

Abstract
Pharmacokinetic/pharmacodynamic (PKPD) modelling is used to describe and quantify dose–concentration–effect relationships. Within paediatric studies in infectious diseases and immunology these methods are often applied to developing guidance on appropriate dosing. In this paper, an introduction to the field of PKPD modelling is given, followed by a review of the PKPD studies that have been undertaken in paediatric infectious diseases and immunology. The main focus is on identifying the methodological approaches used to define the PKPD relationship in these studies. The major findings were that most studies of infectious diseases have developed a PK model and then used simulations to define a dose recommendation based on a pre-defined PD target, which may have been defined in adults or in vitro. For immunological studies much of the modelling has focused on either PK or PD, and since multiple drugs are usually used, delineating the relative contributions of each is challenging. The use of dynamical modelling of in vitro antibacterial studies, and paediatric HIV mechanistic PD models linked with the PK of all drugs, are emerging methods that should enhance PKPD-based recommendations in the future.
Keywords: Pharmacokinetics/pharmacodynamics (PKPD), Non-linear mixed effects (NLME), Paediatrics, Antimicrobial, Antibacterial, Antifungal, Antiviral (and antiretrovirals), HIV viral and T-cell dynamics, Immune reconstitution
Graphical abstract

1. Introduction
Pharmacokinetic/pharmacodynamic (PKPD) modelling seeks to quantify the dose–concentration–effect relationship with a mathematical model. It can be used in both pre-clinical and clinical study designs. The statistical aspect to PKPD modelling seeks to quantify variability in the data, and following suitable evaluation, simulate the predicted behaviour of the system. This so-called pharmacometric approach is increasingly recognised as being an important supplement to randomised control trial (RCT) data [1], particularly in patient groups where recruitment to RCTs is problematic, such as in children and neonates.
During model development, when fitting a PKPD model to data from in vitro and/or in vivo sources, there will always be some element of ‘noise’ whereby the model does not fit the exact data points, since by definition a mathematical model is a simplification of reality. The mathematical PKPD model is therefore extended to include a statistical component which models this noise; typically the central tendency of the noise is assumed to have a mean of zero (so that model predictions go through the middle of the data) and the noise magnitude (its variance) is estimated. Typical uses of a PKPD model are to make inferences on whether and to what extent a drug's dose and concentrations are associated with markers of disease or disease resolution.
One difficulty in properly conducting clinical PKPD studies is the fact that the experimental units (patients) are not homogenous. For this reason, care is needed when modelling data from more than one subject, because it is known a priori that individuals differ from one another; this is true for both PK and PD outcomes. Indeed, fitting a single model to all data (the naïve pooled or data averaging approach), and to a lesser extent fitting the model to each individual and then averaging parameter estimates (two-stage approach) are methods known to bias parameter estimates [2]. Fortunately, over the past 30 years clinical pharmacology has been at the forefront of implementing the so-called ‘population approach’ using the statistical method known as non-linear mixed effects (NLME) modelling. By fitting a single model to all individuals simultaneously, whilst allowing for two levels of random effects (noise), estimates can be made of both the model parameter typical values and their inter-individual variability, in addition to the residual variability. By splitting the variability in this way, and so allowing model parameters to take different values in each individual, unbiased estimates can be obtained. A further benefit is that by taking this population-level approach, individual subjects can contribute differing amounts of data, making opportunistic sampling designs a possibility. An introduction to the field of population PKPD modelling is described elsewhere [2–7].
For the majority of antimicrobial agents there tends to be some published PK which, although may not cover all paediatric age groups, is usually sufficient to predict paediatric PK across ages using knowledge of developmental differences [8]. Knowledge of PK alone is, however, insufficient to define a dose recommendation. In order to develop guidance on dosing, the dose-concentration-effect relationship needs to be understood and this means going beyond PK, to make extrapolations as to how these concentrations link with effect (PD). There are two main approaches that make the link from PK to PD in infectious diseases research. The most common approach uses simulations from the PK model to determine a dose which yields a pre-defined endpoint, which is thought to translate to the desired effect. Such endpoints are often derived from in vitro concentration-effect experiments [9], although in vivo animal PKPD outcomes or previous clinical PKPD studies can be used [10]. A major disadvantage of this approach is that assumptions have to be made about factors such as the circulating drug concentration being proportional to that at the site of pathogen load, or the transferability of outcomes between different studies. The second, and more challenging approach, is to collect and model both PK and PD information in the same patient, thereby eliminating the need for extrapolations. Whilst the data generated from this approach could be considered gold standard, the difficulty lies in defining a suitable PD endpoint, recruiting patients to such studies (e.g. many neonates treated with antimicrobials do not have proven infection) and obtaining stable estimates of a joint PKPD model. This review is focused on summarising the PKPD information available in paediatric infectious diseases and immunology, the modelling approaches taken, and identifying potential avenues for future PKPD research. The PubMed database search terms included the generic drug names and classes of antimicrobials, pharmacokinetics, pharmacodynamics, paediatric/pediatric and neonatal, and disease states where relevant: titles and abstracts of papers available in English were reviewed, and selected for inclusion if they contained relevant or new information.
2. Infectious diseases
2.1. Bacterial infections
The PKPD indices of most antibacterial agents have been defined from both in vitro and animal and human in vivo studies. A summary of these is given in Table 1. Typically a breakpoint for susceptibility will be set based on the minimum inhibitory concentration (MIC) of the antibacterial, that is to say the minimal concentration required to inhibit growth in strains considered sensitive. This breakpoint is then compared to either circulating maximum (Cmax) concentrations, area under the curve (AUC), or circulating time above MIC (T>MIC) depending on the antibacterial mode of action.
Table 1.
PKPD indices for major classes of antibacterial agents.
| Pharmacodynamics | Class of antibacterial | Specific drug (where applicable) | PD parameter | PD target of therapy |
|---|---|---|---|---|
| Time-dependent | Beta-lactams | – | %T>MIC [9] | Max %T in the dosing interval > MIC |
| Macrolides | Conventional macrolides | %T>MIC [192] | Max %T in the dosing interval > MIC | |
| Azithromycin | AUC/MIC [9] | Optimal daily amount | ||
| Glycopeptides | – | AUC/MIC [57] | Optimal daily amount | |
| Tetracyclines | – | AUC/MIC [191] | Optimal daily amount | |
| Oxazolidinones | Conventional oxazolidinones | %T>MIC [192] | Max %T in the dosing interval > MIC | |
| Linezolid | AUC/MIC [191] | Optimal daily amount | ||
| Concentration-dependent | Aminoglycosides | – | Cmax/MIC or AUC/MIC [10] | Max peak concentration or optimal daily amount |
| (Fluoro)quinolones | – | Cmax/MIC or AUC/MIC [66] | Max peak concentration or optimal daily amount | |
| Metronidazole | – | Cmax/MIC or AUC/MIC [192] | Max peak concentration or optimal daily amount |
2.1.1. Beta-lactam antibiotics
The beta-lactam agents act principally by causing irreversible inhibition of bacterial cell wall synthesis, achieved by covalent binding to penicillin-binding proteins [11,12]. Beta-lactams are renally cleared, and have a wide therapeutic index [13]. On the basis of in vitro and in vivo data, the pharmacodynamic target for beta-lactam therapy is the fraction of time per dosing interval that the free drug concentration exceeds the minimum inhibitory concentration (MIC) of the organism (denoted by %T>MIC) [14]. This is thought to be due to the fact that the penicillin binding protein requires near saturation before cytotoxicity occurs, implying that the minimum concentration required for effect is close to the maximum possible effect. The standard therapeutic goal for penicillins is %T>MIC of 30–40% which has generally been derived from observing that this value leads to decrease in bacterial load either in vitro or in tissues of interest during animal in vivo studies [15]; although neonates are considered functionally immunocompromised and so 40–50% or more is often recommended [16].
2.1.1.1. Penicillins
Neonatal studies on benzylpenicillin (penicillin G) [17], intravenous flucloxacillin [18], intravenous amoxicillin [19,20] and piperacillin/tazobactam [21] have been conducted, each of which involved estimating the parameters of a population PK model and using them to simulate dosing regimens that attain in vitro-derived circulating %T>MIC values.
Another approach to defining dose regimens can be seen with the example of ticarcillin/clavulanate in paediatric cystic fibrosis (CF) patients who received high-dose intermittent ticarcillin-clavulanate (above the maximum dose approved by the FDA (United States Food and Drug Administration) [22]. The authors had PD data and obtained PK parameter estimates for intravenous ticarcillin in children and adults with CF from the literature [23], which were used to simulate serum free drug concentrations.
No paediatric penicillin study was found which used microbiological or clinical PD outcomes observed in the patients undergoing PK sampling, and so it would appear that dosing guidelines for these agents are exclusively based on simulations or even simple extrapolations to reach PD targets derived in adult and in vitro studies. The use of different PD targets and breakpoints between studies is notable; importantly, some breakpoints have since changed (such as the piperacillin breakpoint for Pseudomonas) [24,25], and will be expected to increase further as increasing antimicrobial resistance is encountered.
2.1.1.2. Cephalosporins
A PK study on cefuroxime was undertaken in 15 paediatric cardiovascular surgery patients undergoing cardiopulmonary bypass who received prophylaxis against surgical site infection [26]. Simulations were used to compare two dosing regimens; single and double dose. Both regimens were found to result in a free drug concentration exceeding the MIC value for staphylococcal species of 8 μg/mL (taken from the literature) for the whole dosing interval, so therefore there was no benefit in a second dose.
Ceftriaxone was studied using published serum concentrations of ceftriaxone from 78 Japanese paediatric patients from 8 previous studies; simulation was then used to assess %T>MIC in different dosing regimens [27]. The PD target of %T>MIC used MICs for several pathogens taken from the literature and the analysis suggested that a once daily dosing regimen (of 20 mg/kg) was appropriate in most cases.
The PK of cefotaxime and its active metabolite, desacetylcefotaxime (DACT), were estimated in a study of 37 neonates on extracorporeal membrane oxygenation (ECMO) [28]. It is known that ECMO may alter drug disposition in various ways, for example by increasing the apparent volume of distribution, or by reducing drug clearance [29]. In the neonatal cefotaxime study the PD target was set at > 50%T>MIC, and using MIC values from the literature for pathogens that may cause neonatal meningitis, with consideration of drug protein binding, simulated concentration-time curves were constructed for each individual.
As with the penicillins, a heavy reliance on linking circulating in vitro derived breakpoints has been used to define cephalosporin dose recommendations.
2.1.1.3. Carbapenems
There have been several population PK studies of meropenem involving neonates, infants and/or children [30–37]. A population PK study was conducted in 37 infants following a single dose of meropenem (10 or 20 mg/kg) [30]. Simulation was then used to evaluate the PD target attainment of %T>MIC ≥ 60%, using MIC distributions for relevant common pathogens. In another study, neonates were randomly allocated to receive a single intravenous dose of 10, 20, or 40 mg/kg [33]. Simulation was used to estimate probability of target attainment with different doses, infusion durations and dose intervals, using MIC values from the literature. The final neonatal study investigated the impact of short versus long infusion duration of meropenem in 19 very-low-birth-weight neonates [35]. While shorter infusions gave a higher Cmax, the %T>MIC was higher with prolonged infusions.
Meropenem plasma and cerebrospinal fluid (CSF) PK were investigated in a study of 188 infants with suspected/complicated intra-abdominal infections using sparse sampling [37]. The PD targets were > 4 μg/mL for 50% of the dose interval and > 2 μg/mL for 75% of the dose interval, chosen due to the reduced immunocompetence of neonates. CSF was obtained from 6 participants, but the reported 70% penetration of meropenem into the CSF was erroneous since CSF:plasma ratio was calculated rather than AUCCSF:AUCplasma [37]. Two further studies in children developed a population PK model and then used simulation to recommend an optimal infusion length [34] or absolute dose [32] for infections with common bacteria without further specifying an indication.
The only meropenem study to link PK with PD in the recruited patients used data from 99 individuals to develop a population PK model [31]. The validated model was used for simulation to predict meropenem plasma concentrations in 37 paediatric meningitis patients, following 40 mg/kg meropenem, with MICs of the causative bacteria known. The causative pathogens were eradicated in all cases, so no break points were identified.
A paediatric population PK imipenem meta-analysis in NONMEM has recently been undertaken using pooled individual patient data from 15 previous studies [38]. The dataset included PK samples from 60 neonates and 39 children. The PD target was 40%T>MIC, using MIC distributions from the literature for 5 common pathogens. Simulations were conducted to assess the probability of target attainment with different doses and infusion lengths.
In summary the population PK of the carbapenems have been comprehensively studied in children, with dose recommendations largely being derived by simulation to attain an in vitro derived target.
2.1.2. Aminoglycosides
Aminoglycosides exhibit antibacterial activity by binding to the ribosome and interrupting protein synthesis. This intra-cellular mode of action is the reason for the observed post-antibiotic effect, whereby bacterial death continues to occur after aminoglycoside concentrations have fallen to low levels. Furthermore, many pathogens exhibit aminoglycoside adaptive resistance whereby the transporters responsible for internalising the drug are transiently down-regulated [39,40]. Thus the PD target for aminoglycosides is high peak relative to the MIC (to penetrate the bacteria) and low trough (to provide a drug-free period and minimise adaptive resistance and toxicity), and this strategy has been shown to correlate with clinical response in adults (Fig. 1) [10].
Fig. 1.
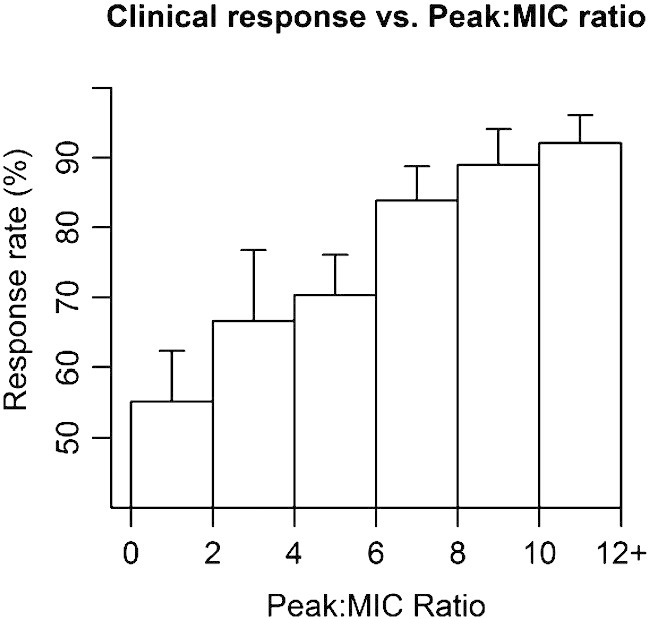
Relationship found by Moore et al. relating clinical response to Cmax:MIC ratio of aminoglycosides in a meta-analysis of adult studies [10].
