et al.20 and Li et al.21 may also be partly blamed.
4βHC has been proposed as a potentially useful biomarker for early detection of CYP3A4 inhibition and induction in clinical development. The PK/PD model we have developed based on the clinical study in healthy subjects allows comparison of 4βHC vs. MDZ, the “gold standard” of CYP3A4 probe substrates, in realistic and clinically relevant scenarios. The simulations performed with the model suggest that dynamic range of 4βHC for detection of CYP3A4 induction or inhibition is significantly narrower compared with that of MDZ. In particular, 4βHC is rather insensitive for the detection of CYP3A4 inhibitors; for example, an inhibitor that increases MDZ AUC by 20-fold would result in only a 20% decrease in 4βHC after 14 days of dosing. Therefore, 4βHC is unlikely to detect CYP3A4 inhibition for the majority of new chemical entities that are routinely developed, unless the sample size is sufficiently large to detect small changes in 4βHC.
In deciding to incorporate 4βHC as a biomarker for either inducers or inhibitors in clinical development, the sample size of the study must be taken into account. This is particularly true for inhibitors, due to the lower sensitivity of 4βHC for detection of CYP3A4 inhibition. We have used the PK/PD model to simulate 4βHC in trials with different sample sizes, with both inducers and inhibitors of CYP3A4. For very strong inducers such as rifampin, there would be a high probability of detecting 4βHC elevation, even by comparing a single arm (N ~ 6) of a typical multiple ascending dose study. Detection of the signal could be enhanced by combining data from several panels of a multiple ascending dose study. For detecting the weak inducers (e.g., 1.5-fold induction), a much larger sample size (n > 60) would be needed to detect the 4βHC elevation with high probability. These simulations suggest that 4βHC could be a useful biomarker for detection of CYP3A4 induction for a new chemical entity early in the clinical development program. However, the utility of 4βHC to detect CYP3A4 inhibition is much more limited. For an inhibitor similar to ketoconazole, with an [I]/Ki at Cmin of 0.11 (Figure 5), administered with a dosing regimen of 200 mg b.i.d., it would be feasible to expect detection of 4βHC reduction in a typical multiple ascending dose study. Not surprisingly, the same inhibitor administered as a 400 mg q.d. regimen (Figure 5) would require a larger sample size. Most CYP3A4 inhibitors that are now in clinical development are likely to be weaker than ketoconazole. In addition, it is likely that the accuracy and precision of the 4βHC assay would make the identification of these relatively small changes difficult to determine with any degree of confidence, making the potential for detecting false-negatives or false-positives unacceptably high. Therefore, the utility of 4βHC to be used for detection of CYP3A4 inhibition early in clinical development is low.
There have been several clinical trials conducted to evaluate the clinical utility of 4βHC as a biomarker for inducers or inhibitors of CYP3A4. These trials have utilized commonly known perpetrators of CYP3A4 interactions; inducers such as rifampin, efavirenz, and carbamazepine;6,7,9 or inhibitors such as ketoconazole, itraconazole, and ritonavir.5,9,22 Reports of the utility of 4βHC in the early detection of CYP3A4 induction or inhibition in clinical development have yet to be published. The simulations performed in this study demonstrate the limited utility of 4βHC for detection of CYP3A4 inhibition in early clinical development. However, detection of CYP3A4 induction appears to be possible with the sample sizes typically used in phase I clinical trials. In either case, incorporation of 4βHC in phase I studies should be relatively straightforward, and significant changes detected in these studies could be used to trigger a more definitive MDZ interaction study later in clinical development.
Methods
Development of the PK/PD model for 4βHC
PK and PD data from a clinical study of the effects of rifampin and ketoconazole on the metabolism of endogenous markers of CYP3A4 activity, and on MDZ PK, in 34 healthy subjects aged between 18 and 45 years, were used in the analysis.23 All subjects were randomized to receive oral (2 mg) and i.v. (0.4 mg) MDZ on day 1 or 2, and on day 15 or 16 after receiving q.d. rifampin 600 mg, ketoconazole 400 mg, or placebo for 2 weeks. Subjects were followed up until day 30. Serial blood samples for MDZ PK were collected on days 1, 2, 15, and 16. Predose blood samples were collected from day 1 to 30 for assessment of 4βHC, 4αHC, and total cholesterol. The data used in the PK/PD analysis included 1,600 MDZ plasma concentrations, 606 plasma total cholesterol concentrations, 606 plasma 4βHC concentrations, and 605 plasma 4αHC concentrations. The lower limit of quantification was 0.3 nmol/l (0.1 ng/ml) for MDZ plasma concentration and 2 ng/ml for total cholesterol (52 µmol/l), 4βHC (5 nmol/l), and 4αHC (5 nmol/l). Observations below the lower limit of quantification were ignored in the analysis.
