

Abstract
High-dimensional single-cell technologies are revolutionizing the way we understand biological systems. Technologies such as mass cytometry measure dozens of parameters simultaneously in individual cells, making interpretation daunting. We developed viSNE, a tool to map high-dimensional cytometry data onto 2D while conserving high-dimensional structure. We integrated mass cytometry with viSNE to map healthy and cancerous bone marrow samples. Healthy bone marrow maps into a canonical shape that separates between immune subtypes. In leukemia, however, the shape is malformed: the maps of cancer samples are distinct from the healthy map and from each other. viSNE highlights structure in the heterogeneity of surface phenotype expression in cancer, traverses the progression from diagnosis to relapse, and identifies a rare leukemia population in minimal residual disease settings. As several new technologies raise the number of simultaneously measured parameters in each cell to the hundreds, viSNE will become a mainstay in analyzing and interpreting such experiments.
Introduction
Emerging single-cell technologies have been pivotal in uncovering an extensive degree of heterogeneity between and within tissues [1]. Analysis of single-cell data has shed light on many different cellular processes [2–7] and recent technological advances have enabled the study of large number of parameters in single cells at unparalleled resolution. Mass cytometry [8] can measure up to 45 parameters simultaneously in tens of thousands of individual cells. Additionally, high-resolution microscopy [9, 10] and single-cell RNA quantification [11–14] are raising the number of parameters to one hundred in dozens and soon hundreds of individual cells. These innovations promise to revolutionize the way we think about development, differentiation, and disease [1, 15, 16].
However, we are hindered from the optimal use of this data due to the difficulties in visualizing such a high number of dimensions in a meaningful manner. Single-cell data is often examined in two dimensions at a time in the form of a scatter plot [17]. Yet, as the number of parameters increases, the number of pairs becomes overwhelming: a typical mass cytometry dataset will have several hundred pairwise combinations. Additionally, a pairwise viewpoint could miss biologically meaningful multivariate relationships that cannot be discerned in two dimensions. Several computational tools, such as SPADE [18], have been developed to address these problems [19, 20]. However, these approaches typically cluster cells and examine the average of each cluster, resulting in the loss of single-cell resolution of the data. Principal component analysis (PCA), another computational tool, has been applied to mass cytometry datasets [21] and can be used to project data into two dimensions while maintaining single-cell resolution. The caveat is that PCA is a linear transformation that cannot faithfully capture nonlinear relationships, a hallmark of many single cell datasets. Therefore, there remains a critical need for new tools to visualize and interpret high-dimensional single-cell data such as that produced by mass cytometry. An informative tool should enable visualization at single-cell resolution, preserve the geometry and nonlinearity of the data, represent both abundant and rare populations, and provide a robust, interpretable view of the data.
We therefore developed viSNE, a visualization tool for high-dimensional single-cell data based on the t-Distributed Stochastic Neighbor Embedding (t-SNE) algorithm [22]. viSNE finds a 2D representation of single cell data that best preserves its local and global geometry. The resulting viSNE map provides a visual representation of the single-cell data that is similar to a biaxial plot, with the positions of cells reflecting their proximity in high-dimensional space. We utilize color as a third dimension by which we can interactively visualize features of these cells. We demonstrate how viSNE can be used to interpret mass cytometry data derived from healthy and leukemic bone marrow, revealing the cellular relationships and structure contained within the data. We show that the healthy immune system can be mapped into two dimensions while preserving separation between immune subtypes and capturing the heterogeneity of markers within populations, thus highlighting transitional cell populations. We demonstrate that viSNE empowers the exploration of domains such as uncharted heterogeneity in cancer, cancer progression from diagnosis to relapse, or identification of rare aberrant cells in the clinical assessment of disease status (minimal residual disease). viSNE is a flexible approach for exploring the high-dimensional structure of single-cell data, giving us a unique insight into healthy systems and disease.