Several population PK studies have been conducted on gentamicin in children of different ages, with dose recommendations usually being made using simulation [41–46]. A recent paper by Mohamed et al. has sought to use in vitro data to more fully investigate gentamicin PD by developing a semi-mechanistic PKPD model [47]. In vitro experiments using E. coli (with MIC of 2 mg/L) were performed in order to define the time-kill curve. In order to characterise a model that would take into account the development of adaptive resistance, two compartments were defined: one for drug-susceptible, growing bacteria, and the other for insusceptible, resting bacteria. They found that when describing the killing effect of gentamicin, a saturable logistic Emax model was significantly better than a linear model. Adaptive resistance was incorporated in the model by using a binding function. When gentamicin was present and thus adaptive resistance could develop, the binding function reduced Emax and resulted in lower killing rates. It was also found that the half-life of the development of adaptive resistance is concentration-dependent, and it takes longer to develop at lower drug concentrations. In this study the rate of returning to susceptibility was fixed (to 50 h, the lowest value that did not reduce the fit of the model) due to uninformative data. The estimated maximal killing rate of 51/h confirmed the fast bactericidal effect of gentamicin. It was also found that even though the dosing regimen 4 mg/kg every 24 h produces lower peaks in preterm neonates, the bactericidal effect may be sufficient. By linking dynamic in vitro experiments with rigorous clinical PK data, this study may point to a better way of incorporating PD data when making dosing recommendations in situations where clinical PD data are difficult to obtain.
Amikacin PK has also been extensively studied in children [48–50], and one of these by Sherwin et al. did attempt to incorporate clinical PD endpoints [51]. This retrospective study included 80 neonates, 26 of whom had 35 confirmed septic episodes between them. A population PK model was developed and the PD was a logistic regression approach with a dichotomous outcome i.e. success/failure of the treatment. It was found that therapeutic failure depends only on amikacin peak concentration/MIC ratio.
Tobramycin is frequently used for treatment of pulmonary infections in CF, as it is highly effective against Pseudomonas aeruginosa. Several PK studies have been published in children without attempting to model PKPD relationships within the included patients [52–55]. One study looked at all the aminoglycosides in 45 HIV-infected children and reported whether peak concentrations reached a target level for the given dosage regime [56].
Overall the aminoglycosides have been the focus of numerous PK studies, which is probably the result of them being the subject of therapeutic drug monitoring (TDM) due to a narrow therapeutic index. PD modelling has focussed on efficacy and some particularly interesting work on integrating dynamic in vitro data has been done [47], but no studies that we found investigated the PKPD relationship with the development of nephrotoxicity (except some animal studies [57] and reviews [58] that linked aminoglycoside dose with nephrotoxicity); this may become increasingly important to understand, as the pressure to increase doses to meet rising pathogen MICs continues.
2.1.3. Glycopeptides
Glycopeptides bind to alanine during bacterial cell wall synthesis thus interrupting this process by preventing the membranes cross-linking. The dynamics of this effect are time dependent and the relationship between dose and effect is best characterised by AUC:MIC ratio [59]. Several studies have investigated the PK of vancomycin (mainly in neonates and infants) [60–64] and also teicoplanin [65–67]. However, the PD link in these studies has been through simulations to achieve target concentrations rather than looking at the PD in the included subjects.
2.1.4. Quinolones
Quinolone antibiotics inhibit DNA gyrase and, in common with the aminoglycosides, this would suggest that peak:MIC ratio would be the key PD endpoint. During the development of levofloxacin, which remains a gold standard against which other clinical antimicrobial studies ought to be measured, the peak:MIC ratio at the site of infection was shown to correlate with clinical response in adults [68]. The use of quinolones in children has been limited due to fears of the potential to cause cartilage damage, as demonstrated in a study where Beagle dogs developed arthropathy after ciprofloxacin administration [69].
Despite this, quinolones are increasingly used in children, and population PK studies of ciprofloxacin have been published [70–72]. No linked PKPD studies have yet been performed in paediatric patients to reflect the seminal adult papers by Ambrose et al. [73] and Forrest et al. [74], which supported the use of AUC/MIC ratio as the most clinically relevant PD target. However, further PKPD studies will be required to verify which PKPD index is most suitable for targeting quinolone therapy in children.
2.1.5. Macrolides
One population-PK study involving macrolides has been conducted, focusing on the PK of single-dose azithromycin in 12 preterm neonates at risk of Ureaplasma colonisation [75]. The pharmacodynamic target was to maintain azithromycin plasma concentrations above the MIC50 (1 μg/mL); this value was obtained from Ureaplasma isolates from 25 neonates at the local institution. A single dose (10 mg/kg/day) or multiple doses of this amount was simulated to achieve this target.
2.1.6. Other antibacterial agents
Metronidazole is used to treat anaerobic infections and has an intracellular mechanism of action. Recently there have been studies looking at the population PK of metronidazole in neonates, and dose recommendations have been made based on checking whether the predefined PD targets (steady-state plasma concentration of metronidazole>MIC of anaerobic microorganism) were met [76–78], and whether there were any anaerobic infections present or not [76]. A lack of clarity on optimal metronidazole PKPD endpoints means that there is a need to obtain further clinical dose–response data in children.
Linezolid is a synthetic antibiotic of the oxazolidinone class, effective against Gram-negative microorganisms. The PD target for linezolid is a maximised ratio of AUC:MIC [191]. To our knowledge there are no published population PK studies of linezolid in children, only a non-compartmental PK analysis [79].
2.2. Fungal infections
2.2.1. Azoles
The azole antifungals inhibit ergosterol synthesis in the fungal cell wall. Clinical data on resolution of Candida infection with measured MIC values regressed against typical exposures for dose showed AUC:MIC ratio to predict effect in adults [80]. Azole antifungals appear to display post antimicrobial effects whereby cytotoxic effects continue even once concentrations have fallen below the MIC [81].
The dose recommendations of fluconazole in neonates are based on preliminary PK studies that suggested a dose of 6 mg/kg daily results in serum concentrations that were above the MIC of Candida parapsilosis 72 h post dose [82]. Clearance of fluconazole rises with gestational age, approximately doubling in the first 4 weeks of life, meaning prolonged dosing intervals (i.e. 48–72 h) are often used in clinical practice [83,84]. In the treatment of invasive candidiasis, a higher neonatal dosage of 12 mg/kg daily in neonates < 29 weeks gestation results in a median AUC of ~ 700 mg/L h (which approach drug exposures required for successful treatment in animals studies) [85]. Loading doses enable AUC:MIC targets to be achieved rapidly [86]. TDM for fluconazole is not generally performed, but assays are available and some have argued it may be beneficial particularly in the neonatal population [87]. No linked PKPD or modelling studies have been carried out to inform fluconazole dosing in infants and older children. The pharmacokinetics appear linear, with a dosage of 8 mg/kg daily producing an AUC of ~ 200 mg/L h, which is comparable to an average adult receiving a dose of 200 mg daily [88]. Higher exposures are usually required for the treatment of disseminated candidiasis in adults, suggesting that 8 mg/kg may be inadequate in children, especially against organisms with reduced susceptibility.
Itraconazole is available in a capsular form, a cyclodextrin-based oral solution, and an intravenous preparation (not widely available in the U.S. or Europe). Doses of 2.5-5.0 mg/kg daily have been used with apparent efficacy [89–92]; however, there are no PKPD studies available to guide dosing. Limited data suggest the PK of itraconazole are non-linear and highly variable, and that weight-based dosing leads to sub-therapeutic dosing in smaller children [93,94]. When itraconazole is used, TDM is generally advocated with a target trough levels of > 0.5 mg/L (using HPLC) and 5–17 mg/L (using bioassay) are extrapolated from studies in adults [95–97].
Voriconazole is, at present, infrequently used in neonates, its main use being in the treatment of invasive aspergillosis and in fluconazole-resistant candidiasis [98–100]. Extreme variability in serum concentrations is apparent, with no clear relationship between drug exposure and weight-based dosages [99]. Voriconazole PK in older children and adults is characterised by significant variability in concentration–time profiles and non-linear clearance [101]. Population PK models fitted to paediatric datasets have been used to define regimens for intravenous voriconazole that produce drug exposures comparable with those seen in adults [102]. Population PK analyses have consistently identified considerable PK variability in children, usually making dosing recommendations based on simulation to achieve adult exposures, and providing the basis for TDM [103–107]. A trough concentration target of 1–5 mg/L is used in adults receiving voriconazole [108]. In children, trough plasma concentrations < 1 mg/L are associated with higher probability of death and extrapolation of the adult upper boundary of 5 mg/L to paediatric populations is disputed given the lack of hepatotoxicity seen in children [103,106]. One population PK model has attempted to provide TDM guidance by showing that an increase in dose of 1 mg/kg results in an increase in the median trough concentrations by 0.5 mg/L [106].
Posaconazole is only currently available as an oral suspension, and is only licenced for use in children over twelve years. No studies were found that examine posaconazole PK linked with PD in children. Twelve children aged 8–17 years receiving posaconazole 800 mg daily in divided doses had mean serum concentrations and clinical outcomes comparable with adults [109]. Further studies are required to define appropriate regimens for posaconazole use in children.
2.2.2. Echinocandins
Echinocandins inhibit fungal cell wall synthesis and have activity against Candida and Aspergillus species. This concentration-dependent effect has translated into the PKPD target being circulating Cmax:MIC ratio or tissue AUC:MIC ratio which have been defined in animal models [110]. Echinocandins display an apparent post-antimicrobial effect, which is probably due to tissue accumulation of the drug resulting in antifungal activity after plasma concentrations have fallen [110].
The pharmacokinetics of caspofungin have been described in 18 neonates and mean peak and trough concentrations were compared to adults receiving 50 mg, although clinical efficacy was not described [111]. In 39 neutropenic children receiving antifungal prophylaxis, daily doses of 1 mg/kg daily resulted in significantly lower drug exposures than those observed in adults receiving a standard 50 mg daily. Surface area-adjusted dosing (70 mg/m2 loading followed by 50 mg/m2 maintenance) resulted in drug exposures more comparable with adults [112,113]. No linked PKPD modelling studies were found for caspofungin.
Micafungin has been studied in a dose-ranging study on neonates using population PK modelling; this study identified an optimal dose of 10 mg/kg daily, which resulted in AUCs approaching near maximal clearance of the fungal burden within the CNS (an important target tissue compartment in this age) [114]. Population PK modelling of micafungin in older children showed doses of 2 mg/kg achieve comparable exposure with adults [115,116].
There are few data for PKPD of anidulafungin in neonates and children. The drug has a unique mechanism of clearance, whereby the parent compound undergoes spontaneous non-enzymatic hydrolysis. In neutropenic children aged 2–17 years dosages of 0.75 and 1.5 mg/kg daily resulted in drug exposures that are comparable with adults receiving 50 and 100 mg daily, respectively [117,118].
2.2.3. Polyene antimycotics
Amphotericin B is the only systemically available polyene, which binds to ergosterol causing membrane depolarisation and giving a broad spectrum of activity against the majority of pathogenic yeasts and moulds. Solubilisation of amphotericin B for parenteral administration was initially achieved using the bile salt deoxycholate (amphotericin B deoxycholate) and subsequently by incorporation into various lipid formulations (e.g. amphotericin B lipid complex and liposomal amphotericin B). The PKPD profiles of each of these formulations are distinct and remain relatively poorly understood, particularly in children. The pharmacokinetics of amphotericin B deoxycholate are highly variable in neonates, and this may lead to treatment failure or toxicity. Amphotericin B doses of 0.25–1 mg/kg administered daily to infants results in lower serum concentrations compared with larger children and adults [119,120]. Thirteen premature neonates treated for invasive C. albicans infection with this dosing range had peak unbound drug concentrations below reported MIC values in 25% of cases, resulting in death attributed to fungal sepsis in two cases. Correlations between clinical outcomes, serum levels and pre-defined MIC targets have not, however, been studied in larger cohorts of neonates or children [121].
The various lipid formulations of amphotericin B are increasingly used in place of amphotericin B deoxycholate, predominantly because of their more favourable toxicity profiles. Liposomal amphotericin B and amphotericin B lipid complex (ABLC) are the most commonly used compounds in the U.S. and Europe. Despite extensive information on clinical usage and safety, the PK of these compounds are poorly understood in neonates and children. PK modelling of data describing 28 neonates suggested that 2.5 to 5 mg/kg is safe and effective in the treatment of invasive candidiasis [122]. Population PK modelling of liposomal amphotericin B has been used to describe data from 39 children with haematological malignancy. These analyses suggest that doses of 4 and 7.5 mg/kg daily achieve drug exposures that exceed target MIC values for Candida and Aspergillus species, respectively. Children < 10 kg probably required higher doses, although these have not been defined [123]. Pharmacokinetic studies that enable equivalence of drug exposure with these formulations to be established are urgently required.
2.3. Viral infections
Whilst bacteria in particular present potential drug targets that are usually quite distinct from host substrates, viral replication relies on the sequestration of host cell machinery, and all infections have an intra-cellular component, which both make developing effective antiviral therapies more difficult. Due to this, antivirals tend to have a narrower therapeutic index than antibacterials, which provides a compelling case for using PKPD modelling to optimise dosing. Antiviral agents generally utilise the following mechanisms: interrupting viral nucleic acid replication, inhibiting attachment to host cells and viral entry, or blocking viral un-coating once in the cell. A comprehensive review of antiviral (and antiretroviral) pharmacology and PK available for children is given by Kimberlin [124].
2.3.1. Nucleoside and nucleotide analogues
Aciclovir is a purine nucleoside which is selectively phosphorylated by viral thymidine kinase; subsequently, the active aciclovir triphosphate preferentially inhibits viral DNA polymerase. Due to its low oral bioavailability, a prodrug of aciclovir called valaciclovir has been developed. Valaciclovir undergoes conversion to aciclovir during first-pass metabolism, thus enhancing bioavailability.
The PK of aciclovir have been extensively studied in children, and in many cases an attempt to link circulating concentrations, either measured or predicted from a model, has been made. In one of the earliest published PK studies, dose escalation of intravenous aciclovir (5, 10 and 15 mg/kg) was performed in neonates infected with herpes virus, and measured PK was compared against in vitro IC50 values [125]. A more sophisticated approach was taken by Sullender et al. [126], who used simulation (without accounting for uncertainty) from a model built to describe the PK of an oral aciclovir suspension to show that intravenous dosing would be required to achieve target IC50 values although full details on the in vitro methods and PD endpoints are lacking in these studies. Three further studies reported PK metrics [127–129] and linked them to a measure of clinical outcomes (e.g. clinical response: yes/no [127]) in the included patients, but did not then go on to attempt to model this relationship.
The first population PK study to implement an NLME approach was published by Tod et al. in 2001 [130]. This large study in 102 neonates and infants developed dosing guidelines based on achieving time greater than in vitro IC50 values by simulating from the PK model. More recent population PK modelling has looked at bioavailability differences between aciclovir and valaciclovir in children [131], and again the linking to PD has been done using simulation and comparison with in vitro inhibition data [132].
Penciclovir and its oral prodrug famciclovir have a similar spectrum of activity to aciclovir; both non-compartmental [133,134] and population PK models [135] have been published in children, but none have sought to model the link between PK and PD endpoints.
Ganciclovir is a guanine analogue with inhibitory activity against human herpes viruses. It has been used in children mainly for cytomegalovirus (CMV) infections which are not susceptible to aciclovir due to the lack of expression of the unique thymidine kinase. Possibly due to its use against more serious infections, there are more PKPD studies involving ganciclovir and its oral prodrug valganciclovir in children.