Development of the nonlinear mixed effects model was conducted through a MCMC Bayesian estimation approach with both informative priors and noninformative priors (Table 1) using the NONMEM software system (version VII, level 2.0, ICON Development Solutions, Ellicott City, MD). Intersubject variability was assumed to be log normally distributed for all fixed effect parameters except for the bioavailability parameters, which were assumed to follow a logit distribution constrained between 0 and 1. A normal distribution was assumed for the prior fixed effects and an Inverse-Wishart distribution for the prior intersubject variance. No prior distributions were used for the residual variances. The PK models and prior distributions for fixed and random effects for ketoconazole and MDZ are based on previously published meta-analyses.20,21 Key model equations have been included in Figure 1, and descriptions of model parameters can be found in Table 1. All model equations and parameters can be found in the NONMEM control file included in the Supplementary Data.
Thirty thousand iterations of each MCMC chain were used during the burn-in phase and 1,000 iterations for the stationary phase. Plasma ketoconazole concentration was not measured in the clinical study; therefore, highly informative priors for ketoconazole PK parameters were used (Table 1). The effect of CYP3A44 induction was incorporated as previously described: as an exponential function that increases with time and would be expected to reach steady state by ~20 days of treatment, based on half-life of CYP3A4 (70 h).11 Therefore, the dose response of inducers is not incorporated in the model, but rather the fold induction can be used as input to predict the change in 4βHC as a function of time. Convergence of the model parameters and objective function values was evaluated by visual inspection of the “caterpillar” plots for three independent MCMC chains and by examination of the Gelman and Rubin statistics.24 Gelman and Rubin convergence statistics for the three MCMC chains were generated using the Bayesian Output Analysis Program (BOA) version 1.12 (ref. 25) in S-Plus (TIBCO Spotfire S+ 8.2; TIBCO Software, Princeton, NJ).
Evaluation of the PK/PD model for 4βHC
The posterior predictive performance of the PK/PD model was evaluated by generating predictions for the 34 subjects included in the analysis using 500 of the posterior sets of population mean parameter estimates. The 500 sets of parameter estimates were randomly sampled from the 1,000 sets generated during the stationary phase of the MCMC distribution. Predictions were generated using NONMEM. AUCs were calculated for the observed and predicted concentrations using the trapezoidal method. Summary statistics and plots were produced using S-Plus (TIBCO Spotfire S+ 8.2; TIBCO Software).
Simulation of dynamic range of 4βHC vs. MDZ
Simulations to compare the dynamic range for 4βHC vs. MDZ were performed by assuming 14 days of treatment with either inhibitors or inducers of CYP3A4. Two hundred and fifty inhibitors were simulated with CYP3A4 Ki values ranging from 1 x 10−7 nmol/l to 10 µmol/l. Inhibitors were administered as 400 mg q.d. regimens with population mean PK parameters identical to those for ketoconazole. Two hundred and fifty inducers were simulated that ranged in induction potential for CYP3A4 from 0- to 100-fold and were assumed to achieve maximal induction with the same rate constant as rifampin. It should be noted that a onefold induction is equivalent to a 100% increase in CYP3A4 enzyme activity. All simulations were conducted using R version 3.0 (http://www.r-project.org/).
Clinical trial simulations of CYP3A4 induction and inhibition
Clinical trials were simulated to evaluate the sample sizes needed to detect changes in 4βHC after 14 days of q.d. dosing of either CYP3A4 inhibitors or CYP3A4 inducers. The change in 4βHC was evaluated relative to a placebo group of equivalent sample size. Five different CYP3A4 inhibitors with different [I]/Ki at Cmin were simulated with sample sizes from 6 to 100. To evaluate the effect of [I]/Ki at Cmin on the sample size for 80% power, different [I]/Ki properties were created by varying the dose and regimen of the five different CYP3A4 inhibitors. Trial simulations for inducers were conducted in a similar manner, with inducers that increase CYP3A4 activity by 1.5- to 4.6-fold. The probability of successfully detecting a significant change in 4βHC was defined as the percentage of 500 simulated trials in which the 95% confidence interval for the treatment group does not overlap with 95% confidence interval in the placebo group. Type 1 error was estimated by evaluating the probability that the 95% confidence intervals between two groups treated with placebo do not overlap with sample sizes of eight subjects. All simulations, summary statistics, and plots were generated using R version 3.0 (http://www.r-project.org/).
