

Abstract
Cryptococcus neoformans is a significant cause of fungal meningitis in patients with impaired T cell-mediated immunity (CMI). Experimental pulmonary infection with a C. neoformans strain engineered to produce IFN-γ, H99γ, results in the induction of Th1-type CMI, resolution of the acute infection, and protection against challenge with WT Cryptococcus. Given that individuals with suppressed CMI are highly susceptible to pulmonary C. neoformans infection, we sought to determine whether antimicrobial peptides were produced in mice inoculated with H99γ. Thus, we measured levels of antimicrobial peptides lipocalin-2, S100A8, S100A9, calprotectin (S100A8/A9 heterodimer), serum amyloid A-3 (SAA3), and their putative receptors Toll-like receptor 4 (TLR4) and the receptor for advanced glycation end products (RAGE) in mice during primary and recall responses against C. neoformans infection. Results showed increased levels of IL-17A and IL-22, cytokines known to modulate antimicrobial peptide production. We also observed increased levels of lipocalin-2, S100A8, S100A9 and SAA3 as well as TLR4+ and RAGE+ macrophages and dendritic cells in mice inoculated with H99γ compared with WT H99. Similar results were observed in the lungs of H99γ-immunized, compared with heat-killed C. neoformans-immunized, mice following challenge with WT yeast. However, IL-22-deficient mice inoculated with H99γ demonstrated antimicrobial peptide production and no change in survival rates compared with WT mice. These studies demonstrate that protection against cryptococcosis is associated with increased production of antimicrobial peptides in the lungs of protected mice that are not solely in response to IL-17A and IL-22 production and may be coincidental rather than functional.
Introduction
Cryptococcus neoformans, the aetiological agent of cryptococcosis, is an opportunistic fungal pathogen that typically affects individuals with impaired T-cell function (i.e. individuals with AIDS and lymphoid malignancies, and recipients of immunosuppressive therapies) (Levitz, 1991; Mitchell & Perfect, 1995; Shoham & Levitz, 2005; Singh et al., 1997, 2008). Previous studies have shown that protective immunity against this organism is dependent upon the induction of Th1-type cytokine responses (Blasi et al., 1994; Buchanan & Doyle, 2000; Chuck & Sande, 1989; Hill & Harmsen, 1991; Huffnagle et al., 1991; Mody et al., 1990; Shoham & Levitz, 2005; Singh et al., 2008; Wormley et al., 2007; Wozniak et al., 2009). Pulmonary infection in BALB/c mice using a C. neoformans strain engineered to produce murine IFN-γ (H99γ) results in induction of Th1-type cytokines, IL-17A, and significantly reduced fungal burden. Mice immunized with H99γ are completely protected against a subsequent pulmonary challenge with an otherwise lethal WT C. neoformans strain (Wormley et al., 2007; Wozniak et al., 2009). IL-17A and IL-22 function cooperatively to induce the production of antimicrobial peptides and acute phase proteins such as β-defensins, S100A8, S100A9 and lipocalin-2 in bronchial epithelial cells (Aujla et al., 2008; Gessner et al., 2012; Kolls et al., 2008; Liang et al., 2006; Wolk et al., 2011). Studies have shown the importance of IL-17A and IL-22 clinically to mediate clearance in patients with chronic mucocutaneous candidiasis (Eyerich et al., 2008) and experimentally to protect mice against disseminated candidiasis (Huang et al., 2004), pulmonary aspergillosis (Gessner et al., 2012) and pulmonary Klebsiella pneumoniae infection (Aujla et al., 2008).
S100A8 and S100A9, which can collectively form a heterodimer called calprotectin (Lagasse & Clerc, 1988; Odink et al., 1987; Zwadlo et al., 1988), are involved in neutrophil recruitment and are known to be produced by epithelial cells following induction by IL-22 (Aujla et al., 2008; Kolls et al., 2008; Liang et al., 2006). S100 proteins are calcium-binding cytosolic proteins that can be expressed by multiple cell types, including alveolar epithelial cells, endothelial cells, monocytes, macrophages, neutrophils and dendritic cells (DCs) (Ehrchen et al., 2009; Foell et al., 2007; Ramirez-Ortiz et al., 2011). Importantly, calprotectin may have direct antifungal and fungistatic activity against fungi, including C. neoformans (Mambula et al., 2000; McCormick et al., 2010; Urban et al., 2009).
Lipocalin-2, an acute phase protein that is a component of neutrophil granules, is involved in iron sequestration and is a biomarker for inflammation and sepsis (Bachman et al., 2009; Borregaard & Cowland, 2006; Li & Chan, 2011). Secretion of lipocalin-2 involves signalling via Toll-like receptor 4 (TLR4) and subsequent NF-κB binding to consensus sequences upstream of the lipocalin-2 promoter (Flo et al., 2004; Li & Chan, 2011), and its production is regulated via IL-1β, IL-17A and other cytokines (Chan et al., 2009; Kolls et al., 2008; Li & Chan, 2011). Although lipocalin-2 is increased in the lungs of mice during infection with Aspergillus fumigatus, lipocalin-2-deficient mice do not demonstrate impaired fungal clearance (Gessner et al., 2012), and lipocalin-2 does not bind to fungal siderophores (Flo et al., 2004). Thus, lipocalin-2 may only serve as a biomarker for inflammatory immune responses against fungi and may not have any direct fungicidal effects, although its role against C. neoformans remains to be determined.
Serum amyloid A-3 (SAA3) is a member of the SAA family of acute phase proteins and has been long considered a biomarker for inflammatory diseases (Uhlar & Whitehead, 1999). SAA3 is induced via a TLR4/NF-κB-dependent pathway, and SAA3 in turn induces chemotactic proteins, pro-inflammatory cytokines (including IL-1β) and nitric oxide from neutrophils, macrophages and epithelial cells (Ather et al., 2011; Hiratsuka et al., 2008). SAA3 instillation into the lungs elicits robust pro-inflammatory cytokine production and phagocyte recruitment into the lungs (Ather et al., 2011). Our previous data show significant IL-1β increases in the lungs of H99γ-immunized mice that are protected against experimental pulmonary C. neoformans infection (Wozniak et al., 2009), which could be induced by mediators such as SAA3. Therefore, SAA3 may serve as more than just a ‘biomarker’ during the protective immune response to C. neoformans infection.
Receptors for these antimicrobial peptides include the receptor for advanced glycation end products (RAGE) and TLR4 (Ehrchen et al., 2009; Foell et al., 2007; Herold et al., 2007). RAGE is expressed on endothelial cells as well as macrophages, neutrophils, DCs, T cells, B cells, alveolar type II cells and alveolar epithelial cells (Fehrenbach et al., 1998; Katsuoka et al., 1997). S100A8, S100A9 and SAA3 are TLR4 agonists and induce the activation of MyD88-dependent pathways as well as ERK1/2, p38, mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK) and protein kinase C (PKC). Stimulation of TLR4 induces the release of proinflammatory cytokines, chemokines and reactive oxygen species necessary for the activation of potent host defences (Ehrchen et al., 2009; Hiratsuka et al., 2008).