Results
viSNE preserves high-dimensional relationships when displaying single-cell data
In viSNE, each cell is represented as a point in high-dimensional space (each coordinate is one parameter, i.e, the expression level of one protein). An optimization algorithm searches for a projection of the points from the high-dimensional space into 2D (or 3D) so that pairwise distances between the points are best conserved between the high-dimensional space and the low-dimensional space (see Materials and Methods). The resulting low-dimensional projection, the viSNE map, is visualized as a scatter plot, where a cell’s location in the plot represents information from all of the original dimensions. We developed cyt, an interactive tool for the visualization of viSNE maps. cyt has multiple features, including plotting the maps, coloring cells by marker expression, sample or subtype, gating and others. Figure 1A demonstrates how viSNE works on a synthetic example: the algorithm identifies the global structure of the embedded manifold (a 1D line, along with its curvature, embedded in 3D space) and the local structure (pairwise distances between points along the line are conserved).
Figure 1.
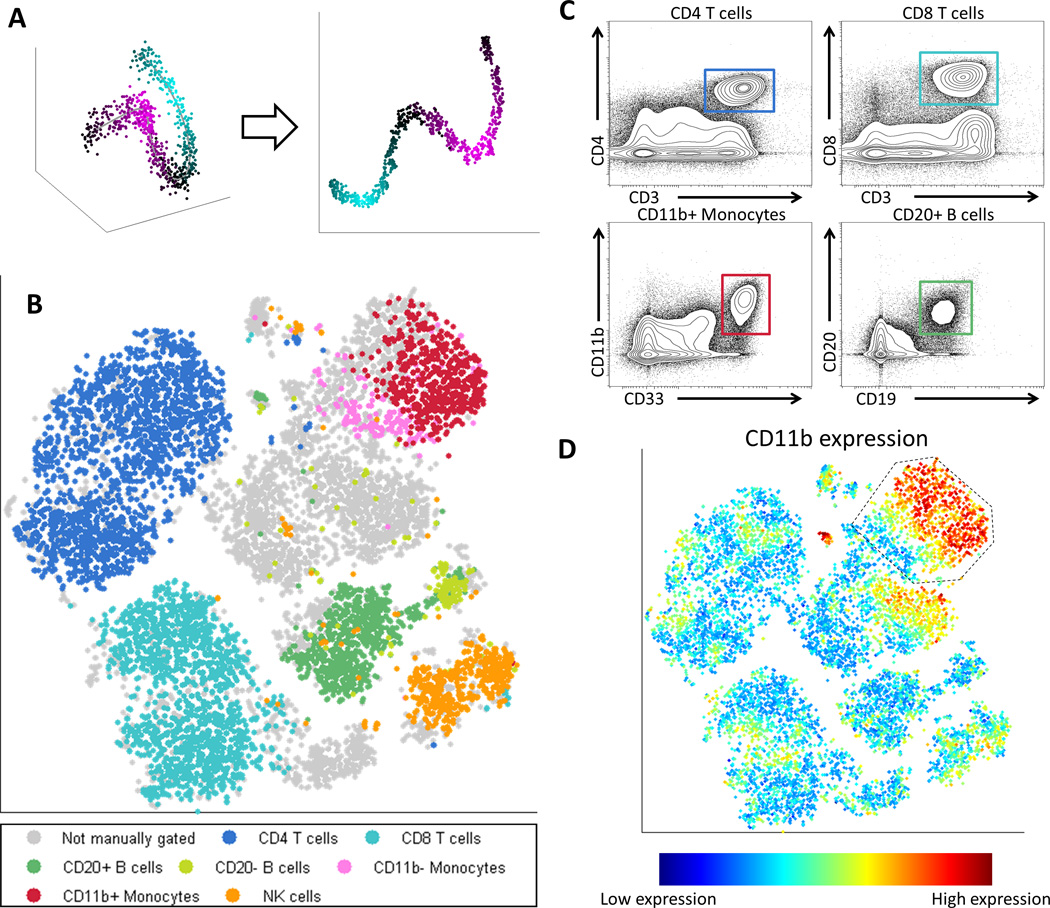
viSNE, a Nonlinear Dimensionality Reduction algorithm, creates a map of the immune system. (A) In this toy example, viSNE projects a one-dimensional curve embedded in 3D (left) to 2D (right). The color gradient demonstrates that nearby points in 3D remain close in 2D. (B) Application of viSNE to a healthy human bone marrow sample [21]. viSNE automatically separates cells based on their subtype (see Supplementary Figure 1 for further analysis). Each point in the viSNE map represents an individual cell and its color represents its immune subtype based on independent manual gating. The axes are in arbitrary units. (C) Biaxial plots representing the same data shown in 1B, select subpopulations are shown with canonical markers, square color matches subtype in 1B. The actual gating used is more complex and uses a series of biaxial plots for each population [21]. Note, unlike 1B, these plots do not separate between all subtypes in a single viewpoint. (D) The same viSNE map represented in 1B, this time each cell is colored based on CD11b expression. Gated cells are all CD33 high and show a CD11b (maturity) gradient. Many of these cells were not classified as monocytes by manual gating (grey cells 1B).