Several studies have used both traditional and population PK modelling to describe ganciclovir and valganciclovir concentrations and then link these with in vitro-derived targets or clinical response in an attempt to define a suitable dose [136–141]. The problem with this approach was highlighted by Veniza et al. [142], who showed that out of eight patients with measured ganciclovir concentrations (following oral valganciclovir administration) which exceeded the in vitro IC50 for Epstein Barr Virus (EBV), four of them did not experience viral suppression. This hints at the problem outlined by Luck et al. that the target circulating concentration for ganciclovir is often unknown, and further understanding of PKPD relationships is required [143].
In 36 HIV-infected children with CMV infection, oral ganciclovir PK was defined in ascending doses by a non-compartmental approach and followed up over up to 168 weeks with CMV being qualitatively analysed and tested for susceptibility [144]. A study on congenital CMV infection looked at intravenous ganciclovir and oral valganciclovir pharmacokinetics and their link with viral load over a 42 week treatment course [145]. This study looked at the change of viral load from baseline, showing that antiviral therapy significantly reduced viral load especially in patients with higher baseline, although the authors did not develop a full linked PKPD model for this relationship. Many of the ganciclovir and valganciclovir studies reported haematological toxicities, in particular neutropenia, and further work on this by modelling the PKPD link between ganciclovir PK and neutrophil count may point the way to defining a maximum tolerable dose.
Ribavirin is a structural analogue of guanosine and has a broad spectrum of antiviral activity. Several studies have characterised the PK of ribavirin in children via inhaled, intravenous and oral routes, although no model for both PK and PD has been presented [146–149]. Finally, in this group of agents, the nucleotide analogue cidofovir is used in children for indications such as ganciclovir-resistant CMV, but again no linked PKPD studies have been published in this population.
2.3.2. Interferons
Interferons induce the production of a family of proteins that inhibit viral protein synthesis, cause viral RNA deactivation, and can enhance phagocytosis, therefore providing broad non-specific antiviral effects. Interferon-alpha is commonly used in combination with oral ribavirin for the treatment of hepatitis C virus (HCV). One PKPD study investigated the combination of interferon-alpha and ribavirin in children with chronic HCV infection [150]. This study measured detailed PD outcomes including quantitative PCR for viral loads, but did not model their time course. There is increasing interest in fitting data of this type to mechanistic PD models, which can then be used to extrapolate the combination of dose and course length required to suppress or cure the infection.
2.3.3. Neuraminidase inhibitors
The neuraminidase inhibitors are used to counter influenza A and B through blockage of viral entry into host cells, as well as inhibiting the release of new virions [151]. A detailed review of oseltamivir PKPD data in children found that dosing guidelines were mainly based on extrapolations of circulating PK to in vitro inhibitory concentrations [152], although recent data on PK and influenza viral loads, and the development of resistance, may in the future yield a PKPD model [153]. No PKPD studies for zanamivir or laninamivir were found, but a large study including 117 children receiving peramivir did not find a link between AUC or Cmax versus markers of infection resolution, although this was not done with a joint PKPD approach [154].
3. Immunology
3.1. PKPD modelling in HIV infection
The PK of most commonly used antiretrovirals have been studied individually in children. How best to link these PK data with important PD markers is only gradually becoming clear, as combination therapy means that knowledge of single drug PK does not give the whole picture. Antiretroviral therapy (ART) has two important PD effects. Firstly, by interrupting the viral life cycle, it suppresses the viral load to levels undetectable by most assays. Secondly, this reduction in the viral burden allows patients' own homeostatic mechanisms to replenish the CD4 T-cell pool. Both effects are important aims of therapy. A number of models have been proposed for both processes, and for joint descriptions of viral and T-cell responses, although most have only been applied to adult data, and very few have also incorporated drug PK.
3.1.1. Modelling viral dynamics in children
Various approaches have been taken to quantifying and modelling viral suppression in HIV-infected children on ART. The simplest pharmacodynamic studies assess correlations between measures of drug exposure (AUC, peak or trough plasma concentration) and viral suppression (typically time to undetectable viral load, or reduction in viral load by a specified time point). Studies of this kind were comprehensively reviewed in 2011 by Neely and Rakhmanina [155].
More advanced mechanistic models of the virological activity of ART predict a biphasic decay in viral load [156], and models of this kind have been successfully fitted in a mixed-effects framework to data from infants (< 2 years) starting ART [157]. This study showed no significant difference between the rates of viral eradication in different age groups, but did find faster decay in the viral load (in both phases) where children had received a higher dose of ritonavir, the antiretroviral in question.
3.1.2. Modelling T-cell dynamics in HIV-infected children
In some studies, investigators have ignored the role of viral suppression altogether, focussing instead on T-cell reconstitution. A simple exponential model has been employed in a mixed-effects statistical framework to describe recovery of CD4% in HIV-infected children starting ART, and to identify explanatory factors, which included mean viral load on ART and age at ART initiation [158]. The same function was applied to CD4-for-age in European and Brazilian children to construct predictive models of long-term CD4 counts, given characteristics measurable at ART initiation (again including age and viral load) [159]. Models of this type have great potential for informing guidelines for ART initiation and individual patient management, as these children increasingly survive into adulthood and long-term immunological health becomes a more pressing question. Finally, the same simple exponential model was also used to demonstrate contrasts between recovery in children and in adults, which are likely to be due to the importance of the thymus and the naïve T-cell pool to children's T-cell dynamics [160].
3.1.3. Joint modelling of viral and T-cell dynamics
A number of joint models of viral and T-cell dynamics in HIV have been developed and applied in a mixed-effects framework, many of which use adult data. While adult models provide an important starting point, epidemiological evidence suggests that both virological and immunological responses to HIV infection and ART are different in adults and children [161]. In the simplest joint models, CD4 count and viral load following ART initiation are described using piecewise linear mixed-effects models, allowing correlation between random effects in the intercepts and slopes of the viral and CD4 responses with time on ART [162]. While not a pharmacodynamic model in the conventional sense, this approach explores and describes the response to ART in an empirical way while acknowledging the existence of a biological relationship. In adults, the model identified predictors of virological and immunological responses to ART, and also showed that a greater decrease in viral load was associated with better immunological response.
Most joint models of CD4 count and viral load used for population pharmacodynamic studies have been extensions of the model proposed by Perelson et al. [156]. The original model described four compartments: for non-infected and infected T-cells and infectious and non-infectious virus (Fig. 2). This model has been used for parameter estimation, with population data from adults recently infected with HIV or starting ART [163,164]. One extension of the same model included a compartment for quiescent cells not liable to viral infection [165], while another modelled a compartment of latently-infected cells which, though infected, do not produce further virus [166].
Fig. 2.

Model for HIV-T-cell dynamics model proposed by Perelson et al. [156]. The numbers of uninfected CD4+ T-cells were assumed to remain constant throughout (grey box). Outlined boxes indicate quantities modelled by differential equations; solid arrows represent first-order rate constants, and the dashed arrow indicates the effect of viral load on the infection rate. Before ART, all new virions produced are infectious. T-cells are infected at a rate proportional to the number of infectious virions, kVI, die at rate δ and produce new virus at rate Nδ (i.e. N new virions per cell death). Virus is cleared at rate c. On ART, all newly produced virions are non-infectious. As infectious virus is cleared the T-cell infection rate decreases, numbers of infected T-cells fall and fewer new HIV particles are produced. The reducing viral production rate combines with constant clearance to give a falling viral load. Joint models of viral and T-cell dynamics have usually allowed for T-cell reconstitution by adding a differential equation for uninfected cells, T, with a constant zero-order input representing T-cell production [164,166].
The major strength of the Perelson-type models as a basis for population-based statistical models is that they propose a realistic mechanistic basis for changing viral loads and CD4 counts, such that their parameter values quantify meaningful biological quantities including, importantly, drug effect. However, it is usually impossible to estimate all of a model's parameters and some must be fixed at values estimated by other methods. Extensive and complete analysis of the sensitivity of results to these assumed values is not always carried out or fully reported, and inference methods do not usually quantify and describe parameter collinearities or uncertainty in model structure. This is probably because the most obvious tools for doing so are Bayesian methods, which are often prohibitively computationally expensive. Another criticism is that while this class of models impressively reproduce dynamics in states of change — for example, immediately following HIV infection or ART initiation — they fail to explain the slow decline in CD4 count over several years, which is characteristic of chronic HIV infection. Thus, while they are good representations and helpful descriptions of dynamic states, they are not able to shed light on the effects of HIV on healthy T-cell homeostasis — effects which may be responsible for important phenomena such as incomplete T-cell reconstitution even on long-term ART [167].
3.1.4. PKPD models of HIV infection
Most modelling studies which incorporate a drug effect on HIV and/or CD4 dynamics do so using a single parameter which is “switched on” for the period over which the drug is assumed to be effective [165,166]. Models incorporating pharmacokinetics are less common, and limited mainly to theoretical rather than data-based studies [168,169]. Nevertheless, these models could prove very helpful in understanding the effects of poor adherence which can be a particular issue in children because of intolerability and social considerations.
Another interesting avenue for investigation might be the incorporation of PK models into mechanistic models of other drug effects in HIV. In one example of such a study, modelling was used to quantify effects of IL-7 therapy on CD4 T-cell survival and proliferation in HIV [170]. Another example is evidence that the integrase inhibitor raltegravir increases production of naïve T-cells, possibly via an effect on thymic activity, and modelling might also prove a productive way to investigate this possibility further [171].
In summary, a number of separate and joint PD models of viral and CD4 T-cell dynamics on ART have been developed and applied to data. Some models have incorporated a drug effect, though usually as a simple modification of one of the parameters. There is still much scope for the development of these models by including realistic pharmacokinetics and this might produce important conclusions about the effects of planned or unplanned treatment interruptions. Recently, a joint PKPD model including mechanistic viral dynamics, CD4 count and the PK of each of three antiretrovirals was published on data from children aged 2 to 15 years using NLME [172]. The authors of this study were able to separate the relative effects of each drug and develop predictive models of virological failure based on individual estimates of drug inhibition scores. The use of this approach for secondary analysis of paediatric data already collected as part of routine care or clinical trials, could be valuable for elucidating differences between children and adults, understanding immunological development in HIV and explaining differences between individual children's responses to HIV and ART [173,174]. Models also have potential as clinical tools for predicting long-term effects of potential ART strategies and tuning drug doses [159,175].
3.2. Haematopoietic stem cell transplant
3.2.1. Introduction
Haematopoietic stem cell transplants (HSCTs) are given as treatment for many immune system disorders. The stem cell source defines treatment classification, which can be either bone marrow transplants (BMTs), peripheral blood stem cell transplants (PBSCTs) or umbilical cord blood transplants (CBTs). A further layer of complexity is added by the donors also coming in many different categories (e.g. related/unrelated, matched/unmatched). Before an HSCT, a child will usually be given a conditioning regimen to reduce or ablate the host immune system in order to prevent graft rejection, decrease the incidence and severity of graft versus host disease (GvHD), and, in the case of leukaemia, prevent relapse. The conditioning can be radiotherapy, chemotherapy, anti-lymphocyte antibodies, or a mixture of these. This leaves the patient severely immunocompromised, and therefore vulnerable to opportunistic infections. Following HSCT, short-term complications and long-term successful outcomes are associated with the rate and extent of recovery in the child's immune system. Understanding the dynamics of immune reconstitution both immediately after the HSCT and in the long term is therefore vitally important. Table 2 gives an overview of the mechanism of action and references to the PK for some drug therapies used in conditioning and post-transplant GvHD prophylaxis. Clearly, the multi-faceted management approach, which includes non-drug therapies, means that linked PKPD studies of immune reconstitution are difficult to interpret. In addition to PK studies on individual drugs, there is a parallel, almost separate literature on the PD of interest: immune reconstitution post-transplant. In this section these analyses are reviewed, together with consideration of how PKPD in HSCT could be linked in the future.
Table 2.
A summary of the mechanisms of action of drugs used for conditioning and prophylaxis in HSCT patients, with references to paediatric PK studies.
| Drug type | Type | Mode of action | PK Studies |
|---|---|---|---|
| Conditioning | |||
| Fludarabine | Purine analogue | Interferes with ribonucleotide reductase and DNA polymerase, preventing DNA synthesis. | Plunket et al. [195], Salinger et al. [196]. |
| Cyclophosphamide | Nitrogen mustard alkylating agent | Attaches alkyl group to guanine bases in DNA, thus inducing cell apoptosis. Attacks resting and dividing cells. | Tasso et al. [197], Van Hasselt et al. [198]. |
| Melphalan | Nitrogen mustard alkylating agent | Attaches alkyl group to guanine bases in DNA, thus inducing cell apoptosis. Attacks resting and dividing cells. | Van Hasselt et al. [198]. |
| Busulphan | Alkylating anti-neoplastic agent | Alkylation causes adenine–guanine cross-links, causing cell apoptosis. Attacks resting and dividing cells. | Van Hasselt et al. [198]. |
| Treosulphan | Alkylating anti-neoplastic agent | Alkylation causes adenine–guanine cross-links, causing cell apoptosis. Attacks resting and dividing cells. Less toxicity than with busulphan therapy. | Główka et al. [199]. |
| Alemtuzumab | Monoclonal antibody | Binds to CD54, a protein found on the surface of mature lymphocytes but not on haematopoietic stem cells. | Elter et al. [200]. |
| Anti-thymocyte globulin | Polyclonal antibody | Rabbit derived antibodies against human T-cells, formed by injecting human lymphatic cells into a rabbit and harvesting the antibodies produced. | Call et al. [201], Seidel et al. [202]. |
| Prophylaxis | |||
| Cyclosporin | Immuno-suppressant | Lowers immune response and activity of T-cells. Binds to lymphocyte cyclophilin, and the resulting complex inhibits calcineurin, preventing transcription of IL-2. | Willemze et al. [203]. |
| Methotrexate | Antimetabolite | Inhibits metabolism of folic acid, thereby inhibiting purine base synthesis. Mainly inhibits rapidly proliferating cells. | Van Hasselt et al. [198]. |
| Mycophenolate | Immuno-suppressant | Inhibits inosine monophosphate dehydrogenase, the enzyme controlling the rate of synthesis of guanine monophosphate in purine base synthesis in proliferating B- and T-cells. | Downing et al. [204]. |
3.2.2. Assessing immune reconstitution
Studies to date have tended to use two methods to assess reconstitution: (1) the concentration of lymphocyte subsets at certain fixed time points after the HSCT; and (2) the time taken for lymphocyte subsets to reach fixed concentrations.
The first method is used to assess differences in the extent of reconstitution. One such study found that B cell reconstitution is faster after CBT than BMT [176]. Another found that reconstitution was faster with matched sibling donors rather than family mismatched or unrelated donors [177], whilst others found T-cell reconstitution at 6 months was significantly delayed in patients having autologous PBSCT compared with allogeneic BMT or PBSCT [178]. A study comparing reconstitution at 90 days found that unrelated CBTs had faster B-cell and Natural Killer (NK) cell reconstitution, but delayed T-cell reconstitution when compared to matched sibling BMTs and unrelated BMTs. They also found that patients with consistently higher CD4 T-cell counts had a better rate of survival [179]. A study on allogeneic HSCT patients found at one month that younger patients reconstituted CD8 T-cells more slowly, and that at one year measurable EBV in the blood had a negative impact on the reconstitution of CD3 T-cells [180]. A study assessing the effects of reduced intensity conditioning found that there is more extensive recovery of CD3 T-cells and NK cells by four months after HSCT compared with those given myeloablative conditioning, and that therefore reduced conditioning accelerates immune reconstitution [181].