The studies described herein were designed to examine the effect of high levels of IL-17A and IL-22 on the presence of antimicrobial peptides during protective cryptococcal infection and the recruitment of TLR4+ and RAGE+ effector cells in these mice. We hypothesized that IL-17A and IL-22 in protected mice would lead to increased production of antimicrobial peptides and increased recruitment of RAGE+ and TLR4+ antigen-presenting cells (APCs), and reveal a role for those cells and mediators in protection against pulmonary cryptococcal infection.
Methods
Mice.
Female BALB/c (H-2d) mice (National Cancer Institute/Charles River Laboratories) with a mean weight of 20–25 g and IL-22 knockout (IL-22−/−) mice and their controls, ‘129 mice’ (Mutant Mouse Resource Center), which were subsequently bred in our facility, were used throughout these studies. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Mice were housed at The University of Texas at San Antonio Small Animal Laboratory Vivarium. These animal experiments were approved by The University of Texas at San Antonio Institutional Animal Care and Use Committee (IACUC), and mice were handled according to IACUC guidelines. All efforts were made to minimize animal suffering.
Strains and media.
C. neoformans strains H99 (serotype A, Mat α) and H99γ (an INF-γ-producing C. neoformans strain derived from H99) were recovered from 15 % glycerol stocks stored at −80 °C prior to use in the experiments described herein. The strains were maintained on yeast-extract-peptone-glucose (YPD) media. Yeast cells were grown for 16–18 h at 30 °C with shaking in YPD broth, collected by centrifugation and washed three times with sterile PBS, and viable cells were quantified using trypan blue dye exclusion in a haemacytometer.
Pulmonary inoculations.
Pulmonary immunizations or inoculations were initiated by nasal inhalation as previously described (Wormley et al., 2007; Wozniak & Levitz, 2009). Mice were anaesthetized with 2 % isoflurane using a rodent anaesthesia device (Eagle Eye Anaesthesia) and then given a yeast inoculum of 1×104 c.f.u. of C. neoformans strain H99 or H99γ in 50 µl of sterile PBS pipetted directly into the nares. The inocula used were verified by quantitative culture on YPD agar. The mice were fed ad libitum and were monitored by inspection twice daily. Mice were killed at specific time points post-inoculation by CO2 inhalation followed by cervical dislocation, and lung tissues were excised using an aseptic technique. Tissues were homogenized in 1 ml of sterile PBS, followed by culture of 10-fold dilutions of each tissue on YPD agar supplemented with chloramphenicol (Mediatech). Colony-forming units were enumerated following incubation at 30 °C for 48 h. For secondary challenge studies, mice were immunized with either 1×104 c.f.u. of C. neoformans strain H99γ or heat-killed C. neoformans (HK C.n.) in 50 µl of sterile PBS and were allowed 60 days to resolve the infection. Subsequently, the immunized mice received a second experimental pulmonary challenge with 1×104 c.f.u. of C. neoformans strain H99 in 50 µl of sterile PBS and were killed at specific time points post-inoculation. Alternatively, mice intended for survival analysis were monitored by inspection twice daily and killed if they appeared to be in pain or moribund. Mice were killed using CO2 inhalation, followed by cervical dislocation.
Cytokine and antimicrobial peptide analysis.
Cytokine and antimicrobial peptide levels in lung tissues were analysed using ELISAs (R & D Systems or EMD Millipore) performed according to the manufacturers’ instructions. Briefly, lung tissue was excised and homogenized in ice-cold sterile PBS (1 ml). An aliquot (50 µl) was taken to quantify the pulmonary fungal burden and an anti-protease buffer solution (1 ml) containing PBS, protease inhibitors (inhibiting cysteine, serine and other metalloproteinases) and 0.05 % Triton X-100 were added to the homogenate. Samples were then clarified by centrifugation (800 g) for 5 min. Supernatants from pulmonary homogenates were assayed for the presence of IL-22, IL-17A, SAA3, lipocalin-2, S100A8 and S100A9 by ELISA.
Flow cytometry.
Standard methodology was employed for the direct immunofluorescence of pulmonary leukocytes. Briefly, in 96-well U-bottom plates, 100 µl containing 1×106 cells in PBS+2 % FBS (FACS buffer) were incubated with 50 µl Fc Block (BD Biosciences) diluted in FACS buffer for 5 min to block non-specific binding of antibodies to cellular Fc receptors. Subsequently, an optimal concentration of fluorochrome-conjugated antibodies (eBioscience and Molecular Probes) (0.06–0.5 µg/1×106 cells in 50 µl FACS buffer) were added in various combinations to allow for dual or triple staining experiments, and plates were incubated for 30 min at 4 °C. Following incubation, the cells were washed three times with FACS buffer and cells were fixed in 200 µl of 2 % ultrapure formaldehyde (Polysciences) diluted in FACS buffer (fixation buffer). Cells incubated with either FACS buffer alone or single fluorochrome-conjugated antibodies were used to determine positive staining and spillover/compensation calculations. The samples were analysed using BD FACSArray software on a BD FACSArray flow cytometer (BD Biosciences). Dead cells were excluded on the basis of forward angle and 90° light scatter. For data analyses, 30 000 events (cells) were evaluated from a predominantly leukocytic population identified by backgating from CD45+-stained cells. The absolute number of total leukocytes was quantified by multiplying the total number of cells observed by haemacytometer counting by the percentage of CD45+ cells determined by flow cytometry. The absolute number of each leukocyte subset (F4/80+ macrophages, CD11c+/CD11bint DCs) expressing TLR4 or RAGE was determined by multiplying the percentage of each gated population by the total number of CD45+ cells.
Antimicrobial gene expression.
Total RNA was isolated from lung homogenates using TRIzol reagent (Invitrogen), and then treated with DNase to remove possible traces of contaminating DNA according to the manufacturer’s instructions (Qiagen). Total RNA was subsequently recovered using the Qiagen RNeasy kit. cDNA was synthesized from 1 µg of total RNA using the oligo dT primer and reagents supplied in the SuperScript III RT kit (Invitrogen) according to the manufacturer’s instructions. The cDNA was used as a template for real-time PCR analysis using the TaqMan Gene Expression Assay (Applied Biosystems) according to the manufacturer’s instructions. All real-time PCRs were performed using the 7300 Real-Time PCR System (Applied Biosystems). For each real-time PCR, a master mix was prepared on ice with TaqMan Gene Expression Assays specific for S100A8, S100A9, SAA3, β-defensins 1 and 2, and LCN-2 (Applied Biosystems). Each reaction was normalized to the endogenous control GAPDH, and relative mRNA expression was derived using the comparative Ct method. The results shown represent the fold increase or decrease in gene expression in BALB/c H99γ-infected compared with H99-infected mice or IL-22−/− mice compared with WT 129 mice.
In vitro antimicrobial assays.