Author Contributions
T.A.L. wrote the manuscript. T.A.L., S.K., D.W.B., and F.L. designed the research. T.A.L., S.K., D.W.B., and F.L. performed the research. T.A.L. analyzed the research.
Conflict of Interest
All authors are employees of Bristol-Myers Squibb.
Study Highlights

Supplementary Material
References
- Wynn G.H., Oesterheld J.R., Cozza K.L., Armstrong S.C. Manual of Drug Interaction Principles for Medical Practice: The P450 System. American Psychiatric Publishing; Arlington, VA; 2009. [Google Scholar]
- Center for Drug Evaluation and Research . Guidance for Industry Drug Interaction Studies — Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. Food and Drug Administration - U.S. Department of Health and Human Services; Silver Spring, MD; 2012. p. 75. [Google Scholar]
- Diczfalusy U., Nylén H., Elander P., Bertilsson L. 4β-Hydroxycholesterol, an endogenous marker of CYP3A4/5 activity in humans. Br. J. Clin. Pharmacol. 2011;71:183–189. doi: 10.1111/j.1365-2125.2010.03773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanebratt K.P., et al. Cytochrome P450 induction by rifampicin in healthy subjects: determination using the Karolinska cocktail and the endogenous CYP3A4 marker 4beta-hydroxycholesterol. Clin. Pharmacol. Ther. 2008;84:589–594. doi: 10.1038/clpt.2008.132. [DOI] [PubMed] [Google Scholar]
- Lütjohann D., et al. 4beta-hydroxycholesterol as a marker of CYP3A4 inhibition in vivo - effects of itraconazole in man. Int. J. Clin. Pharmacol. Ther. 2009;47:709–715. doi: 10.5414/cpp47709. [DOI] [PubMed] [Google Scholar]
- Josephson F., et al. CYP3A induction and inhibition by different antiretroviral regimens reflected by changes in plasma 4beta-hydroxycholesterol levels. Eur. J. Clin. Pharmacol. 2008;64:775–781. doi: 10.1007/s00228-008-0492-8. [DOI] [PubMed] [Google Scholar]
- Bjorkhem-Bergman L., et al. Comparison of endogenous 4beta-hydroxycholesterol with midazolam as markers for CYP3A4 induction by rifampicin. Drug Metab. Dispos. 2013;41:1488–1493. doi: 10.1124/dmd.113.052316. [DOI] [PubMed] [Google Scholar]
- Habtewold A., et al. Pharmacogenetic and pharmacokinetic aspects of CYP3A induction by efavirenz in HIV patients. Pharmacogenomics J. 2013;13:484–489. doi: 10.1038/tpj.2012.46. [DOI] [PubMed] [Google Scholar]
- Wide K., Larsson H., Bertilsson L., Diczfalusy U. Time course of the increase in 4beta-hydroxycholesterol concentration during carbamazepine treatment of paediatric patients with epilepsy. Br. J. Clin. Pharmacol. 2008;65:708–715. doi: 10.1111/j.1365-2125.2007.03078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diczfalusy U., Kanebratt K.P., Bredberg E., Andersson T.B., Böttiger Y., Bertilsson L. 4beta-hydroxycholesterol as an endogenous marker for CYP3A4/5 activity. Stability and half-life of elimination after induction with rifampicin. Br. J. Clin. Pharmacol. 2009;67:38–43. doi: 10.1111/j.1365-2125.2008.03309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Rodrigues A.D. Does the long plasma half-life of 4beta-hydroxycholesterol impact its utility as a cytochrome P450 3A (CYP3A) metric. J. Clin. Pharmacol. 2010;50:1330–1338. doi: 10.1177/0091270009360041. [DOI] [PubMed] [Google Scholar]
- Nong A., et al. Bayesian calibration of a physiologically based pharmacokinetic/pharmacodynamic model of carbaryl cholinesterase inhibition. J. Toxicol. Environ. Health. A. 2008;71:1363–1381. doi: 10.1080/15287390802271608. [DOI] [PubMed] [Google Scholar]