This method is also used to find indicators for improved long-term survival, taking white blood cell counts above certain thresholds at certain time points. One study uses a CD4 count over 115 cells/μL at 20 days after HSCT [182], whilst another used a CD4 count of 86 cells/μL at 35 days [177]. A study in adults used an absolute lymphocyte count of 150 cells/μL at 30 days for better non-relapse mortality and improved survival [183]. A more sophisticated method involved forming an ellipsoid reference domain for the normal values of a combination of three lymphocyte subtypes, and classifying people into high- and low-risk groups. Those in the low-risk group had a higher chance of survival [184].
The second method is generally used to compare the rates of reconstitution. One study found unrelated CBT leads to faster B-cell reconstitution and slower CD8 T-cell reconstitution compared with unrelated BMT [185], while another that PBSCT led to faster reconstitution than BMT [186]. Work looking at the effects of the conditioning drug anti-thymocyte globulin (ATG) found that time to reach normal CD4 T-cell counts were almost doubled in those who received ATG, but that CD8 T-cells were unaffected [187]. The high dose ATG group were also more likely to experience life-threatening infections. Another investigation found that those who received a CBT dose high in CD45 had improved reconstitution [188]. Finally, one group found a negative correlation for CD4 T-cell reconstitution with age [185].
Some studies also use this method as an indicator for long-term survival. Research in allogeneic HSCT demonstrated that the rate of reconstitution of CD8 T-cells and the time to reach the 10th percentile of normal CD4 T-cell counts conditioned survival [189]; a separate analysis showed that those who reconstituted to above the 5th percentile of normal CD4 T-cell counts within a year were significantly more likely to survive than those who never did [186].
3.2.3. The role of modelling immune reconstitution following HSCT
Whilst the approaches discussed above have sought to answer specific questions relating to the rate or extent of immune reconstitution, mechanistic modelling in the statistical framework of NLME has the potential to do both. The heterogeneity in HSCTs means it is difficult to find any overarching phenomena in the reconstitution, and so mixed-effects modelling could provide more informative analyses. By taking into account the inter-individual variability, caused in part by different donor types, stem cells sources, and conditioning regimens, it is possible to find the fundamental determinants of the immune reconstitution. Mechanistic modelling uses equations that represent the underlying biology and thus give biologically relevant parameters, allowing a more meaningful covariate analysis.
CD4 T-cells can be used as an indicator for overall immune reconstitution. However, modelling CD4 T-cell reconstitution in children is challenging for three reasons. (1) CD4 T-cell reconstitution is slow (taking months to years), so the rapidly developing immune system, which results in variation of expected CD4 T-cell counts of as much as three-fold, must be taken into account [190]. (2) There is a body of evidence suggesting that the thymus, where T cells are formed, has impaired function in the months after transplant [176,191–193]. (3) A model would need to include the effects of homeostatic mechanisms for T-cells such as competition for resources and space constraints and their effects on cell-proliferation and cell-death rates [194]. A mechanistic model for T-cells such as this could then form the PD element, which would be the basis for investigating whether, and to what extent, the PK of the drugs used in HSCT affect reconstitution.
4. Conclusions
PKPD modelling in paediatric infectious diseases and immunology is well-developed, in that PKPD methods have been applied in all therapeutic areas, albeit to varying degrees. In the case of antibacterial and antifungal agents in particular, most studies to date have used the method of collecting PK data and then deriving dosing recommendations based on pre-defined PD targets. The potential problems with this approach are two-fold: firstly one must assume that the pre-defined PD target is applicable to the patient group studied for PK; secondly, whilst an absolute dose may be defined, no study that we found used the PKPD model to recommend treatment duration. Recommending an absolute dose and duration requires the time-course of infection to be modelled as the PD element, and this is usually not feasible in clinical studies. More sophisticated in vitro systems which simulate multiple dose PK profiles as used in the neonatal gentamicin example [47], may direct the way to making recommendations for both dosing and duration of treatment. In immunology, the PD endpoints are more straight-forward to measure in the target patient groups; HIV infection, which crosses the boundary of infectious diseases and immunology, is a prime example of how PD models can be developed. The integration of these mechanistic PD models with PK has begun in children [172], and applying such methods for studying immune reconstitution following HSCT and transplants indicate a potential approach towards optimising treatment in these patients.
Acknowledgements
C.I.S.B. is funded as a Clinical Research Fellow by the Global Research in Paediatrics — Network of Excellence (GRiP), part of the European Union Seventh Framework Programme (FP7/2007–2013, Grant Agreement number 261060).
J.M.L. is funded as an Academic Clinical Fellow by the National Institute for Health Research and has received financial support from Gilead.
J.F.S. received funding from a United Kingdom Medical Research Council Fellowship (grant number G1002305).
Footnotes
This review is part of the Advanced Drug Delivery Reviews theme issue on "Drug delivery and the paediatric population: where are we at?".
References
- 1.Lee J.Y., Garnett C.E., Gobburu J.V., Bhattaram V.A., Brar S., Earp J.C., Jadhav P.R., Krudys K., Lesko L.J., Li F., Liu J., Madabushi R., Marathe A., Mehrotra N., Tornoe C., Wang Y., Zhu H. Impact of pharmacometric analyses on new drug approval and labelling decisions: a review of 198 submissions between 2000 and 2008. Clin. Pharmacokinet. 2011;50:627–635. doi: 10.2165/11593210-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 2.Ette E.I., Williams P.J. Population pharmacokinetics II: estimation methods. Ann. Pharmacother. 2004;38:1907–1915. doi: 10.1345/aph.1E259. [DOI] [PubMed] [Google Scholar]
- 3.Ette E.I., Williams P.J. Population pharmacokinetics I: background, concepts, and models. Ann. Pharmacother. 2004;38:1702–1706. doi: 10.1345/aph.1D374. [DOI] [PubMed] [Google Scholar]
- 4.Ette E.I., Williams P.J., Lane J.R. Population pharmacokinetics III: design, analysis, and application of population pharmacokinetic studies. Ann. Pharmacother. 2004;38:2136–2144. doi: 10.1345/aph.1E260. [DOI] [PubMed] [Google Scholar]
- 5.Bonate P.L. Recommended reading in population pharmacokinetic pharmacodynamics. AAPS J. 2005;7:E363–E373. doi: 10.1208/aapsj070237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson B.J., Allegaert K., Holford N.H. Population clinical pharmacology of children: general principles. Eur. J. Pediatr. 2006;165:741–746. doi: 10.1007/s00431-006-0188-y. [DOI] [PubMed] [Google Scholar]
- 7.Anderson B.J., Allegaert K., Holford N.H. Population clinical pharmacology of children: modelling covariate effects. Eur. J. Pediatr. 2006;165:819–829. doi: 10.1007/s00431-006-0189-x. [DOI] [PubMed] [Google Scholar]
- 8.Tod M., Jullien V., Pons G. Facilitation of drug evaluation in children by population methods and modelling. Clin. Pharmacokinet. 2008;47:231–243. doi: 10.2165/00003088-200847040-00002. [DOI] [PubMed] [Google Scholar]
- 9.Drusano G.L. Antimicrobial pharmacodynamics: critical interactions of ‘bug and drug’. Nat. Rev. Microbiol. 2004;2:289–300. doi: 10.1038/nrmicro862. [DOI] [PubMed] [Google Scholar]
- 10.Moore R.D., Lietman P.S., Smith C.R. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimal inhibitory concentration. J. Infect. Dis. 1987;155:93–99. doi: 10.1093/infdis/155.1.93. [DOI] [PubMed] [Google Scholar]
- 11.Waxman D.J., Strominger J.L. Penicillin-binding proteins and the mechanism of action of beta-lactam antibiotics. Annu. Rev. Biochem. 1983;52:825–869. doi: 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- 12.Bayles K.W. The bactericidal action of penicillin: new clues to an unsolved mystery. Trends Microbiol. 2000;8:274–278. doi: 10.1016/s0966-842x(00)01762-5. [DOI] [PubMed] [Google Scholar]
- 13.Doogue M.P., Polasek T.M. Drug dosing in renal disease, the clinical biochemist. Rev. Aust. Assoc. Clin. Biochem. 2011;32:69–73. [PMC free article] [PubMed] [Google Scholar]
- 14.Lodise T.P., Lomaestro B.M., Drusano G.L., P. Society of Infectious Diseases Application of antimicrobial pharmacodynamic concepts into clinical practice: focus on beta-lactam antibiotics: insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy. 2006;26:1320–1332. doi: 10.1592/phco.26.9.1320. [DOI] [PubMed] [Google Scholar]
- 15.Craig W.A. Basic pharmacodynamics of antibacterials with clinical applications to the use of beta-lactams, glycopeptides, and linezolid. Infect. Dis. Clin. N. Am. 2003;17:479–501. doi: 10.1016/s0891-5520(03)00065-5. [DOI] [PubMed] [Google Scholar]
- 16.de Hoog M., Mouton J.W., van den Anker J.N. New dosing strategies for antibacterial agents in the neonate. Semin. Fetal Neonatal. Med. 2005;10:185–194. doi: 10.1016/j.siny.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 17.Muller A.E., DeJongh J., Bult Y., Goessens W.H., Mouton J.W., Danhof M., van den Anker J.N. Pharmacokinetics of penicillin G in infants with a gestational age of less than 32 weeks. Antimicrob. Agents Chemother. 2007;51:3720–3725. doi: 10.1128/AAC.00318-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pullen J., de Rozario L., Stolk L.M., Degraeuwe P.L., van Tiel F.H., Zimmermann L.J. Population pharmacokinetics and dosing of flucloxacillin in preterm and term neonates. Ther. Drug Monit. 2006;28:351–358. doi: 10.1097/01.ftd.0000211831.96102.91. [DOI] [PubMed] [Google Scholar]
- 19.Charles B.G., Preechagoon Y., Lee T.C., Steer P.A., Flenady V.J., Debuse N. Population pharmacokinetics of intravenous amoxicillin in very low birth weight infants. J. Pharm. Sci. 1997;86:1288–1292. doi: 10.1021/js970068l. [DOI] [PubMed] [Google Scholar]
- 20.Muller A.E., Oostvogel P.M., DeJongh J., Mouton J.W., Steegers E.A., Dorr P.J., Danhof M., Voskuyl R.A. Pharmacokinetics of amoxicillin in maternal, umbilical cord, and neonatal sera. Antimicrob. Agents Chemother. 2009;53:1574–1580. doi: 10.1128/AAC.00119-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z., Chen Y., Li Q., Cao D., Shi W., Cao Y., Wu D., Zhu Y., Wang Y., Chen C. Population pharmacokinetics of piperacillin/tazobactam in neonates and young infants. Eur. J. Clin. Pharmacol. 2013;69:1223–1233. doi: 10.1007/s00228-012-1413-4. [DOI] [PubMed] [Google Scholar]
- 22.Zobell J.T., Stockmann C., Young D.C., Cash J., McDowell B.J., Korgenski K., Sherwin C.M., Spigarelli M., Chatfield B.A., Ampofo K. Population pharmacokinetic and pharmacodynamic modeling of high-dose intermittent ticarcillin-clavulanate administration in pediatric cystic fibrosis patients. Clin. Ther. 2011;33:1844–1850. doi: 10.1016/j.clinthera.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 23.de Groot R., Hack B.D., Weber A., Chaffin D., Ramsey B., Smith A.L. Pharmacokinetics of ticarcillin in patients with cystic fibrosis: a controlled prospective study. Clin. Pharmacol. Ther. 1990;47:73–78. doi: 10.1038/clpt.1990.11. [DOI] [PubMed] [Google Scholar]
- 24.Dudley M.N., Ambrose P.G., Bhavnani S.M., Craig W.A., Ferraro M.J., Jones R.N., C. Antimicrobial Susceptibility Testing Subcommittee of the, I. Laboratory Standards Background and rationale for revised clinical and laboratory standards institute interpretive criteria (breakpoints) for Enterobacteriaceae and Pseudomonas aeruginosa: I. Cephalosporins and Aztreonam. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2013;56:1301–1309. doi: 10.1093/cid/cit017. [DOI] [PubMed] [Google Scholar]
- 25.Tamma P.D., Turnbull A.E., Milstone A.M., Hsu A.J., Carroll K.C., Cosgrove S.E. Does the piperacillin minimum inhibitory concentration for Pseudomonas aeruginosa influence clinical outcomes of children with pseudomonal bacteremia? Clin. Infect. Dis. 2012;55:799–806. doi: 10.1093/cid/cis545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knoderer C.A., Saft S.A., Walker S.G., Rodefeld M.D., Turrentine M.W., Brown J.W., Healy D.P., Sowinski K.M. Cefuroxime pharmacokinetics in pediatric cardiovascular surgery patients undergoing cardiopulmonary bypass. J. Cardiothorac. Vasc. Anesth. 2011;25:425–430. doi: 10.1053/j.jvca.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 27.Iida S., Kawanishi T., Hayashi M. Indications for a ceftriaxone dosing regimen in Japanese paediatric patients using population pharmacokinetic/pharmacodynamic analysis and simulation. J. Pharm. Pharmacol. 2011;63:65–72. doi: 10.1111/j.2042-7158.2010.01179.x. [DOI] [PubMed] [Google Scholar]