For these experiments, purified S100A8, S100A9, SAA3 or lipocalin-2 (R&D Systems) were incubated with C. neoformans strain H99 at a concentration of 500 ng ml−1, the maximum concentration available for these peptides (Hole et al., 2012; Miyakawa et al., 1996). Cryptococci were resuspended in 10 mM phosphate buffer with 2 % RPMI medium, pH 5.5 (phosphate buffer). Fungi were added to a 96-well plate in a volume of 50 ml (2.5×105 ml−1). Phosphate buffer was added to control wells, and 50 ml of antimicrobial peptide was added to test wells, for a total volume of 100 ml per well in duplicate wells. Plates were incubated for 24 h at 37 °C, with 5 % CO2. Cryptococci were diluted in sterile PBS and plated on YPD agar to determine c.f.u. numbers. Killing was defined as c.f.u. significantly below the inoculum, and inhibition was defined as c.f.u. significantly below that of growth in phosphate buffer alone (Hole et al., 2012). All peptides were commercially manufactured and are ≥95 % pure.
Statistical analysis.
The unpaired Student’s t-test (two-tailed) was used to analyse fungal burden, pulmonary cell populations and cytokine/antimicrobial peptide data using GraphPad Prism version 5.00 for Windows (GraphPad Prism Software). Survival data were analysed using the log-rank test (GraphPad Software). Significant differences were defined as P<0.05.
Results
Protection against C. neoformans is associated with increased IL-17A and IL-22 production in lungs
Experimental pulmonary inoculation in mice using a C. neoformans strain engineered to express IFN-γ, H99γ, results in the induction of Th1-type cell-mediated immunity responses, resolution of the acute infection and complete protection against a second pulmonary challenge with a pathogenic C. neoformans strain (Wormley et al., 2007; Wozniak et al., 2009). Additionally, we observed significant increases in IL-17A production in the lungs of mice following inoculation with H99γ and in H99γ-immunized mice challenged with WT C. neoformans, which does not express IFN-γ (Wormley et al., 2007; Wozniak et al., 2009). IL-17A and IL-22 are crucial regulators of antimicrobial peptide production in the lungs (Liang et al., 2006). Thus, we determined the levels of IL-17A and IL-22 present in lung homogenates derived from mice infected with WT C. neoformans strain H99 compared with H99γ on days 1, 3, 5 and 7 post-inoculation. We observed significantly increased levels of IL-17A (Fig. 1a) and IL-22 (Fig. 1b) on days 5 and 7 post-inoculation in the lungs of mice infected with H99γ compared with those infected with WT C. neoformans. We then determined IL-17A and IL-22 production in mice protectively immunized with C. neoformans strain H99γ or non-protectively immunized with HK C.n. and subsequently challenged with WT C. neoformans. A significant increase in IL-17A production was observed in lung homogenates prepared from protectively immunized mice (H99γ immunized) compared with non-protectively immunized mice (HK C.n. immunized) on days 1, 3, 5 and 7 post-secondary challenge (Fig. 1c). Also, significantly greater IL-22 production was observed in lung homogenates prepared from protectively immunized mice compared with non-protectively immunized mice on days 3, 5 and 7 post-secondary challenge (Fig. 1d). Thus, IL-17A and IL-22 production is increased during protective anti-cryptococcal immune responses.
Fig. 1.

Pulmonary levels of IL-17A and IL-22 following inoculation with C. neoformans strain H99γ or H99 and during secondary challenge of protectively and non-protectively immunized mice. BALB/c mice were intranasally inoculated with 1×104 c.f.u. of C. neoformans strain H99 or H99γ (a, b). Alternatively, BALB/c mice received an initial immunization with HK C.n. yeasts or C. neoformans strain H99γ and were allowed 60 days to resolve the infection. Mice then received a second intranasal challenge with 1×104 c.f.u. of C. neoformans strain H99 (c, d). Lung homogenates were prepared from lungs excised on days 1, 3, 5 and 7 post-inoculation and assayed for IL-17A and IL-22 production by ELISA. Data are cumulative of three experiments utilizing four mice per group per time point for each experiment. Asterisks indicate significant differences (*P<0.05).
Enhanced antimicrobial peptide and acute phase protein production correlates with protective anti-C. neoformans immune responses
We next determined if the increased pulmonary IL-17A and IL-22 production observed in mice infected with C. neoformans H99γ was associated with enhanced antimicrobial peptide and/or acute phase protein production. Mice were intranasally inoculated with C. neoformans strain H99γ or WT C. neoformans strain H99. RNA was then extracted from lungs excised from mice in each group at days 1, 3, 5 and 7 post-inoculation to measure the levels of S100A8, S100A9, β-defensins 1 and 2, lipocalin-2, and SAA3 transcripts. Results showed an increased expression of transcripts for S100A8 and S100A9 at days 5 and 7 post-inoculation and for lipocalin-2 and SAA3 at days 3, 5 and 7 post-inoculation within the lungs of mice inoculated with the H99γ strain compared with H99 (Fig. 2). We observed no difference in protein levels of cathlecidins or gene expression of β-defensins 1 and 2 between each group of mice on all days tested (data not shown).
Fig. 2.
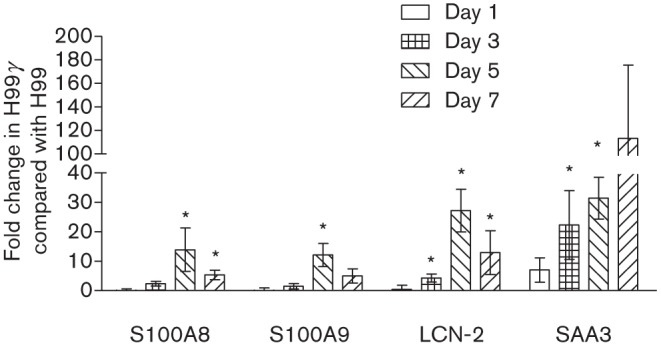
Antimicrobial peptide gene expression in mice following inoculation with H99γ compared with H99. BALB/c mice were intranasally inoculated with 1×104 c.f.u. of C. neoformans strain H99γ or H99 in 50 µl sterile PBS. At days 1, 3, 5 and 7 post-inoculation, mice were killed and pulmonary RNA was isolated from whole lung tissues and homogenized in Trizol reagent. cDNA was prepared from total RNA and real-time PCR was conducted to measure S100A8, S100A9, lipocalin-2 (LCN-2) and SAA3 expression. Bars represent the fold change in gene expression in pulmonary tissues from H99γ-infected mice compared with tissues from H99-infected mice. Data shown are cumulative of three independent experiments utilizing individual tissues from four mice per group per time point per experiment and are reported as means±sem (*P<0.05).