- Weijs L., Yang R.S., Das K., Covaci A., Blust R. Application of Bayesian population physiologically based pharmacokinetic (PBPK) modeling and Markov chain Monte Carlo simulations to pesticide kinetics studies in protected marine mammals: DDT, DDE, and DDD in harbor porpoises. Environ. Sci. Technol. 2013;47:4365–4374. doi: 10.1021/es400386a. [DOI] [PubMed] [Google Scholar]
- Jonsson F., Jonsson E.N., Bois F.Y., Marshall S. The application of a Bayesian approach to the analysis of a complex, mechanistically based model. J. Biopharm. Stat. 2007;17:65–92. doi: 10.1080/10543400600851898. [DOI] [PubMed] [Google Scholar]
- Kathman S.J., Williams D.H., Hodge J.P., Dar M. A Bayesian population PK-PD model of ispinesib-induced myelosuppression. Clin. Pharmacol. Ther. 2007;81:88–94. doi: 10.1038/sj.clpt.6100021. [DOI] [PubMed] [Google Scholar]
- Kathman S.J., Williams D.H., Hodge J.P., Dar M. A Bayesian population PK-PD model for ispinesib/docetaxel combination-induced myelosuppression. Cancer Chemother. Pharmacol. 2009;63:469–476. doi: 10.1007/s00280-008-0760-4. [DOI] [PubMed] [Google Scholar]
- Bauer R.J., Guzy S., Ng C. A survey of population analysis methods and software for complex pharmacokinetic and pharmacodynamic models with examples. AAPS J. 2007;9:E60–E83. doi: 10.1208/aapsj0901007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm M.F., Busse D., Kroemer H.K., Eichelbaum M. Differential induction of prehepatic and hepatic metabolism of verapamil by rifampin. Hepatology. 1996;24:796–801. doi: 10.1002/hep.510240407. [DOI] [PubMed] [Google Scholar]
- Tomalik-Scharte D., Lütjohann D., Doroshyenko O., Frank D., Jetter A., Fuhr U. Plasma 4beta-hydroxycholesterol: an endogenous CYP3A metric. Clin. Pharmacol. Ther. 2009;86:147–153. doi: 10.1038/clpt.2009.72. [DOI] [PubMed] [Google Scholar]
- Yu M., Kim S., Wang Z., Hall S., Li L. A Bayesian meta-analysis on published sample mean and variance pharmacokinetic data with application to drug-drug interaction prediction. J. Biopharm. Stat. 2008;18:1063–1083. doi: 10.1080/10543400802369004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Yu M., Chin R., Lucksiri A., Flockhart D.A., Hall S.D. Drug-drug interaction prediction: a Bayesian meta-analysis approach. Stat. Med. 2007;26:3700–3721. doi: 10.1002/sim.2837. [DOI] [PubMed] [Google Scholar]
- Goodenough A.K., et al. Quantification of 4-beta-hydroxycholesterol in human plasma using automated sample preparation and LC-ESI-MS/MS analysis. Chem. Res. Toxicol. 2011;24:1575–1585. doi: 10.1021/tx2001898. [DOI] [PubMed] [Google Scholar]
- Kasichayanula S., et al. Assessment of 4β-hydroxycholesterol as a CYP3A activity marker in humans after ketoconazole, rifampin and placebo treatment. Clin. Pharmacol. Ther. 2013;93:S87. [Google Scholar]
- Gelman A., Rubin D.B. Inference from iterative simulation using multiple sequences. Stat. Sci. 1992;7:457–511. [Google Scholar]
- Smith B. Bayesian Output Analysis Program (BOA) Version 1.1.5 for R. Vol. 2013. University of Iowa, Iowa City, IA; 2013. [Google Scholar]
- Bodin K., et al. Antiepileptic drugs increase plasma levels of 4beta-hydroxycholesterol in humans: evidence for involvement of cytochrome p450 3A4. J. Biol. Chem. 2001;276:38685–38689. doi: 10.1074/jbc.M105127200. [DOI] [PubMed] [Google Scholar]
- Tomalik-Scharte D.et al., Plasma 4beta-hydroxycholesterol: an endogenous CYP3A metric Clin Pharmacol Ther 200986(2147–53. [DOI] [PubMed] [Google Scholar]
- London I.M, Rittenberg D. Deuterium studies in normal man. I. The rate of synthesis of serum cholesterol. II. Measurement of total body water and water absorption. J Biol Chem. 1950;184 (2:687–91. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.