- 28.Ahsman M.J., Wildschut E.D., Tibboel D., Mathot R.A. Pharmacokinetics of cefotaxime and desacetylcefotaxime in infants during extracorporeal membrane oxygenation. Antimicrob. Agents Chemother. 2010;54:1734–1741. doi: 10.1128/AAC.01696-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buck M.L. Pharmacokinetic changes during extracorporeal membrane oxygenation: implications for drug therapy of neonates. Clin. Pharmacokinet. 2003;42:403–417. doi: 10.2165/00003088-200342050-00001. [DOI] [PubMed] [Google Scholar]
- 30.Bradley J.S., Sauberan J.B., Ambrose P.G., Bhavnani S.M., Rasmussen M.R., Capparelli E.V. Meropenem pharmacokinetics, pharmacodynamics, and Monte Carlo simulation in the neonate. Pediatr. Infect. Dis. J. 2008;27:794–799. doi: 10.1097/INF.0b013e318170f8d2. [DOI] [PubMed] [Google Scholar]
- 31.Du X., Li C., Kuti J.L., Nightingale C.H., Nicolau D.P. Population pharmacokinetics and pharmacodynamics of meropenem in pediatric patients. J. Clin. Pharmacol. 2006;46:69–75. doi: 10.1177/0091270005283283. [DOI] [PubMed] [Google Scholar]
- 32.Ikawa K., Morikawa N., Ikeda K., Miki M., Kobayashi M. Population pharmacokinetics and pharmacodynamics of meropenem in Japanese pediatric patients. J. Infect. Chemother. 2010;16:139–143. doi: 10.1007/s10156-009-0025-0. [DOI] [PubMed] [Google Scholar]
- 33.van den Anker J.N., Pokorna P., Kinzig-Schippers M., Martinkova J., de Groot R., Drusano G.L., Sorgel F. Meropenem pharmacokinetics in the newborn. Antimicrob. Agents Chemother. 2009;53:3871–3879. doi: 10.1128/AAC.00351-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohata Y., Tomita Y., Nakayama M., Kozuki T., Sunakawa K., Tanigawara Y. Optimal dosage regimen of meropenem for pediatric patients based on pharmacokinetic/pharmacodynamic considerations. Drug Metab. Pharmacokinet. 2011;26:523–531. doi: 10.2133/dmpk.dmpk-11-rg-027. [DOI] [PubMed] [Google Scholar]
- 35.Padari H., Metsvaht T., Korgvee L.T., Germovsek E., Ilmoja M.L., Kipper K., Herodes K., Standing J.F., Oselin K., Lutsar I. Short versus long infusion of meropenem in very-low-birth-weight neonates. Antimicrob. Agents Chemother. 2012;56:4760–4764. doi: 10.1128/AAC.00655-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parker E.M., Hutchison M., Blumer J.L. The pharmacokinetics of meropenem in infants and children: a population analysis. J. Antimicrob. Chemother. 1995;36(Suppl. A):63–71. doi: 10.1093/jac/36.suppl_a.63. [DOI] [PubMed] [Google Scholar]
- 37.Smith P.B., Cohen-Wolkowiez M., Castro L.M., Poindexter B., Bidegain M., Weitkamp J.H., Schelonka R.L., Ward R.M., Wade K., Valencia G., Burchfield D., Arrieta A., Bhatt-Mehta V., Walsh M., Kantak A., Rasmussen M., Sullivan J.E., Finer N., Brozanski B.S., Sanchez P., van den Anker J., Blumer J., Kearns G.L., Capparelli E.V., Anand R., Benjamin D.K., Jr., T. Meropenem Study Population pharmacokinetics of meropenem in plasma and cerebrospinal fluid of infants with suspected or complicated intra-abdominal infections. Pediatr. Infect. Dis. J. 2011;30:844–849. doi: 10.1097/INF.0b013e31822e8b0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshizawa K., Ikawa K., Ikeda K., Ohge H., Morikawa N. Population pharmacokinetic–pharmacodynamic target attainment analysis of imipenem plasma and urine data in neonates and children. Pediatr. Infect. Dis. J. 2013;32:1208–1216. doi: 10.1097/INF.0b013e31829b5880. [DOI] [PubMed] [Google Scholar]
- 39.Barclay M.L., Begg E.J., Chambers S.T. Adaptive resistance following single doses of gentamicin in a dynamic in vitro model. Antimicrob. Agents Chemother. 1992;36:1951–1957. doi: 10.1128/aac.36.9.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barclay M.L., Begg E.J. Aminoglycoside adaptive resistance: importance for effective dosage regimens. Drugs. 2001;61:713–721. doi: 10.2165/00003495-200161060-00001. [DOI] [PubMed] [Google Scholar]
- 41.Nielsen E.I., Sandstrom M., Honore P.H., Ewald U., Friberg L.E. Developmental pharmacokinetics of gentamicin in preterm and term neonates: population modelling of a prospective study. Clin. Pharmacokinet. 2009;48:253–263. doi: 10.2165/00003088-200948040-00003. [DOI] [PubMed] [Google Scholar]
- 42.Garcia B., Barcia E., Perez F., Molina I.T. Population pharmacokinetics of gentamicin in premature newborns. J. Antimicrob. Chemother. 2006;58:372–379. doi: 10.1093/jac/dkl244. [DOI] [PubMed] [Google Scholar]
- 43.Botha J.H., du Preez M.J., Adhikari M. Population pharmacokinetics of gentamicin in South African newborns. Eur. J. Clin. Pharmacol. 2003;59:755–759. doi: 10.1007/s00228-003-0663-6. [DOI] [PubMed] [Google Scholar]
- 44.Vervelde M.L., Rademaker C.M., Krediet T.G., Fleer A., van Asten P., van Dijk A. Population pharmacokinetics of gentamicin in preterm neonates: evaluation of a once-daily dosage regimen. Ther. Drug Monit. 1999;21:514–519. doi: 10.1097/00007691-199910000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Izquierdo M., Lanao J.M., Cervero L., Jimenez N.V., Dominguez-Gil A. Population pharmacokinetics of gentamicin in premature infants. Ther. Drug Monit. 1992;14:177–183. doi: 10.1097/00007691-199206000-00001. [DOI] [PubMed] [Google Scholar]
- 46.Jensen P.D., Edgren B.E., Brundage R.C. Population pharmacokinetics of gentamicin in neonates using a nonlinear, mixed-effects model. Pharmacotherapy. 1992;12:178–182. [PubMed] [Google Scholar]
- 47.Mohamed A.F., Nielsen E.I., Cars O., Friberg L.E. Pharmacokinetic–pharmacodynamic model for gentamicin and its adaptive resistance with predictions of dosing schedules in newborn infants. Antimicrob. Agents Chemother. 2012;56:179–188. doi: 10.1128/AAC.00694-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Assael B.M., Parini R., Rusconi F., Cavanna G. Influence of intrauterine maturation on the pharmacokinetics of amikacin in the neonatal period. Pediatr. Res. 1982;16:810–815. doi: 10.1203/00006450-198210000-00002. [DOI] [PubMed] [Google Scholar]
- 49.Sardemann H., Colding H., Hendel J., Kampmann J.P., Hvidberg E.F., Vejlsgaard R. Kinetics and dose calculations of amikacin in the newborn. Clin. Pharmacol. Ther. 1976;20:59–66. doi: 10.1002/cpt197620159. [DOI] [PubMed] [Google Scholar]
- 50.Kafetzis D.A., Sinaniotis C.A., Papadatos C.J., Kosmidis J. Pharmacokinetics of amikacin in infants and pre-school children. Acta Paediatr. Scand. 1979;68:419–422. doi: 10.1111/j.1651-2227.1979.tb05030.x. [DOI] [PubMed] [Google Scholar]
- 51.Sherwin C.M., Svahn S., Van der Linden A., Broadbent R.S., Medlicott N.J., Reith D.M. Individualised dosing of amikacin in neonates: a pharmacokinetic/pharmacodynamic analysis. Eur. J. Clin. Pharmacol. 2009;65:705–713. doi: 10.1007/s00228-009-0637-4. [DOI] [PubMed] [Google Scholar]
- 52.Hennig S., Norris R., Kirkpatrick C.M. Target concentration intervention is needed for tobramycin dosing in paediatric patients with cystic fibrosis—a population pharmacokinetic study. Br. J. Clin. Pharmacol. 2008;65:502–510. doi: 10.1111/j.1365-2125.2007.03045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Hoog M., Schoemaker R.C., Mouton J.W., van den Anker J.N. Tobramycin population pharmacokinetics in neonates. Clin. Pharmacol. Ther. 1997;62:392–399. doi: 10.1016/S0009-9236(97)90117-X. [DOI] [PubMed] [Google Scholar]
- 54.Lam W., Tjon J., Seto W., Dekker A., Wong C., Atenafu E., Bitnun A., Waters V., Yau Y., Solomon M., Ratjen F. Pharmacokinetic modelling of a once-daily dosing regimen for intravenous tobramycin in paediatric cystic fibrosis patients. J. Antimicrob. Chemother. 2007;59:1135–1140. doi: 10.1093/jac/dkm097. [DOI] [PubMed] [Google Scholar]
- 55.Touw D.J., Knox A.J., Smyth A. Population pharmacokinetics of tobramycin administered thrice daily and once daily in children and adults with cystic fibrosis. J. Cyst. Fibros. 2007;6:327–333. doi: 10.1016/j.jcf.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 56.Rodriguez J.C., Schoenike S., Scott G.B., Rossique-Gonzalez M.T., Gomez-Marin O. An evaluation of gentamicin, tobramycin, and amikacin pharmacokinetic/pharmacodynamic parameters in HIV-infected children. J. Pediatr. Pharmacol. Ther. 2003;8:274–283. doi: 10.5863/1551-6776-8.4.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giuliano R.A., Verpooten G.A., Verbist L., Wedeen R.P., De Broe M.E. In vivo uptake kinetics of aminoglycosides in the kidney cortex of rats. J. Pharmacol. Exp. Ther. 1986;236:470–475. [PubMed] [Google Scholar]
- 58.Tulkens P.M. Nephrotoxicity of aminoglycoside antibiotics. Toxicol. Lett. 1989;46:107–123. doi: 10.1016/0378-4274(89)90121-5. [DOI] [PubMed] [Google Scholar]
- 59.Vandecasteele S.J., De Vriese A.S., Tacconelli E. The pharmacokinetics and pharmacodynamics of vancomycin in clinical practice: evidence and uncertainties. J. Antimicrob. Chemother. 2013;68:743–748. doi: 10.1093/jac/dks495. [DOI] [PubMed] [Google Scholar]
- 60.Anderson B.J., Allegaert K., Van den Anker J.N., Cossey V., Holford N.H. Vancomycin pharmacokinetics in preterm neonates and the prediction of adult clearance. Br. J. Clin. Pharmacol. 2007;63:75–84. doi: 10.1111/j.1365-2125.2006.02725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Hoog M., Schoemaker R.C., Mouton J.W., van den Anker J.N. Vancomycin population pharmacokinetics in neonates. Clin. Pharmacol. Ther. 2000;67:360–367. doi: 10.1067/mcp.2000.105353. [DOI] [PubMed] [Google Scholar]
- 62.Grimsley C., Thomson A.H. Pharmacokinetics and dose requirements of vancomycin in neonates. Arch. Dis. Child. Fetal Neonatal Ed. 1999;81:F221–F227. doi: 10.1136/fn.81.3.f221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marques-Minana M.R., Saadeddin A., Peris J.E. Population pharmacokinetic analysis of vancomycin in neonates. A new proposal of initial dosage guideline. Br. J. Clin. Pharmacol. 2010;70:713–720. doi: 10.1111/j.1365-2125.2010.03736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao W., Lopez E., Biran V., Durrmeyer X., Fakhoury M., Jacqz-Aigrain E. Vancomycin continuous infusion in neonates: dosing optimisation and therapeutic drug monitoring. Arch. Dis. Child. 2013;98:449–453. doi: 10.1136/archdischild-2012-302765. [DOI] [PubMed] [Google Scholar]
- 65.Tarral E., Jehl F., Tarral A., Simeoni U., Monteil H., Willard D., Geisert J. Pharmacokinetics of teicoplanin in children. J. Antimicrob. Chemother. 1988;21(Suppl. A):47–51. doi: 10.1093/jac/21.suppl_a.47. [DOI] [PubMed] [Google Scholar]
- 66.Terragna A., Ferrea G., Loy A., Danese A., Bernareggi A., Cavenaghi L., Rosina R. Pharmacokinetics of teicoplanin in pediatric patients. Antimicrob. Agents Chemother. 1988;32:1223–1226. doi: 10.1128/aac.32.8.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lukas J.C., Karikas G., Gazouli M., Kalabalikis P., Hatzis T., Macheras P. Pharmacokinetics of teicoplanin in an ICU population of children and infants. Pharm. Res. 2004;21:2064–2071. doi: 10.1023/b:pham.0000048198.56873.d8. [DOI] [PubMed] [Google Scholar]
- 68.Preston S.L., Drusano G.L., Berman A.L., Fowler C.L., Chow A.T., Dornseif B., Reichl V., Natarajan J., Corrado M. Pharmacodynamics of levofloxacin: a new paradigm for early clinical trials. JAMA. 1998;279:125–129. doi: 10.1001/jama.279.2.125. [DOI] [PubMed] [Google Scholar]
- 69.Burkhardt J.E., Hill M.A., Carlton W.W., Kesterson J.W. Histologic and histochemical changes in articular cartilages of immature beagle dogs dosed with difloxacin, a fluoroquinolone. Vet. Pathol. 1990;27:162–170. doi: 10.1177/030098589002700303. [DOI] [PubMed] [Google Scholar]
- 70.Payen S., Serreau R., Munck A., Aujard Y., Aigrain Y., Bressolle F., Jacqz-Aigrain E. Population pharmacokinetics of ciprofloxacin in pediatric and adolescent patients with acute infections. Antimicrob. Agents Chemother. 2003;47:3170–3178. doi: 10.1128/AAC.47.10.3170-3178.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rajagopalan P., Gastonguay M.R. Population pharmacokinetics of ciprofloxacin in pediatric patients. J. Clin. Pharmacol. 2003;43:698–710. [PubMed] [Google Scholar]
- 72.Schaefer H.G., Stass H., Wedgwood J., Hampel B., Fischer C., Kuhlmann J., Schaad U.B. Pharmacokinetics of ciprofloxacin in pediatric cystic fibrosis patients. Antimicrob. Agents Chemother. 1996;40:29–34. doi: 10.1128/aac.40.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ambrose P.G., Grasela D.M., Grasela T.H., Passarell J., Mayer H.B., Pierce P.F. Pharmacodynamics of fluoroquinolones against Streptococcus pneumoniae in patients with community-acquired respiratory tract infections. Antimicrob. Agents Chemother. 2001;45:2793–2797. doi: 10.1128/AAC.45.10.2793-2797.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Forrest A., Nix D.E., Ballow C.H., Goss T.F., Birmingham M.C., Schentag J.J. Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob. Agents Chemother. 1993;37:1073–1081. doi: 10.1128/aac.37.5.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hassan H.E., Othman A.A., Eddington N.D., Duffy L., Xiao L., Waites K.B., Kaufman D.A., Fairchild K.D., Terrin M.L., Viscardi R.M. Pharmacokinetics, safety, and biologic effects of azithromycin in extremely preterm infants at risk for ureaplasma colonization and bronchopulmonary dysplasia. J. Clin. Pharmacol. 2011;51:1264–1275. doi: 10.1177/0091270010382021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suyagh M., Collier P.S., Millership J.S., Iheagwaram G., Millar M., Halliday H.L., McElnay J.C. Metronidazole population pharmacokinetics in preterm neonates using dried blood-spot sampling. Pediatrics. 2011;127:e367–e374. doi: 10.1542/peds.2010-0807. [DOI] [PubMed] [Google Scholar]