Subsequently, we determined if increased gene expression of the various antimicrobial peptides and acute phase proteins tested would translate to increased protein production. For this, BALB/c mice were intranasally inoculated with either C. neoformans strain H99γ or WT C. neoformans. We then measured S100A8, S100A9, β-defensins 1 and 2, lipocalin-2, and SAA3 production in total lung homogenates derived from each mouse on days 1, 3, 5 and 7 post-inoculation by ELISA. We observed significantly increased levels of SAA3 on days 3, 5 and 7 (Fig. 3d) and S100A8 (Fig. 3a), S100A9 (Fig. 3b) and lipocalin-2 (Fig. 3c) on days 5 and 7 post-inoculation in the lungs of mice inoculated with H99γ (P<0.05 for each) compared with mice inoculated with WT yeast. However, we observed no difference in β-defensins 1 and 2 expression between each group of mice on all days tested (data not shown).
Fig. 3.

Pulmonary antimicrobial peptide production following inoculation with C. neoformans strain H99γ or WT C. neoformans. BALB/c mice were intranasally inoculated with 1×104 c.f.u. of C. neoformans strain H99 or H99γ. Lung homogenates were prepared from lungs excised on days 1, 3, 5 and 7 post-inoculation and assayed for S100A8 (a), S100A9 (b), lipocalin-2 (c) and SAA3 (d) production by ELISA. Data are cumulative of three experiments utilizing four mice per group per time point for each experiment. Asterisks indicate significant differences (*P<0.05).
Experimental pulmonary inoculation with C. neoformans strain H99γ results in the recruitment of TLR4+ and RAGE+ macrophages and DCs to the lungs during infection
TLR4 and RAGE have been identified as receptors for S100A8/A9 and SAA3 (Ehrchen et al., 2009; Foell et al., 2007; Herold et al., 2007). Thus, we determined the expression of TLR4 and RAGE on the surface of pulmonary macrophages and DCs derived from mice inoculated with WT C. neoformans strain H99 or C. neoformans strain H99γ on days 5 and 7 post-inoculation, the time points at which production of antimicrobial peptides and acute phase proteins, respectively, was highest in mice inoculated with C. neoformans strain H99γ. Flow cytometric analysis of leukocyte infiltrates demonstrated an increase in the absolute number of TLR4+ pulmonary macrophages and DCs (Fig. 4a, b, respectively) and RAGE+ pulmonary macrophages and DCs (Fig. 4c, d, respectively).
Fig. 4.

Antimicrobial peptide receptors on macrophages (Macs) and DCs following inoculation with H99γ compared with H99. BALB/c mice were intranasally inoculated with 1×104 c.f.u. of C. neoformans strain H99 or H99γ. Lungs were excised at days 5 and 7 post-inoculation and a single cell suspension was generated using enzymic digestion. The leukocytes were evaluated by flow cytometry for the presence of TLR4 on the surface of macrophages (a) or DCs (b) or RAGE on the surface of macrophages (c) or DCs (d). Flow cytometry data are representative results of two independent experiments using pooled leukocytes from four mice per group per time point for each experiment.
Protective recall responses to pulmonary C. neoformans infection are associated with enhanced antimicrobial peptide and acute phase protein production
We then determined if similar increases in antimicrobial peptide and acute phase protein production in the lungs would be observed during protective recall responses to pulmonary C. neoformans infection. BALB/c mice were intranasally immunized with C. neoformans strain H99γ, which induces protective anti-cryptococcal immune responses, or HK C.n. strain H99 yeasts, which results in non-protective immune responses, and allowed 60 days to resolve the infection. Subsequently, the mice were challenged with WT C. neoformans strain H99, and S100A8, S100A9, lipocalin-2 and SAA3 production in total lung homogenates was determined. We observed significantly increased levels of S100A8 (days 3, 5 and 7), S100A9 (days 1, 3, 5 and 7), lipocalin-2 (days 3 and 5) and SAA3 (days 1, 3, 5 and 7) during the recall response to pulmonary challenge with WT C. neoformans in mice that were protectively immunized compared with non-protectively immunized mice (Fig. 5). Thus, protection against pulmonary C. neoformans infection correlates with increased production of S100A8, S100A9, lipocalin-2 and SAA3.
Fig. 5.

Pulmonary antimicrobial peptide production during the recall response to C. neoformans in protectively and non-protectively immunized mice. BALB/c mice received an initial immunization with HK C.n. (HK) or C. neoformans strain H99γ and were allowed 60 days to resolve the infection. Mice then received a second intranasal challenge with 1×104 c.f.u. of C. neoformans strain H99. Lung homogenates were prepared from lungs excised on days 1, 3, 5 and 7 post-inoculation and assayed for S100A8 (a), S100A9 (b), lipocalin-2 (c) and SAA3 (d) production by ELISA. Data are cumulative of two experiments utilizing four mice per group per time point for each experiment. Asterisks indicate significant differences (*P<0.05).
Protection against pulmonary C. neoformans challenge correlates with recruitment of TLR4+ and RAGE+ pulmonary macrophages and DCs
We next determined the expression of TLR4 and RAGE on pulmonary macrophage and DC infiltrates during the recall response to C. neoformans in mice protectively immunized with C. neoformans strain H99γ or non-protectively immunized with HK C.n. on days 1, 3 and 5 post-secondary inoculations. Flow cytometric analysis of pulmonary leukocyte infiltrates demonstrated dramatic increases in the absolute number of TLR4+ macrophages (Fig. 6a), TLR4+ DCs (Fig. 6b), RAGE+ macrophages (Fig. 6c) and RAGE+ DCs (Fig. 6d) during the recall response to C. neoformans in protected mice compared with non-protected mice. These results are similar to the results obtained during primary infection (Fig. 4), and suggest that macrophages and DCs are trafficking to the lungs in response to increased antimicrobial peptide and acute phase protein production in the pulmonary mucosa.
Fig. 6.

Antimicrobial peptide receptors on macrophages (Macs) and DCs during secondary challenge in protectively compared with non-protectively immunized mice. BALB/c mice received an initial immunization with HK C.n. (HK) or C. neoformans strain H99γ and were allowed 60 days to resolve the infection. Mice then received a second intranasal challenge with 1×104 c.f.u. of C. neoformans strain H99. Lungs were excised at days 1, 3 and 5 post-challenge and a single cell suspension was generated using enzymic digestion. The leukocytes were then evaluated by flow cytometry for the presence of TLR4 on the surface of macrophages (a) or DCs (b) or RAGE on the surface of macrophages (c) or DCs (d). Flow cytometry data are representative results of two independent experiments using pooled leukocytes from four mice per group per time point for each experiment.