- 77.Cohen-Wolkowiez M., Ouellet D., Smith P.B., James L.P., Ross A., Sullivan J.E., Walsh M.C., Zadell A., Newman N., White N.R., Kashuba A.D., Benjamin D.K., Jr. Population pharmacokinetics of metronidazole evaluated using scavenged samples from preterm infants. Antimicrob. Agents Chemother. 2012;56:1828–1837. doi: 10.1128/AAC.06071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cohen-Wolkowiez M., Sampson M., Bloom B.T., Arrieta A., Wynn J.L., Martz K., Harper B., Kearns G.L., Capparelli E.V., Siegel D., Benjamin D.K., Jr., Smith P.B., N. on behalf of the Best Pharmaceuticals for Children Act — Pediatric Trials Determining population and developmental pharmacokinetics of metronidazole using plasma and dried blood spot samples from premature infants. Pediatr. Infect. Dis. J. 2013;32:956–961. doi: 10.1097/INF.0b013e3182947cf8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kearns G.L., Abdel-Rahman S.M., Blumer J.L., Reed M.D., James L.P., Jacobs R.F., Bradley J.A., Welshman I.R., Jungbluth G.L., Stalker D.J., N. Pediatric Pharmacology Research Unit ingle dose pharmacokinetics of linezolid in infants and children. Pediatr. Infect. Dis. J. 2000;19:1178–1184. doi: 10.1097/00006454-200012000-00012. [DOI] [PubMed] [Google Scholar]
- 80.Rodriguez-Tudela J.L., Almirante B., Rodriguez-Pardo D., Laguna F., Donnelly J.P., Mouton J.W., Pahissa A., Cuenca-Estrella M. Correlation of the MIC and dose/MIC ratio of fluconazole to the therapeutic response of patients with mucosal candidiasis and candidemia. Antimicrob. Agents Chemother. 2007;51:3599–3604. doi: 10.1128/AAC.00296-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andes D. In vivo pharmacodynamics of antifungal drugs in treatment of candidiasis. Antimicrob. Agents Chemother. 2003;47:1179–1186. doi: 10.1128/AAC.47.4.1179-1186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saxen H., Hoppu K., Pohjavuori M. Pharmacokinetics of fluconazole in very low birth weight infants during the first two weeks of life. Clin. Pharmacol. Ther. 1993;54:269–277. doi: 10.1038/clpt.1993.147. [DOI] [PubMed] [Google Scholar]
- 83.Kaufman D., Boyle R., Hazen K.C., Patrie J.T., Robinson M., Donowitz L.G. Fluconazole prophylaxis against fungal colonization and infection in preterm infants. N. Engl. J. Med. 2001;345:1660–1666. doi: 10.1056/NEJMoa010494. [DOI] [PubMed] [Google Scholar]
- 84.Kicklighter S.D., Springer S.C., Cox T., Hulsey T.C., Turner R.B. Fluconazole for prophylaxis against candidal rectal colonization in the very low birth weight infant. Pediatrics. 2001;107:293–298. doi: 10.1542/peds.107.2.293. [DOI] [PubMed] [Google Scholar]
- 85.Louie A., Liu Q.F., Drusano G.L., Liu W., Mayers M., Anaissie E., Miller M.H. Pharmacokinetic studies of fluconazole in rabbits characterizing doses which achieve peak levels in serum and area under the concentration–time curve values which mimic those of high-dose fluconazole in humans. Antimicrob. Agents Chemother. 1998;42:1512–1514. doi: 10.1128/aac.42.6.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Piper L., Smith P.B., Hornik C.P., Cheifetz I.M., Barrett J.S., Moorthy G., Hope W.W., Wade K.C., Cohen-Wolkowiez M., Benjamin D.K., Jr. Fluconazole loading dose pharmacokinetics and safety in infants. Pediatr. Infect. Dis. J. 2011;30:375–378. doi: 10.1097/INF.0b013e318202cbb3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu D., Wade K.C., Paul D.J., Barrett J.S. A rapid and sensitive LC–MS/MS method for determination of fluconazole in human plasma and its application in infants with Candida infections. Ther. Drug Monit. 2009;31:703–709. doi: 10.1097/FTD.0b013e3181b20b40. [DOI] [PubMed] [Google Scholar]
- 88.Lee J.W., Seibel N.L., Amantea M., Whitcomb P., Pizzo P.A., Walsh T.J. Safety and pharmacokinetics of fluconazole in children with neoplastic diseases. J. Pediatr. 1992;120:987–993. doi: 10.1016/s0022-3476(05)81975-4. [DOI] [PubMed] [Google Scholar]
- 89.Bhandari V., Narang A., Kumar B., Singh M., Nair P.M., Bhakoo O.N. Itraconazole therapy for disseminated candidiasis in a very low birthweight neonate. J. Paediatr. Child Health. 1992;28:323–324. doi: 10.1111/j.1440-1754.1992.tb02678.x. [DOI] [PubMed] [Google Scholar]
- 90.Abdel-Rahman S.M., Jacobs R.F., Massarella J., Kauffman R.E., Bradley J.S., Kimko H.C., Kearns G.L., Shalayda K., Curtin C., Maldonado S.D., Blumer J.L. Single-dose pharmacokinetics of intravenous itraconazole and hydroxypropyl-beta-cyclodextrin in infants, children, and adolescents. Antimicrob. Agents Chemother. 2007;51:2668–2673. doi: 10.1128/AAC.00297-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmitt C., Perel Y., Harousseau J.L., Lemerle S., Chwetzoff E., le Moing J.P., Levron J.C. Pharmacokinetics of itraconazole oral solution in neutropenic children during long-term prophylaxis. Antimicrob. Agents Chemother. 2001;45:1561–1564. doi: 10.1128/AAC.45.5.1561-1564.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Foot A.B., Veys P.A., Gibson B.E. Itraconazole oral solution as antifungal prophylaxis in children undergoing stem cell transplantation or intensive chemotherapy for haematological disorders. Bone Marrow Transplant. 1999;24:1089–1093. doi: 10.1038/sj.bmt.1702023. [DOI] [PubMed] [Google Scholar]
- 93.Groll A.H., Wood L., Roden M., Mickiene D., Chiou C.C., Townley E., Dad L., Piscitelli S.C., Walsh T.J. Safety, pharmacokinetics, and pharmacodynamics of cyclodextrin itraconazole in pediatric patients with oropharyngeal candidiasis. Antimicrob. Agents Chemother. 2002;46:2554–2563. doi: 10.1128/AAC.46.8.2554-2563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de Repentigny L., Ratelle J., Leclerc J.M., Cornu G., Sokal E.M., Jacqmin P., De Beule K. Repeated-dose pharmacokinetics of an oral solution of itraconazole in infants and children. Antimicrob. Agents Chemother. 1998;42:404–408. doi: 10.1128/aac.42.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Andes D., Pascual A., Marchetti O. Antifungal therapeutic drug monitoring: established and emerging indications. Antimicrob. Agents Chemother. 2009;53:24–34. doi: 10.1128/AAC.00705-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lestner J.M., Roberts S.A., Moore C.B., Howard S.J., Denning D.W., Hope W.W. Toxicodynamics of itraconazole: implications for therapeutic drug monitoring. Clin. Infect. Dis. 2009;49:928–930. doi: 10.1086/605499. [DOI] [PubMed] [Google Scholar]
- 97.Hope W.W., Billaud E.M., Lestner J., Denning D.W. Therapeutic drug monitoring for triazoles. Curr. Opin. Infect. Dis. 2008;21:580–586. doi: 10.1097/QCO.0b013e3283184611. [DOI] [PubMed] [Google Scholar]
- 98.Groll A.H., Jaeger G., Allendorf A., Herrmann G., Schloesser R., von Loewenich V. Invasive pulmonary aspergillosis in a critically ill neonate: case report and review of invasive aspergillosis during the first 3 months of life. Clin. Infect. Dis. 1998;27:437–452. doi: 10.1086/514717. [DOI] [PubMed] [Google Scholar]
- 99.Doby E.H., Benjamin D.K., Jr., Blaschke A.J., Ward R.M., Pavia A.T., Martin P.L., Driscoll T.A., Cohen-Wolkowiez M., Moran C. Therapeutic monitoring of voriconazole in children less than three years of age: a case report and summary of voriconazole concentrations for ten children. Pediatr. Infect. Dis. J. 2012;31:632–635. doi: 10.1097/INF.0b013e31824acc33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kohli V., Taneja V., Sachdev P., Joshi R. Voriconazole in newborns. Indian Pediatr. 2008;45:236–238. [PubMed] [Google Scholar]
- 101.Hope W.W. Population pharmacokinetics of voriconazole in adults. Antimicrob. Agents Chemother. 2012;56:526–531. doi: 10.1128/AAC.00702-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Herbrecht R., Denning D.W., Patterson T.F., Bennett J.E., Greene R.E., Oestmann J.W., Kern W.V., Marr K.A., Ribaud P., Lortholary O., Sylvester R., Rubin R.H., Wingard J.R., Stark P., Durand C., Caillot D., Thiel E., Chandrasekar P.H., Hodges M.R., Schlamm H.T., Troke P.F., de Pauw B., R. Invasive Fungal, Infections Group of the European Organisation for, C. Treatment of, G. the Global Aspergillus Study Voriconazole versus amphotericin B for primary therapy of invasive aspergillosis. N. Engl. J. Med. 2002;347:408–415. doi: 10.1056/NEJMoa020191. [DOI] [PubMed] [Google Scholar]
- 103.Driscoll T.A., Yu L.C., Frangoul H., Krance R.A., Nemecek E., Blumer J., Arrieta A., Graham M.L., Bradfield S.M., Baruch A., Liu P. Comparison of pharmacokinetics and safety of voriconazole intravenous-to-oral switch in immunocompromised children and healthy adults. Antimicrob. Agents Chemother. 2011;55:5770–5779. doi: 10.1128/AAC.00531-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Driscoll T.A., Frangoul H., Nemecek E.R., Murphey D.K., Yu L.C., Blumer J., Krance R.A., Baruch A., Liu P. Comparison of pharmacokinetics and safety of voriconazole intravenous-to-oral switch in immunocompromised adolescents and healthy adults. Antimicrob. Agents Chemother. 2011;55:5780–5789. doi: 10.1128/AAC.05010-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karlsson M.O., Lutsar I., Milligan P.A. Population pharmacokinetic analysis of voriconazole plasma concentration data from pediatric studies. Antimicrob. Agents Chemother. 2009;53:935–944. doi: 10.1128/AAC.00751-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Neely M., Rushing T., Kovacs A., Jelliffe R., Hoffman J. Voriconazole pharmacokinetics and pharmacodynamics in children. Clin. Infect. Dis. 2010;50:27–36. doi: 10.1086/648679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Friberg L.E., Ravva P., Karlsson M.O., Liu P. Integrated population pharmacokinetic analysis of voriconazole in children, adolescents, and adults. Antimicrob. Agents Chemother. 2012;56:3032–3042. doi: 10.1128/AAC.05761-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pascual A., Calandra T., Bolay S., Buclin T., Bille J., Marchetti O. Voriconazole therapeutic drug monitoring in patients with invasive mycoses improves efficacy and safety outcomes. Clin. Infect. Dis. 2008;46:201–211. doi: 10.1086/524669. [DOI] [PubMed] [Google Scholar]
- 109.Krishna G., Sansone-Parsons A., Martinho M., Kantesaria B., Pedicone L. Posaconazole plasma concentrations in juvenile patients with invasive fungal infection. Antimicrob. Agents Chemother. 2007;51:812–818. doi: 10.1128/AAC.00454-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lewis R.E. Pharmacodynamic implications for use of antifungal agents. Curr. Opin. Pharmacol. 2007;7:491–497. doi: 10.1016/j.coph.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 111.Saez-Llorens X., Macias M., Maiya P., Pineros J., Jafri H.S., Chatterjee A., Ruiz G., Raghavan J., Bradshaw S.K., Kartsonis N.A., Sun P., Strohmaier K.M., Fallon M., Bi S., Stone J.A., Chow J.W. Pharmacokinetics and safety of caspofungin in neonates and infants less than 3 months of age. Antimicrob. Agents Chemother. 2009;53:869–875. doi: 10.1128/AAC.00868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Walsh T.J., Adamson P.C., Seibel N.L., Flynn P.M., Neely M.N., Schwartz C., Shad A., Kaplan S.L., Roden M.M., Stone J.A., Miller A., Bradshaw S.K., Li S.X., Sable C.A., Kartsonis N.A. Pharmacokinetics, safety, and tolerability of caspofungin in children and adolescents. Antimicrob. Agents Chemother. 2005;49:4536–4545. doi: 10.1128/AAC.49.11.4536-4545.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Li C.C., Sun P., Dong Y., Bi S., Desai R., Dockendorf M.F., Kartsonis N.A., Ngai A.L., Bradshaw S., Stone J.A. Population pharmacokinetics and pharmacodynamics of caspofungin in pediatric patients. Antimicrob. Agents Chemother. 2011;55:2098–2105. doi: 10.1128/AAC.00905-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hope W.W., Smith P.B., Arrieta A., Buell D.N., Roy M., Kaibara A., Walsh T.J., Cohen-Wolkowiez M., Benjamin D.K., Jr. Population pharmacokinetics of micafungin in neonates and young infants. Antimicrob. Agents Chemother. 2010;54:2633–2637. doi: 10.1128/AAC.01679-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hope W.W., Seibel N.L., Schwartz C.L., Arrieta A., Flynn P., Shad A., Albano E., Keirns J.J., Buell D.N., Gumbo T., Drusano G.L., Walsh T.J. Population pharmacokinetics of micafungin in pediatric patients and implications for antifungal dosing. Antimicrob. Agents Chemother. 2007;51:3714–3719. doi: 10.1128/AAC.00398-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Seibel N.L., Schwartz C., Arrieta A., Flynn P., Shad A., Albano E., Keirns J., Lau W.M., Facklam D.P., Buell D.N., Walsh T.J. Safety, tolerability, and pharmacokinetics of Micafungin (FK463) in febrile neutropenic pediatric patients. Antimicrob. Agents Chemother. 2005;49:3317–3324. doi: 10.1128/AAC.49.8.3317-3324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Benjamin D.K., Jr., Driscoll T., Seibel N.L., Gonzalez C.E., Roden M.M., Kilaru R., Clark K., Dowell J.A., Schranz J., Walsh T.J. Safety and pharmacokinetics of intravenous anidulafungin in children with neutropenia at high risk for invasive fungal infections. Antimicrob. Agents Chemother. 2006;50:632–638. doi: 10.1128/AAC.50.2.632-638.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dowell J.A., Knebel W., Ludden T., Stogniew M., Krause D., Henkel T. Population pharmacokinetic analysis of anidulafungin, an echinocandin antifungal. J. Clin. Pharmacol. 2004;44:590–598. doi: 10.1177/0091270004265644. [DOI] [PubMed] [Google Scholar]
- 119.Starke J.R., Mason E.O., Jr., Kramer W.G., Kaplan S.L. Pharmacokinetics of amphotericin B in infants and children. J. Infect. Dis. 1987;155:766–774. doi: 10.1093/infdis/155.4.766. [DOI] [PubMed] [Google Scholar]
- 120.Benson J.M., Nahata M.C. Pharmacokinetics of amphotericin B in children. Antimicrob. Agents Chemother. 1989;33:1989–1993. doi: 10.1128/aac.33.11.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baley J.E., Meyers C., Kliegman R.M., Jacobs M.R., Blumer J.L. Pharmacokinetics, outcome of treatment, and toxic effects of amphotericin B and 5-fluorocytosine in neonates. J. Pediatr. 1990;116:791–797. doi: 10.1016/s0022-3476(05)82674-5. [DOI] [PubMed] [Google Scholar]
- 122.Wurthwein G., Groll A.H., Hempel G., Adler-Shohet F.C., Lieberman J.M., Walsh T.J. Population pharmacokinetics of amphotericin B lipid complex in neonates. Antimicrob. Agents Chemother. 2005;49:5092–5098. doi: 10.1128/AAC.49.12.5092-5098.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hong Y., Shaw P.J., Nath C.E., Yadav S.P., Stephen K.R., Earl J.W., McLachlan A.J. Population pharmacokinetics of liposomal amphotericin B in pediatric patients with malignant diseases. Antimicrob. Agents Chemother. 2006;50:935–942. doi: 10.1128/AAC.50.3.935-942.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kimberlin D.W. Antiviral therapies in children: has their time arrived? Pediatr. Clin. N. Am. 2005;52:837–867. doi: 10.1016/j.pcl.2005.02.006. (vii) [DOI] [PubMed] [Google Scholar]
- 125.Hintz M., Connor J.D., Spector S.A., Blum M.R., Keeney R.E., Yeager A.S. Neonatal acyclovir pharmacokinetics in patients with herpes virus infections. Am. J. Med. 1982;73:210–214. doi: 10.1016/0002-9343(82)90093-6. [DOI] [PubMed] [Google Scholar]
- 126.Sullender W.M., Arvin A.M., Diaz P.S., Connor J.D., Straube R., Dankner W., Levin M.J., Weller S., Blum M.R., Chapman S. Pharmacokinetics of acyclovir suspension in infants and children. Antimicrob. Agents Chemother. 1987;31:1722–1726. doi: 10.1128/aac.31.11.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fletcher C.V., Englund J.A., Bean B., Chinnock B., Brundage D.M., Balfour H.H., Jr. Continuous infusion of high-dose acyclovir for serious herpesvirus infections. Antimicrob. Agents Chemother. 1989;33:1375–1378. doi: 10.1128/aac.33.8.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Meszner Z., Nyerges G., Bell A.R. Oral acyclovir to prevent dissemination of varicella in immunocompromised children. J. Infect. 1993;26:9–15. doi: 10.1016/0163-4453(93)96648-a. [DOI] [PubMed] [Google Scholar]
- 129.Rudd C., Rivadeneira E.D., Gutman L.T. Dosing considerations for oral acyclovir following neonatal herpes disease. Acta Paediatr. 1994;83:1237–1243. doi: 10.1111/j.1651-2227.1994.tb13004.x. [DOI] [PubMed] [Google Scholar]
- 130.Tod M., Lokiec F., Bidault R., De Bony F., Petitjean O., Aujard Y. Pharmacokinetics of oral acyclovir in neonates and in infants: a population analysis. Antimicrob. Agents Chemother. 2001;45:150–157. doi: 10.1128/AAC.45.1.150-157.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bomgaars L., Thompson P., Berg S., Serabe B., Aleksic A., Blaney S. Valacyclovir and acyclovir pharmacokinetics in immunocompromised children. Pediatr. Blood Cancer. 2008;51:504–508. doi: 10.1002/pbc.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zeng L., Nath C.E., Blair E.Y., Shaw P.J., Stephen K., Earl J.W., Coakley J.C., McLachlan A.J. Population pharmacokinetics of acyclovir in children and young people with malignancy after administration of intravenous acyclovir or oral valacyclovir. Antimicrob. Agents Chemother. 2009;53:2918–2927. doi: 10.1128/AAC.01138-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Saez-Llorens X., Yogev R., Arguedas A., Rodriguez A., Spigarelli M.G., De Leon Castrejon T., Bomgaars L., Roberts M., Abrams B., Zhou W., Looby M., Kaiser G., Hamed K. Pharmacokinetics and safety of famciclovir in children with herpes simplex or varicella-zoster virus infection. Antimicrob. Agents Chemother. 2009;53:1912–1920. doi: 10.1128/AAC.01054-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Blumer J., Rodriguez A., Sanchez P.J., Sallas W., Kaiser G., Hamed K. Single-dose pharmacokinetics of famciclovir in infants and population pharmacokinetic analysis in infants and children. Antimicrob. Agents Chemother. 2010;54:2032–2041. doi: 10.1128/AAC.01508-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ogungbenro K., Matthews I., Looby M., Kaiser G., Graham G., Aarons L. Population pharmacokinetics and optimal design of paediatric studies for famciclovir. Br. J. Clin. Pharmacol. 2009;68:546–560. doi: 10.1111/j.1365-2125.2009.03479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pescovitz M.D., Brook B., Jindal R.M., Leapman S.B., Milgrom M.L., Filo R.S. Oral ganciclovir in pediatric transplant recipients: a pharmacokinetic study. Clin. Transpl. 1997;11:613–617. [PubMed] [Google Scholar]
- 137.Filler G., Lampe D., von Bredow M.A., Lappenberg-Pelzer M., Rocher S., Strehlau J., Ehrich J.H. Prophylactic oral ganciclovir after renal transplantation-dosing and pharmacokinetics. Pediatr. Nephrol. 1998;12:6–9. doi: 10.1007/s004670050391. [DOI] [PubMed] [Google Scholar]
- 138.Zhang D., Lapeyraque A.L., Popon M., Loirat C., Jacqz-Aigrain E. Pharmacokinetics of ganciclovir in pediatric renal transplant recipients. Pediatr. Nephrol. 2003;18:943–948. doi: 10.1007/s00467-003-1226-x. [DOI] [PubMed] [Google Scholar]
- 139.Jacqz-Aigrain E., Macher M.A., Sauvageon-Marthe H., Brun P., Loirat C. Pharmacokinetics of ganciclovir in renal transplant children. Pediatr. Nephrol. 1992;6:194–196. doi: 10.1007/BF00866315. [DOI] [PubMed] [Google Scholar]
- 140.Vaudry W., Ettenger R., Jara P., Varela-Fascinetto G., Bouw M.R., Ives J., Walker R., Valcyte W.V.S.G. Valganciclovir dosing according to body surface area and renal function in pediatric solid organ transplant recipients. Am. J. Transplant. 2009;9:636–643. doi: 10.1111/j.1600-6143.2008.02528.x. [DOI] [PubMed] [Google Scholar]
- 141.Pescovitz M.D., Ettenger R.B., Strife C.F., Sherbotie J.R., Thomas S.E., McDiarmid S., Bartosh S., Ives J., Bouw M.R., Bucuvalas J. Pharmacokinetics of oral valganciclovir solution and intravenous ganciclovir in pediatric renal and liver transplant recipients. Transpl. Infect. Dis. 2010;12:195–203. doi: 10.1111/j.1399-3062.2009.00478.x. [DOI] [PubMed] [Google Scholar]
- 142.Vezina H.E., Brundage R.C., Nevins T.E., Balfour H.H., Jr. The pharmacokinetics of valganciclovir prophylaxis in pediatric solid organ transplant patients at risk for Epstein–Barr virus disease. Clin. Pharmacol. Adv. Appl. 2010;2:1–7. doi: 10.2147/CPAA.S8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Luck S., Lovering A., Griffiths P., Sharland M. Ganciclovir treatment in children: evidence of subtherapeutic levels. Int. J. Antimicrob. Agents. 2011;37:445–448. doi: 10.1016/j.ijantimicag.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 144.Frenkel L.M., Capparelli E.V., Dankner W.M., Xu J., Smith I.L., Ballow A., Culnane M., Read J.S., Thompson M., Mohan K.M., Shaver A., Robinson C.A., Stempien M.J., Burchett S.K., Melvin A.J., Borkowsky W., Petru A., Kovacs A., Yogev R., Goldsmith J., McFarland E.J., Spector S.A. Oral ganciclovir in children: pharmacokinetics, safety, tolerance, and antiviral effects. The Pediatric AIDS Clinical Trials Group. J. Infect. Dis. 2000;182:1616–1624. doi: 10.1086/317600. [DOI] [PubMed] [Google Scholar]
- 145.Kimberlin D.W., Acosta E.P., Sanchez P.J., Sood S., Agrawal V., Homans J., Jacobs R.F., Lang D., Romero J.R., Griffin J., Cloud G.A., Lakeman F.D., Whitley R.J., A. National Institute of, G. Infectious Diseases Collaborative Antiviral Study Pharmacokinetic and pharmacodynamic assessment of oral valganciclovir in the treatment of symptomatic congenital cytomegalovirus disease. J. Infect. Dis. 2008;197:836–845. doi: 10.1086/528376. [DOI] [PubMed] [Google Scholar]
- 146.Englund J.A., Piedra P.A., Jefferson L.S., Wilson S.Z., Taber L.H., Gilbert B.E. High-dose, short-duration ribavirin aerosol therapy in children with suspected respiratory syncytial virus infection. J. Pediatr. 1990;117:313–320. doi: 10.1016/s0022-3476(05)80554-2. [DOI] [PubMed] [Google Scholar]
- 147.Lankester A.C., Heemskerk B., Claas E.C., Schilham M.W., Beersma M.F., Bredius R.G., van Tol M.J., Kroes A.C. Effect of ribavirin on the plasma viral DNA load in patients with disseminating adenovirus infection. Clin. Infect. Dis. 2004;38:1521–1525. doi: 10.1086/420817. [DOI] [PubMed] [Google Scholar]
- 148.Hosoya M., Mori S., Tomoda A., Mori K., Sawaishi Y., Kimura H., Shigeta S., Suzuki H. Pharmacokinetics and effects of ribavirin following intraventricular administration for treatment of subacute sclerosing panencephalitis. Antimicrob. Agents Chemother. 2004;48:4631–4635. doi: 10.1128/AAC.48.12.4631-4635.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McJunkin J.E., Nahata M.C., De Los Reyes E.C., Hunt W.G., Caceres M., Khan R.R., Chebib M.G., Taravath S., Minnich L.L., Carr R., Welch C.A., Whitley R.J. Safety and pharmacokinetics of ribavirin for the treatment of la crosse encephalitis. Pediatr. Infect. Dis. J. 2011;30:860–865. doi: 10.1097/INF.0b013e31821c922c. [DOI] [PubMed] [Google Scholar]
- 150.Gonzalez-Peralta R.P., Kelly D.A., Haber B., Molleston J., Murray K.F., Jonas M.M., Shelton M., Mieli-Vergani G., Lurie Y., Martin S., Lang T., Baczkowski A., Geffner M., Gupta S., Laughlin M., C.T.G. International Pediatric Hepatitis Interferon alfa-2b in combination with ribavirin for the treatment of chronic hepatitis C in children: efficacy, safety, and pharmacokinetics. Hepatology. 2005;42:1010–1018. doi: 10.1002/hep.20884. [DOI] [PubMed] [Google Scholar]
- 151.Oxford J. Oseltamivir in the management of influenza. Expert. Opin. Pharmacother. 2005;6:2493–2500. doi: 10.1517/14656566.6.14.2493. [DOI] [PubMed] [Google Scholar]
- 152.Standing J.F., Tsolia M., Lutsar I. Pharmacokinetics and pharmacodynamics of oseltamivir in neonates, infants and children. Infect. Disord. Drug Targets. 2013;13:6–14. doi: 10.2174/18715265112129990003. [DOI] [PubMed] [Google Scholar]
- 153.Kimberlin D.W., Acosta E.P., Prichard M.N., Sanchez P.J., Ampofo K., Lang D., Ashouri N., Vanchiere J.A., Abzug M.J., Abughali N., Caserta M.T., Englund J.A., Sood S.K., Spigarelli M.G., Bradley J.S., Lew J., Michaels M.G., Wan W., Cloud G., Jester P., Lakeman F.D., Whitley R.J., A. National Institute of, G. Infectious Diseases Collaborative Antiviral Study Oseltamivir pharmacokinetics, dosing, and resistance among children aged < 2 years with influenza. J. Infect. Dis. 2013;207:709–720. doi: 10.1093/infdis/jis765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sugaya N., Kohno S., Ishibashi T., Wajima T., Takahashi T. Efficacy, safety, and pharmacokinetics of intravenous peramivir in children with 2009 pandemic H1N1 influenza A virus infection. Antimicrob. Agents Chemother. 2012;56:369–377. doi: 10.1128/AAC.00132-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Neely M.N., Rakhmanina N.Y. Pharmacokinetic optimization of antiretroviral therapy in children and adolescents. Clin. Pharmacokinet. 2011;50:143–189. doi: 10.2165/11539260-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 156.Perelson A.S., Neumann A.U., Markowitz M., Leonard J.M., Ho D.D. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 157.Palumbo P., Wu H., Chadwick E., Ruan P., Luzuriaga K., Rodman J., Yogev R., A.C.T.G.I. Pediatric Virologic response to potent antiretroviral therapy and modeling of HIV dynamics in early pediatric infection. J. Infect. Dis. 2007;196:23–29. doi: 10.1086/518508. [DOI] [PubMed] [Google Scholar]
- 158.De Beaudrap P., Rouet F., Fassinou P., Kouakoussui A., Mercier S., Ecochard R., Msellati P. CD4 cell response before and after HAART initiation according to viral load and growth indicators in HIV-1-infected children in Abidjan, Cote d'Ivoire. J. Acquir. Immune Defic. Syndr. 2008;49:70–76. doi: 10.1097/QAI.0b013e3181831847. [DOI] [PubMed] [Google Scholar]
- 159.Lewis J., Walker A.S., Castro H., De Rossi A., Gibb D.M., Giaquinto C., Klein N., Callard R. Age and CD4 count at initiation of antiretroviral therapy in HIV-infected children: effects on long-term T-cell reconstitution. J. Infect. Dis. 2012;205:548–556. doi: 10.1093/infdis/jir787. [DOI] [PubMed] [Google Scholar]
- 160.Lewis J., Walker A.S., Klein N., Callard R. CD31 + cell percentage correlation with speed of CD4+ T-cell count recovery in HIV-infected adults is reversed in children: higher thymic output may be responsible. Clin. Infect. Dis. 2012;55:304–307. doi: 10.1093/cid/cis374. (author reply 307) [DOI] [PubMed] [Google Scholar]
- 161.H.I.V.E.R.E.S.G. Collaboration of Observational. Sabin C.A., Smith C.J., d'Arminio Monforte A., Battegay M., Gabiano C., Galli L., Geelen S., Gibb D., Guiguet M., Judd A., Leport C., Dabis F., Pantazis N., Porter K., Raffi F., Thorne C., Torti C., Walker S., Warszawski J., Wintergerst U., Chene G., Lundgren J. Response to combination antiretroviral therapy: variation by age. Aids. 2008;22:1463–1473. doi: 10.1097/QAD.0b013e3282f88d02. [DOI] [PubMed] [Google Scholar]
- 162.Thiebaut R., Jacqmin-Gadda H., Walker S., Sabin C., Prins M., Del Amo J., Porter K., Dabis F., Chene G., Collaboration C. Determinants of response to first HAART regimen in antiretroviral-naive patients with an estimated time since HIV seroconversion. HIV Med. 2006;7:1–9. doi: 10.1111/j.1468-1293.2005.00332.x. [DOI] [PubMed] [Google Scholar]
- 163.Drylewicz J., Commenges D., Thiebaut R. Score tests for exploring complex models: application to HIV dynamics models. Biom. J. 2010;52:10–21. doi: 10.1002/bimj.200900030. [DOI] [PubMed] [Google Scholar]
- 164.Putter H., Heisterkamp S.H., Lange J.M., de Wolf F. A Bayesian approach to parameter estimation in HIV dynamical models. Stat. Med. 2002;21:2199–2214. doi: 10.1002/sim.1211. [DOI] [PubMed] [Google Scholar]