IL-22−/− mice have less antimicrobial peptide and acute phase protein expression compared with WT mice at the protein, but not transcript, level, during experimental pulmonary infection with C. neoformans strain H99γ
Experimental pulmonary inoculation with C. neoformans strain H99γ in mice results in significantly increased production of IL-22 and various antimicrobial peptides and acute phase proteins. We thus determined whether IL-22 was critical to the production of S100A8, S100A9, lipocalin-2 and SAA3 in H99γ-infected mice. IL-22−/− mice and WT 129 mice were intranasally inoculated with C. neoformans strain H99γ and lungs were excised at day 5 post-inoculation, the time point when the antimicrobial peptide production is at its highest in our model system (data not shown). Gene expression for the various antimicrobial peptides and acute phase proteins was examined. Fig. 7 shows that no significant differences in gene expression of S100A8, S100A9, lipocalin-2, or SAA3 were observed in IL-22−/− mice compared with WT 129 mice. We then performed studies to further examine the importance of IL-22 in modulating protein-level expression of antimicrobial peptides and acute phase proteins in pulmonary tissues of WT 129 mice and IL-22−/− mice following inoculation with C. neoformans strain H99γ at day 5 post-inoculation. Fig. 8 shows that all antimicrobial peptides tested, except S100A9 (Fig. 8b), were significantly reduced in IL-22−/− mice compared with WT mice at day 5 post-inoculation (Fig. 8a, c, d). Interestingly, the overall levels of antimicrobial peptides present in the pulmonary homogenates of the WT and IL-22−/− mice were lower than those in previous experiments (Fig. 3); however, these experiments were performed in 129 mice (the appropriate background strain for the IL-22−/− mice), whereas the previous experiments were performed in BALB/c mice, which may explain this difference. In addition to antimicrobial peptides, we assessed pulmonary fungal burden in mice at day 14 post-inoculation, and observed no significant difference between the IL-22−/− and WT mice (Fig. 8e). We then determined whether IL-17A may compensate for the loss of IL-22 in the IL-22−/− mice, as these cytokines cooperatively induce antimicrobial peptide production. IL-17A depletion in IL-22−/− mice showed no further reduction in antimicrobial peptide production at day 5 post-infection (data not shown).
Fig. 7.
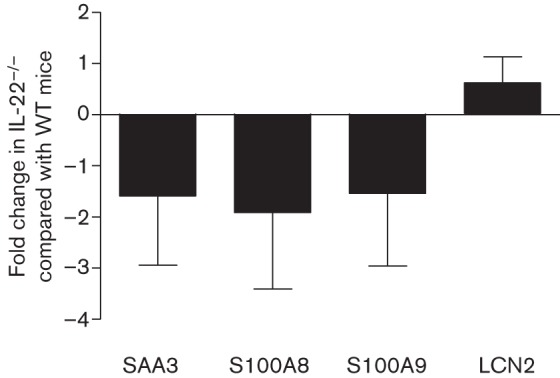
Antimicrobial peptide gene expression in WT and IL-22−/− mice following inoculation with H99γ. BALB/c mice were intranasally inoculated with 1×104 c.f.u. of C. neoformans strain H99γ in 50 µl sterile PBS. At day 5 post-inoculation, mice were killed and pulmonary RNA was isolated from whole lung tissues homogenized in TRIzol reagent. cDNA was prepared from total RNA and real-time PCR was performed to measure S100A8, S100A9, lipocalin-2 (LCN-2) and SAA3 expression. Bars represent the fold change in gene expression in pulmonary tissues from IL-22−/− mice compared with tissues from WT mice. Data shown are cumulative of three independent experiments utilizing individual tissues from four mice per group per time point per experiment and are reported as means±sem (*P<0.05).
Fig. 8.

Pulmonary antimicrobial peptide production and fungal burden in WT and IL-22−/− mice following inoculation with C. neoformans strain H99γ. IL-22−/− mice and WT 129 mice were intranasally inoculated with 1×104 c.f.u. of C. neoformans strain H99γ yeast cells. For pulmonary antimicrobial peptide analysis, lung homogenates were prepared from lungs excised on day 5 post-inoculation and assayed for S100A8 (a), S100A9 (b), lipocalin-2 (c) and SAA3 (d) production by ELISA. For pulmonary fungal burden analysis (e), mice were killed at day 14 post-inoculation and pulmonary homogenates were plated for quantitative culture on YPD agar. Data are cumulative of three experiments utilizing four to five mice per group (for antimicrobial peptides) or two experiments utilizing four mice per group (for fungal burden analysis) per time point for each experiment. Asterisks indicate significant differences (*P<0.05).
IL-22−/− mice have similar survival rates to WT mice during pulmonary C. neoformans strain H99γ infection
To determine the role of IL-22 on survival during cryptococcal infection with H99γ, IL-22−/− mice and WT 129 mice were intranasally inoculated with C. neoformans strain H99γ. After 40 days, all mice (100 %) survived and were killed (Fig. 9). Fungal burden was determined from brain and lungs of surviving mice, and no cryptococci were detected in either group from either organ tested (data not shown).
Fig. 9.
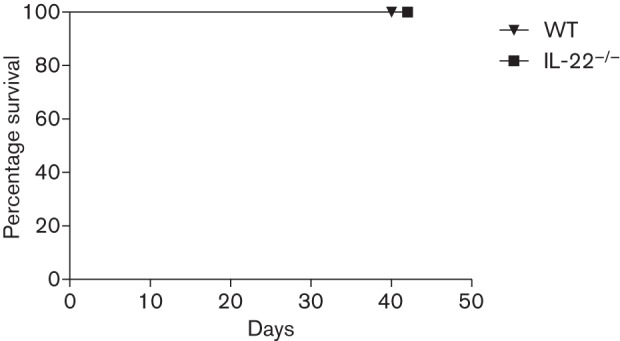
Role of IL-22 in survival following inoculation with C. neoformans strain H99γ. IL-22−/− mice and WT mice were intranasally inoculated with 1×104 c.f.u. of C. neoformans strain H99γ. Mice were monitored for 40 days to determine survival during infection with H99γ. The survival data shown are from one experiment using 10 mice per group.
Experimental pulmonary inoculation of IL-22−/− mice with C. neoformans strain H99γ results in normal recruitment of TLR4+ macrophages and DCs but reduced recruitment of RAGE+ macrophages and DCs to the lungs during infection
We sought to understand why IL-22−/− mice survived infection with C. neoformans strain H99γ by determining the recruitment of TLR4+ and RAGE+ macrophages and DCs during infection in IL-22−/− mice compared with WT 129 mice. Flow cytometric analysis of pulmonary leukocyte infiltrates on day 7 post-inoculation demonstrated similar absolute numbers of total TLR4+ cells, TLR4+ macrophages and TLR4+ DCs (Fig. 10a–c) in IL-22−/− mice compared with WT mice. However, a significant decrease in total RAGE+ cells was found in IL-22−/− mice compared with WT mice (Fig. 10d), and decreased (albeit non-significant) numbers of RAGE+ macrophages and RAGE+ DCs were observed in the lungs of knockout mice compared with WT mice (P = 0.067 and 0.064, respectively). These results suggest that although there is some defect in the recruitment of RAGE+ cells during H99γ pulmonary infection in IL-22−/− mice, all putative receptors for antimicrobial binding are not equally affected and may not uniformly rely on IL-22 for expression and/or activity.
Fig. 10.