- 165.Guedj J., Thiebaut R., Commenges D. Maximum likelihood estimation in dynamical models of HIV. Biometrics. 2007;63:1198–1206. doi: 10.1111/j.1541-0420.2007.00812.x. [DOI] [PubMed] [Google Scholar]
- 166.Lavielle M., Samson A., Karina Fermin A., Mentre F. Maximum likelihood estimation of long-term HIV dynamic models and antiviral response. Biometrics. 2011;67:250–259. doi: 10.1111/j.1541-0420.2010.01422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kelley C.F., Kitchen C.M., Hunt P.W., Rodriguez B., Hecht F.M., Kitahata M., Crane H.M., Willig J., Mugavero M., Saag M., Martin J.N., Deeks S.G. Incomplete peripheral CD4+ cell count restoration in HIV-infected patients receiving long-term antiretroviral treatment. Clin. Infect. Dis. 2009;48:787–794. doi: 10.1086/597093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Rong L., Perelson A.S. Modeling latently infected cell activation: viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy. PLoS Comput. Biol. 2009;5:e1000533. doi: 10.1371/journal.pcbi.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Krakovska O., Wahl L.M. Drug-sparing regimens for HIV combination therapy: benefits predicted for “drug coasting”. Bull. Math. Biol. 2007;69:2627–2647. doi: 10.1007/s11538-007-9234-9. [DOI] [PubMed] [Google Scholar]
- 170.Thiébaut R D.J., Lacabaratz C., Lelievre J.D., Chêne G., Beq S., Croughs T., Commenges D., Delfraissy J.F., Levy Y. Conference Poster at Systems Approaches in Immunology. 2010. In vivo quantification of the effect of IL-7 on proliferation, survival and production of CD4+ T cells: mathematical analysis of one phase I study in HIV-1 infected patients. (Santa Fe, NM, USA) [Google Scholar]
- 171.Garrido C., Rallon N., Soriano V., Lopez M., Zahonero N., de Mendoza C., Benito J.M. Mechanisms involved in CD4 cell gains in HIV-infected patients switched to raltegravir. Aids. 2012;26:551–557. doi: 10.1097/QAD.0b013e3283509826. [DOI] [PubMed] [Google Scholar]
- 172.Bouazza N., Treluyer J.M., Msellati P., Van de Perre P., Diagbouga S., Nacro B., Hien H., Zoure E., Rouet F., Ouiminga A., Blanche S., Hirt D., Urien S. A novel pharmacokinetic approach to predict virologic failure in HIV-1-infected paediatric patients. Aids. 2013;27:761–768. doi: 10.1097/QAD.0b013e32835caad1. [DOI] [PubMed] [Google Scholar]
- 173.Panel on Antiretroviral Therapy and Medical Management of HIV-Infected Children. Guidelines for the Use of Antiretroviral Agents in Pediatric HIV Infection. http://aidsinfo.nih.gov/contentfiles/lvguidelines/pediatricguidelines.pdf Available at. (Last accessed 30th June 2013)
- 174.Committee P.S., Welch S., Sharland M., Lyall E.G., Tudor-Williams G., Niehues T., Wintergerst U., Bunupuradah T., Hainaut M., Della Negra M., Pena M.J., Amador J.T., Gattinara G.C., Compagnucci A., Faye A., Giaquinto C., Gibb D.M., Gandhi K., Forcat S., Buckberry K., Harper L., Konigs C., Patel D., Bastiaans D. PENTA 2009 guidelines for the use of antiretroviral therapy in paediatric HIV-1 infection. HIV Med. 2009;10:591–613. doi: 10.1111/j.1468-1293.2009.00759.x. [DOI] [PubMed] [Google Scholar]
- 175.Prague M., Commenges D., Drylewicz J., Thiebaut R. Treatment monitoring of HIV-infected patients based on mechanistic models. Biometrics. 2012;68:902–911. doi: 10.1111/j.1541-0420.2012.01749.x. [DOI] [PubMed] [Google Scholar]
- 176.Charrier E., Cordeiro P., Brito R.M., Mezziani S., Herblot S., Le Deist F., Duval M. Reconstitution of maturating and regulatory lymphocyte subsets after cord blood and BMT in children. Bone Marrow Transplant. 2013;48:376–382. doi: 10.1038/bmt.2012.176. [DOI] [PubMed] [Google Scholar]
- 177.Berger M., Figari O., Bruno B., Raiola A., Dominietto A., Fiorone M., Podesta M., Tedone E., Pozzi S., Fagioli F., Madon E., Bacigalupo A. Lymphocyte subsets recovery following allogeneic bone marrow transplantation (BMT): CD4+ cell count and transplant-related mortality. Bone Marrow Transplant. 2008;41:55–62. doi: 10.1038/sj.bmt.1705870. [DOI] [PubMed] [Google Scholar]
- 178.Schwinger W., Weber-Mzell D., Zois B., Rojacher T., Benesch M., Lackner H., Dornbusch H.J., Sovinz P., Moser A., Lanzer G., Schauenstein K., Ofner P., Handgretinger R., Urban C. Immune reconstitution after purified autologous and allogeneic blood stem cell transplantation compared with unmanipulated bone marrow transplantation in children. Br. J. Haematol. 2006;135:76–84. doi: 10.1111/j.1365-2141.2006.06244.x. [DOI] [PubMed] [Google Scholar]
- 179.Bartelink I.H., Belitser S.V., Knibbe C.A., Danhof M., de Pagter A.J., Egberts T.C., Boelens J.J. Immune reconstitution kinetics as an early predictor for mortality using various hematopoietic stem cell sources in children. Biol. Blood Marrow Transplant. 2013;19:305–313. doi: 10.1016/j.bbmt.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 180.Kim H.O., Oh H.J., Lee J.W., Jang P.S., Chung N.G., Cho B., Kim H.K. Immune reconstitution after allogeneic hematopoietic stem cell transplantation in children: a single institution study of 59 patients. Korean J. Pediatr. 2013;56:26–31. doi: 10.3345/kjp.2013.56.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chen X., Hale G.A., Barfield R., Benaim E., Leung W.H., Knowles J., Horwitz E.M., Woodard P., Kasow K., Yusuf U., Behm F.G., Hayden R.T., Shurtleff S.A., Turner V., Srivastava D.K., Handgretinger R. Rapid immune reconstitution after a reduced-intensity conditioning regimen and a CD3-depleted haploidentical stem cell graft for paediatric refractory haematological malignancies. Br. J. Haematol. 2006;135:524–532. doi: 10.1111/j.1365-2141.2006.06330.x. [DOI] [PubMed] [Google Scholar]
- 182.Fedele R., Martino M., Garreffa C., Messina G., Console G., Princi D., Dattola A., Moscato T., Massara E., Spiniello E., Irrera G., Iacopino P. The impact of early CD4+ lymphocyte recovery on the outcome of patients who undergo allogeneic bone marrow or peripheral blood stem cell transplantation. Blood Transfus. 2012;10:174–180. doi: 10.2450/2012.0034-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tedeschi S.K., Jagasia M., Engelhardt B.G., Domm J., Kassim A.A., Chinratanalab W., Greenhut S.L., Goodman S., Greer J.P., Schuening F., Frangoul H., Savani B.N. Early lymphocyte reconstitution is associated with improved transplant outcome after cord blood transplantation. Cytotherapy. 2011;13:78–82. doi: 10.3109/14653249.2010.495114. [DOI] [PubMed] [Google Scholar]
- 184.Koenig M., Huenecke S., Salzmann-Manrique E., Esser R., Quaritsch R., Steinhilber D., Radeke H.H., Martin H., Bader P., Klingebiel T., Schwabe D., Schneider G., Lehrnbecher T., Orth A., Koehl U. Multivariate analyses of immune reconstitution in children after allo-SCT: risk-estimation based on age-matched leukocyte sub-populations. Bone Marrow Transplant. 2010;45:613–621. doi: 10.1038/bmt.2009.204. [DOI] [PubMed] [Google Scholar]
- 185.Renard C., Barlogis V., Mialou V., Galambrun C., Bernoux D., Goutagny M.P., Glasman L., Loundou A.D., Poitevin-Later F., Dignat-George F., Dubois V., Picard C., Chabannon C., Bertrand Y., Michel G. Lymphocyte subset reconstitution after unrelated cord blood or bone marrow transplantation in children. Br. J. Haematol. 2011;152:322–330. doi: 10.1111/j.1365-2141.2010.08409.x. [DOI] [PubMed] [Google Scholar]
- 186.Koehl U., Bochennek K., Zimmermann S.Y., Lehrnbecher T., Sorensen J., Esser R., Andreas C., Kramm C., Gruttner H.P., Falkenberg E., Orth A., Bader P., Schwabe D., Klingebiel T. Immune recovery in children undergoing allogeneic stem cell transplantation: absolute CD8+ CD3+ count reconstitution is associated with survival. Bone Marrow Transplant. 2007;39:269–278. doi: 10.1038/sj.bmt.1705584. [DOI] [PubMed] [Google Scholar]
- 187.Duval M., Pedron B., Rohrlich P., Legrand F., Faye A., Lescoeur B., Bensaid P., Larchee R., Sterkers G., Vilmer E. Immune reconstitution after haematopoietic transplantation with two different doses of pre-graft antithymocyte globulin. Bone Marrow Transplant. 2002;30:421–426. doi: 10.1038/sj.bmt.1703680. [DOI] [PubMed] [Google Scholar]
- 188.Barlogis V., Glasman L., Brunet C., Loundou A.D., Lemarie C., Galambrun C., Thuret I., Curtillet C., Le Meignen M., Bernard F., Chambost H., Calmels B., Picard C., Chabannon C., Dignat-George F., Michel G. Impact of viable CD45 cells infused on lymphocyte subset recovery after unrelated cord blood transplantation in children. Biol. Blood Marrow Transplant. 2011;17:109–116. doi: 10.1016/j.bbmt.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 189.Giannelli R., Bulleri M., Menconi M., Casazza G., Focosi D., Bernasconi S., Favre C. Reconstitution rate of absolute CD8+ T lymphocyte counts affects overall survival after pediatric allogeneic hematopoietic stem cell transplantation. J. Pediatr. Hematol. Oncol. 2012;34:29–34. doi: 10.1097/MPH.0b013e3182127add. [DOI] [PubMed] [Google Scholar]
- 190.Huenecke S., Behl M., Fadler C., Zimmermann S.Y., Bochennek K., Tramsen L., Esser R., Klarmann D., Kamper M., Sattler A., von Laer D., Klingebiel T., Lehrnbecher T., Koehl U. Age-matched lymphocyte subpopulation reference values in childhood and adolescence: application of exponential regression analysis. Eur. J. Haematol. 2008;80:532–539. doi: 10.1111/j.1600-0609.2008.01052.x. [DOI] [PubMed] [Google Scholar]
- 191.Olkinuora H., von Willebrand E., Kantele J.M., Vainio O., Talvensaari K., Saarinen-Pihkala U., Siitonen S., Vettenranta K. The impact of early viral infections and graft-versus-host disease on immune reconstitution following paediatric stem cell transplantation. Scand. J. Immunol. 2011;73:586–593. doi: 10.1111/j.1365-3083.2011.02530.x. [DOI] [PubMed] [Google Scholar]
- 192.Clave E., Busson M., Douay C., Peffault de Latour R., Berrou J., Rabian C., Carmagnat M., Rocha V., Charron D., Socie G., Toubert A. Acute graft-versus-host disease transiently impairs thymic output in young patients after allogeneic hematopoietic stem cell transplantation. Blood. 2009;113:6477–6484. doi: 10.1182/blood-2008-09-176594. [DOI] [PubMed] [Google Scholar]
- 193.Fallen P.R., McGreavey L., Madrigal J.A., Potter M., Ethell M., Prentice H.G., Guimaraes A., Travers P.J. Factors affecting reconstitution of the T cell compartment in allogeneic haematopoietic cell transplant recipients. Bone Marrow Transplant. 2003;32:1001–1014. doi: 10.1038/sj.bmt.1704235. [DOI] [PubMed] [Google Scholar]
- 194.Surh C.D., Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 195.Plunkett W., Gandhi V., Huang P., Robertson L.E., Yang L.Y., Gregoire V., Estey E., Keating M.J. Fludarabine: pharmacokinetics, mechanisms of action, and rationales for combination therapies. Semin. Oncol. 1993;20:2–12. [PubMed] [Google Scholar]
- 196.Salinger D.H., Blough D.K., Vicini P., Anasetti C., O'Donnell P.V., Sandmaier B.M., McCune J.S. A limited sampling schedule to estimate individual pharmacokinetic parameters of fludarabine in hematopoietic cell transplant patients. Clin. Cancer. Res. 2009;15:5280–5287. doi: 10.1158/1078-0432.CCR-09-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Tasso M.J., Boddy A.V., Price L., Wyllie R.A., Pearson A.D., Idle J.R. Pharmacokinetics and metabolism of cyclophosphamide in paediatric patients. Cancer Chemother. Pharmacol. 1992;30:207–211. doi: 10.1007/BF00686313. [DOI] [PubMed] [Google Scholar]
- 198.van Hasselt J.G., van Eijkelenburg N.K., Beijnen J.H., Schellens J.H., Huitema A.D. Optimizing drug development of anti-cancer drugs in children using modelling and simulation. Br. J. Clin. Pharmacol. 2013;76:30–47. doi: 10.1111/bcp.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Glowka F.K., Karazniewicz-Lada M., Grund G., Wrobel T., Wachowiak J. Pharmacokinetics of high-dose i.v. treosulfan in children undergoing treosulfan-based preparative regimen for allogeneic haematopoietic SCT. Bone Marrow Transplant. 2008;42(Suppl. 2):S67–S70. doi: 10.1038/bmt.2008.287. [DOI] [PubMed] [Google Scholar]
- 200.Elter T., Molnar I., Kuhlmann J., Hallek M., Wendtner C. Pharmacokinetics of alemtuzumab and the relevance in clinical practice. Leuk. Lymphoma. 2008;49:2256–2262. doi: 10.1080/10428190802475303. [DOI] [PubMed] [Google Scholar]
- 201.Call S.K., Kasow K.A., Barfield R., Madden R., Leung W., Horwitz E., Woodard P., Panetta J.C., Baker S., Handgretinger R., Rodman J., Hale G.A. Total and active rabbit antithymocyte globulin (rATG; Thymoglobulin) pharmacokinetics in pediatric patients undergoing unrelated donor bone marrow transplantation. Biol. Blood Marrow Transplant. 2009;15:274–278. doi: 10.1016/j.bbmt.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Seidel M.G., Fritsch G., Matthes-Martin S., Lawitschka A., Lion T., Potschger U., Rosenmayr A., Fischer G., Gadner H., Peters C. Antithymocyte globulin pharmacokinetics in pediatric patients after hematopoietic stem cell transplantation. J. Pediatr. Hematol. Oncol. 2005;27:532–536. doi: 10.1097/01.mph.0000184575.00717.25. [DOI] [PubMed] [Google Scholar]
- 203.Willemze A.J., Cremers S.C., Schoemaker R.C., Lankester A.C., den Hartigh J., Burggraaf J., Vossen J.M. Ciclosporin kinetics in children after stem cell transplantation. Br. J. Clin. Pharmacol. 2008;66:539–545. doi: 10.1111/j.1365-2125.2008.03217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Downing H.J., Pirmohamed M., Beresford M.W., Smyth R.L. Paediatric use of mycophenolate mofetil. Br. J. Clin. Pharmacol. 2013;75:45–59. doi: 10.1111/j.1365-2125.2012.04305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]