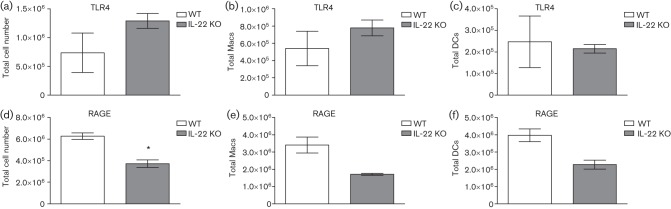
Antimicrobial peptide receptors on macrophages (Macs) and DCs following inoculation of WT and IL-22−/− mice with H99γ. Mice of 129 (WT) and IL-22−/− (KO) strains were intranasally inoculated with 1×104 c.f.u. of C. neoformans strain H99γ. Lungs were excised at day 7 post-inoculation and a single cell suspension was generated using enzymic digestion. The leukocytes were evaluated by flow cytometry for the presence of TLR4 on the surface of all leukocytes (a), macrophages (b) or DCs (c) or RAGE on the surface of all leukocytes (d), macrophages (e) or DCs (f). Flow cytometry data are cumulative of two independent experiments using pooled leukocytes from four mice per group per time point for each experiment and are reported as means±sem. Asterisks indicate significant differences (*P<0.05).
Discussion
Antimicrobial proteins constitute a phylogenetically conserved and highly effective mechanism of protection against invading organisms at the skin and mucosal surfaces (Kolls et al., 2008). IL-17A and IL-22 are considered critical regulators of antimicrobial protein expression in the lungs. Consequently, we sought to determine the contribution of IL-22, IL-17A, and various antimicrobial peptides and acute phase proteins towards providing protection against pulmonary cryptococcosis. We demonstrated that mice inoculated with C. neoformans strain H99γ produce increased pulmonary levels of IL-22 and IL-17A during infection that correlate with increased transcript expression and protein levels of antimicrobial peptides in both primary and secondary infection models. Furthermore, mice inoculated with H99γ have increased expression of receptors TLR4 and RAGE, which interact with these proteins on innate immune cells of the lung. IL-22−/− mice infected with H99γ show no change in antimicrobial gene expression, but do have reduced levels of pulmonary antimicrobial peptides. However, these decreases do not affect fungal burden or survival in this model. Furthermore, IL-22−/− mice show defective recruitment of RAGE+ macrophages and DCs to the lung during infection with C. neoformans strain H99γ, but normal recruitment of TLR4+ macrophages and DCs. These results led us to conclude that although antimicrobial peptides may be important in the initiation of the immune response, reductions in antimicrobial peptides in IL-22−/− mice are not sufficient to abrogate the protective response. In addition, IL-22 and IL-17A do not appear to be solely responsible for the production of antimicrobial peptides, recruitment of TLR4+ macrophages and DCs, and/or protective anti-cryptococcal immune responses.
Several literature examples show that IL-22 and antimicrobial peptides have protective effects during infection, especially in some pulmonary disease models (Aujla et al., 2008; Miyakawa et al., 1996; Pociask et al., 2013). IL-22 has also been shown to be important in influenza infection, where the presence of this cytokine is critical to lung epithelial repair (Pociask et al., 2013). In other pulmonary infections, most notably K. pneumoniae, IL-22 is extremely important in the induction of antimicrobial peptides and resolution of infection (Aujla et al., 2008). However, bacteria are generally more sensitive to the antimicrobial effects of the peptides themselves – including pore formation and iron sequestration (reviewed by Ganz, 2003; Zasloff, 2002) – whereas fungi have not been shown to be as susceptible to these actions. The inherent resistance of fungi to these antimicrobial peptides may be due to their cell-wall structure, which differs greatly from bacterial cell walls. Alternatively, the antimicrobial peptides may act as messengers during fungal infections rather than as effector molecules. In studies of Candida albicans vaginitis, the S100 antimicrobial peptides have been examined, and data have shown that their role is more important in neutrophil recruitment than in direct microbicidal activity against Candida (Johnston et al., 2013; Yano et al., 2010, 2012a, b). However, additional studies by other laboratories have shown that IL-22 is critical to protection in a vaginal model of Candida albicans infection (De Luca et al., 2010, 2013). Furthermore, our in vitro studies did not show anti-fungal activity of any of the purified AMPs against C. neoformans (data not shown).
Although we did not observe a change in survival of IL-22−/− mice compared with WT mice during infection with C. neoformans strain H99γ, it is possible that the antimicrobial peptides are active in early cell recruitment and may serve as biomarkers of disease (Li & Chan, 2011; Uhlar & Whitehead, 1999). As reported elsewhere in the literature, antimicrobial peptides can induce deleterious inflammatory responses if not controlled (Jäger et al., 2010), so it is possible that in our system the antimicrobial peptides are required at early time points, but are ‘turned off’ to control the inflammatory response. Interestingly, IL-22−/− mice still produced antimicrobial peptides during H99γ infection, albeit in lower amounts than that found in WT mice (Fig. 8). Furthermore, IL-22−/− mice had no deficiency in recruitment of TLR4+ macrophages and DCs to the pulmonary tissues, but they did have a deficiency in the recruitment of RAGE+ macrophages and DCs, suggesting that IL-22 signalling may be involved in the expression of RAGE, a receptor that binds multiple antimicrobial peptides (Fig. 10). Therefore, as in many biological systems, there are probably redundancies in pathways leading to antimicrobial peptide production, and IL-22 is not solely responsible for their production. As IL-22 and IL-17A are known to collaborate to induce antimicrobial peptide production, we examined whether IL-17A depletion in IL-22−/− mice would further reduce the amounts of antimicrobial peptides produced. Depletion of IL-17A in IL-22−/− mice did not affect the pulmonary levels of antimicrobial peptides or result in any change in c.f.u. (data not shown). These data correlate with our earlier findings that protection is not dependent on IL-17A in our model system (Hardison et al., 2010; Wozniak et al., 2011).
The upregulation of the receptors for antimicrobial peptides points to a currently unidentified mechanism of antimicrobial action for APCs. Just the presence of antimicrobial peptides may be involved in cryptococcal killing. However, their interaction with receptors on the cell surface of APCs may be necessary for initiation of immune responses by these APCs. It is possible that the production of these antimicrobial peptides leads to a faster, more efficient immune response by these APCs but that IL-22 is not solely responsible for their production, again pointing to redundancy in the induction of antimicrobial peptides.
In conclusion, these data show that IL-22 and IL-17A production are associated with antimicrobial peptide production and the expression of receptors for antimicrobial peptides in mice protected against C. neoformans infection. In addition, the receptors for antimicrobial peptides are increased on macrophages and DCs infiltrating into the pulmonary tissues during infection. However, survival of mice and recruitment of TLR4+ macrophages and DCs during infection with C. neoformans strain H99γ are not significantly affected by the loss of IL-22. Additional studies are in progress to determine what additional factor(s) may be responsible for the induction of antimicrobial peptides and the ultimate resolution of cryptococcal infection.
Acknowledgements
These studies were supported by a grant from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) awarded to F. L .W. (2RO1 AI071752). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Defense or NIAID of the National Institutes of Health.
Abbreviations:
- APC
antigen-presenting cell
- DC
dendritic cell
- HK C.n.
heat-killed Cryptococcus neoformans
- RAGE
receptor for advanced glycation end products
- SAA3
serum amyloid A-3
- TLR4
Toll-like receptor 4
References
- Ather J. L., Ckless K., Martin R., Foley K. L., Suratt B. T., Boyson J. E., Fitzgerald K. A., Flavell R. A., Eisenbarth S. C., Poynter M. E. (2011). Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J Immunol 187, 64–73. 10.4049/jimmunol.1100500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aujla S. J., Chan Y. R., Zheng M., Fei M., Askew D. J., Pociask D. A., Reinhart T. A., McAllister F., Edeal J. & other authors (2008). IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med 14, 275–281. 10.1038/nm1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman M. A., Miller V. L., Weiser J. N. (2009). Mucosal lipocalin 2 has pro-inflammatory and iron-sequestering effects in response to bacterial enterobactin. PLoS Pathog 5, e1000622. 10.1371/journal.ppat.1000622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E., Mazzolla R., Barluzzi R., Mosci P., Bistoni F. (1994). Anticryptococcal resistance in the mouse brain: beneficial effects of local administration of heat-inactivated yeast cells. Infect Immun 62, 3189–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borregaard N., Cowland J. B. (2006). Neutrophil gelatinase-associated lipocalin, a siderophore-binding eukaryotic protein. Biometals 19, 211–215. 10.1007/s10534-005-3251-7 [DOI] [PubMed] [Google Scholar]
- Buchanan K. L., Doyle H. A. (2000). Requirement for CD4+ T lymphocytes in host resistance against Cryptococcus neoformans in the central nervous system of immunized mice. Infect Immun 68, 456–462. 10.1128/IAI.68.2.456-462.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y. R., Liu J. S., Pociask D. A., Zheng M., Mietzner T. A., Berger T., Mak T. W., Clifton M. C., Strong R. K. & other authors (2009). Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol 182, 4947–4956. 10.4049/jimmunol.0803282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuck S. L., Sande M. A. (1989). Infections with Cryptococcus neoformans in the acquired immunodeficiency syndrome. N Engl J Med 321, 794–799. 10.1056/NEJM198909213211205 [DOI] [PubMed] [Google Scholar]
- De Luca A., Zelante T., D’Angelo C., Zagarella S., Fallarino F., Spreca A., Iannitti R. G., Bonifazi P., Renauld J. C. & other authors (2010). IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol 3, 361–373. 10.1038/mi.2010.22 [DOI] [PubMed] [Google Scholar]
- De Luca A., Carvalho A., Cunha C., Iannitti R. G., Pitzurra L., Giovannini G., Mencacci A., Bartolommei L., Moretti S. & other authors (2013). IL-22 and IDO1 affect immunity and tolerance to murine and human vaginal candidiasis. PLoS Pathog 9, e1003486. 10.1371/journal.ppat.1003486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrchen J. M., Sunderkötter C., Foell D., Vogl T., Roth J. (2009). The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol 86, 557–566. 10.1189/jlb.1008647 [DOI] [PubMed] [Google Scholar]
- Eyerich K., Foerster S., Rombold S., Seidl H. P., Behrendt H., Hofmann H., Ring J., Traidl-Hoffmann C. (2008). Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J Invest Dermatol 128, 2640–2645. 10.1038/jid.2008.139 [DOI] [PubMed] [Google Scholar]
- Fehrenbach H., Kasper M., Tschernig T., Shearman M. S., Schuh D., Müller M. (1998). Receptor for advanced glycation endproducts (RAGE) exhibits highly differential cellular and subcellular localisation in rat and human lung. Cell Mol Biol (Noisy-le-grand) 44, 1147–1157. [PubMed] [Google Scholar]
- Flo T. H., Smith K. D., Sato S., Rodriguez D. J., Holmes M. A., Strong R. K., Akira S., Aderem A. (2004). Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432, 917–921. 10.1038/nature03104 [DOI] [PubMed] [Google Scholar]
- Foell D., Wittkowski H., Vogl T., Roth J. (2007). S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol 81, 28–37. 10.1189/jlb.0306170 [DOI] [PubMed] [Google Scholar]
- Ganz T. (2003). The role of antimicrobial peptides in innate immunity. Integr Comp Biol 43, 300–304. 10.1093/icb/43.2.300 [DOI] [PubMed] [Google Scholar]
- Gessner M. A., Werner J. L., Lilly L. M., Nelson M. P., Metz A. E., Dunaway C. W., Chan Y. R., Ouyang W., Brown G. D. & other authors (2012). Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect Immun 80, 410–417. 10.1128/IAI.05939-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison S. E., Wozniak K. L., Kolls J. K., Wormley F. L., Jr (2010). Interleukin-17 is not required for classical macrophage activation in a pulmonary mouse model of Cryptococcus neoformans infection. Infect Immun 78, 5341–5351. 10.1128/IAI.00845-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold K., Moser B., Chen Y., Zeng S., Yan S. F., Ramasamy R., Emond J., Clynes R., Schmidt A. M. (2007). Receptor for advanced glycation end products (RAGE) in a dash to the rescue: inflammatory signals gone awry in the primal response to stress. J Leukoc Biol 82, 204–212. 10.1189/jlb.1206751 [DOI] [PubMed] [Google Scholar]
- Hill J. O., Harmsen A. G. (1991). Intrapulmonary growth and dissemination of an avirulent strain of Cryptococcus neoformans in mice depleted of CD4+ or CD8+ T cells. J Exp Med 173, 755–758. 10.1084/jem.173.3.755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratsuka S., Watanabe A., Sakurai Y., Akashi-Takamura S., Ishibashi S., Miyake K., Shibuya M., Akira S., Aburatani H., Maru Y. (2008). The S100A8–serum amyloid A3–TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol 10, 1349–1355. 10.1038/ncb1794 [DOI] [PubMed] [Google Scholar]
- Hole C. R., Bui H., Wormley F. L., Jr, Wozniak K. L. (2012). Mechanisms of dendritic cell lysosomal killing of Cryptococcus. Sci Rep 2, 739. 10.1038/srep00739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Na L., Fidel P. L., Schwarzenberger P. (2004). Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190, 624–631. 10.1086/422329 [DOI] [PubMed] [Google Scholar]
- Huffnagle G. B., Yates J. L., Lipscomb M. F. (1991). T cell-mediated immunity in the lung: a Cryptococcus neoformans pulmonary infection model using SCID and athymic nude mice. Infect Immun 59, 1423–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S., Stange E. F., Wehkamp J. (2010). Antimicrobial peptides in gastrointestinal inflammation. Int J Inflamm 2010, 910283. 10.4061/2010/910283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D. A., Yano J., Fidel P. L., Jr, Eberle K. E., Palmer G. E. (2013). Engineering Candida albicans to secrete a host immunomodulatory factor. FEMS Microbiol Lett 346, 131–139. 10.1111/1574-6968.12211 [DOI] [PubMed] [Google Scholar]
- Katsuoka F., Kawakami Y., Arai T., Imuta H., Fujiwara M., Kanma H., Yamashita K. (1997). Type II alveolar epithelial cells in lung express receptor for advanced glycation end products (RAGE) gene. Biochem Biophys Res Commun 238, 512–516. 10.1006/bbrc.1997.7263 [DOI] [PubMed] [Google Scholar]
- Kolls J. K., McCray P. B., Jr, Chan Y. R. (2008). Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol 8, 829–835. 10.1038/nri2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagasse E., Clerc R. G. (1988). Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol 8, 2402–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitz S. M. (1991). The ecology of Cryptococcus neoformans and the epidemiology of cryptococcosis. Rev Infect Dis 13, 1163–1169. 10.1093/clinids/13.6.1163 [DOI] [PubMed] [Google Scholar]
- Li C., Chan Y. R. (2011). Lipocalin 2 regulation and its complex role in inflammation and cancer. Cytokine 56, 435–441. 10.1016/j.cyto.2011.07.021 [DOI] [PubMed] [Google Scholar]
- Liang S. C., Tan X. Y., Luxenberg D. P., Karim R., Dunussi-Joannopoulos K., Collins M., Fouser L. A. (2006). Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203, 2271–2279. 10.1084/jem.20061308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mambula S. S., Simons E. R., Hastey R., Selsted M. E., Levitz S. M. (2000). Human neutrophil-mediated nonoxidative antifungal activity against Cryptococcus neoformans. Infect Immun 68, 6257–6264. 10.1128/IAI.68.11.6257-6264.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick A., Heesemann L., Wagener J., Marcos V., Hartl D., Loeffler J., Heesemann J., Ebel F. (2010). NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect 12, 928–936. 10.1016/j.micinf.2010.06.009 [DOI] [PubMed] [Google Scholar]
- Mitchell T. G., Perfect J. R. (1995). Cryptococcosis in the era of AIDS–100 years after the discovery of Cryptococcus neoformans. Clin Microbiol Rev 8, 515–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa Y., Ratnakar P., Rao A. G., Costello M. L., Mathieu-Costello O., Lehrer R. I., Catanzaro A. (1996). In vitro activity of the antimicrobial peptides human and rabbit defensins and porcine leukocyte protegrin against Mycobacterium tuberculosis. Infect Immun 64, 926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody C. H., Lipscomb M. F., Street N. E., Toews G. B. (1990). Depletion of CD4+ (L3T4+) lymphocytes in vivo impairs murine host defense to Cryptococcus neoformans. J Immunol 144, 1472–1477. [PubMed] [Google Scholar]
- Odink K., Cerletti N., Brüggen J., Clerc R. G., Tarcsay L., Zwadlo G., Gerhards G., Schlegel R., Sorg C. (1987). Two calcium-binding proteins in infiltrate macrophages of rheumatoid arthritis. Nature 330, 80–82. 10.1038/330080a0 [DOI] [PubMed] [Google Scholar]
- Pociask D. A., Scheller E. V., Mandalapu S., McHugh K. J., Enelow R. I., Fattman C. L., Kolls J. K., Alcorn J. F. (2013). IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol 182, 1286–1296. 10.1016/j.ajpath.2012.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Ortiz Z. G., Lee C. K., Wang J. P., Boon L., Specht C. A., Levitz S. M. (2011). A nonredundant role for plasmacytoid dendritic cells in host defense against the human fungal pathogen Aspergillus fumigatus. Cell Host Microbe 9, 415–424. 10.1016/j.chom.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoham S., Levitz S. M. (2005). The immune response to fungal infections. Br J Haematol 129, 569–582. 10.1111/j.1365-2141.2005.05397.x [DOI] [PubMed] [Google Scholar]
- Singh N., Gayowski T., Wagener M. M., Marino I. R. (1997). Clinical spectrum of invasive cryptococcosis in liver transplant recipients receiving tacrolimus. Clin Transplant 11, 66–70. [PubMed] [Google Scholar]
- Singh N., Dromer F., Perfect J. R., Lortholary O. (2008). Cryptococcosis in solid organ transplant recipients: current state of the science. Clin Infect Dis 47, 1321–1327. 10.1086/592690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlar C. M., Whitehead A. S. (1999). Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem 265, 501–523. 10.1046/j.1432-1327.1999.00657.x [DOI] [PubMed] [Google Scholar]
- Urban C. F., Ermert D., Schmid M., Abu-Abed U., Goosmann C., Nacken W., Brinkmann V., Jungblut P. R., Zychlinsky A. (2009). Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5, e1000639. 10.1371/journal.ppat.1000639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk K., Warszawska K., Hoeflich C., Witte E., Schneider-Burrus S., Witte K., Kunz S., Buss A., Roewert H. J. & other authors (2011). Deficiency of IL-22 contributes to a chronic inflammatory disease: pathogenetic mechanisms in acne inversa. J Immunol 186, 1228–1239. 10.4049/jimmunol.0903907 [DOI] [PubMed] [Google Scholar]
- Wormley F. L., Jr, Perfect J. R., Steele C., Cox G. M. (2007). Protection against cryptococcosis by using a murine gamma interferon-producing Cryptococcus neoformans strain. Infect Immun 75, 1453–1462. 10.1128/IAI.00274-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak K. L., Levitz S. M. (2009). Isolation and purification of antigenic components of Cryptococcus. Methods Mol Biol 470, 71–83. 10.1007/978-1-59745-204-5_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak K. L., Ravi S., Macias S., Young M. L., Olszewski M. A., Steele C., Wormley F. L. (2009). Insights into the mechanisms of protective immunity against Cryptococcus neoformans infection using a mouse model of pulmonary cryptococcosis. PLoS ONE 4, e6854. 10.1371/journal.pone.0006854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak K. L., Hardison S. E., Kolls J. K., Wormley F. L. (2011). Role of IL-17A on resolution of pulmonary C. neoformans infection. PLoS ONE 6, e17204. 10.1371/journal.pone.0017204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J., Lilly E., Barousse M., Fidel P. L., Jr (2010). Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect Immun 78, 5126–5137. 10.1128/IAI.00388-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J., Noverr M. C., Fidel P. L., Jr (2012a). Cytokines in the host response to Candida vaginitis: identifying a role for non-classical immune mediators, S100 alarmins. Cytokine 58, 118–128. 10.1016/j.cyto.2011.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J., Kolls J. K., Happel K. I., Wormley F., Wozniak K. L., Fidel P. L., Jr (2012b). The acute neutrophil response mediated by S100 alarmins during vaginal Candida infections is independent of the Th17-pathway. PLoS ONE 7, e46311. 10.1371/journal.pone.0046311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. (2002). Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. 10.1038/415389a [DOI] [PubMed] [Google Scholar]
- Zwadlo G., Brüggen J., Gerhards G., Schlegel R., Sorg C. (1988). Two calcium-binding proteins associated with specific stages of myeloid cell differentiation are expressed by subsets of macrophages in inflammatory tissues. Clin Exp Immunol 72, 510–515. [PMC free article] [PubMed] [Google Scholar]