The viSNE map of healthy bone marrow identifies known immune subsets
We evaluated viSNE’s performance on the well-characterized system of human hematopoiesis in bone marrow [23]. The expression of a panel of 13 surface markers was measured in healthy human bone marrow, Marrow1 [21]. viSNE was applied to this dataset, resulting in a map that separates between known immune subsets (Figure 1B). To validate the map, we used an independently derived classification of the cells to immune subtypes, based on manual gating of a series of biaxial plots (Materials and Methods). While viSNE was not provided with this classification or any knowledge of immune subsets, it grouped cells in the same subsets together and separated the subsets from one another (Figure 1B–C). viSNE accurately distinguished CD4+ and CD8+ T cells, mature and immature B cells, mature and immature monocytes and NK cells. Notably, NK cells formed a distinct subset even though CD56, their canonical marker, was not directly measured.
By using all markers simultaneously, viSNE achieves a more accurate grouping of immune subsets. To compare between the expert gating and the viSNE map, we gated the regions corresponding to the different subtypes in the viSNE map (Supplementary Figure 1A) using the cyt feature that allows one to gate subsets directly from the viSNE map. In all cases, the viSNE gate included cells that were not classified with any label (grey cells). Examination of the marker expression of these cells reveals that they are typically just beyond the threshold of one marker, but the viSNE classification is strongly supported based on the expression of all other markers. For example, in Figure 1D, wherein cells are colored for CD11b marker expression, the gated region contains CD33+ cells (canonical monocyte marker, Supplemental Figure 1B). Only 47% of these cells were classified as monocytes by the manual gating (Figure 1B). The marker intensity distributions between the CD11b- monocytes in the viSNE gate and in the manual gate (Supplementary Figure 1C) are similar, supporting the notion that these are indeed CD11b- monocytes.
Traditional gating relies on hard thresholds to classify cells into subtypes. Thus cells whose marker values are slightly below the threshold might not be classified correctly, or classified at all (Supplementary Figure 1D). When dealing with the hematopoietic continuum, this may result in the inability to accurately capture transitional cell types. Using cyt to color cells based on marker expression levels reveals that viSNE organized monocytes based on a gradient: a smooth increase between cells in CD11b expression, a marker of monocyte maturity (Figure 1D). This highlights the continuous and gradual nature of CD11b expression during monocyte maturation and better represents the continuum of normal differentiation [24]. viSNE takes into account all phenotypic markers concurrently instead of relying on hard thresholds and as a result, classifies more cells and captures a more accurate view of the variability within each subtype when compared to biaxial gating. The single cell resolution of the map provides fine detail of each subpopulation, going beyond clustering into subtypes and enabling investigation of the variation, structure and transitions within each subtype.
viSNE is robust and does not require canonical markers to distinguish subsets
We performed a number of analyses to evaluate the robustness of viSNE. The viSNE map in Figure 1B includes 10,000 cells that were subsampled from the complete data set (Marrow1). We independently subsampled multiple subsets of the data and ran viSNE on each, resulting in similar viSNE maps that conserve the separation between subsets (Supplementary Figure 2). This supports the notion that the viSNE map is consistently and reliably representing real structure in the data.
To test viSNE’s reliance on the specific markers to identify immune subtypes, multiple viSNE maps were generated, excluding some of the markers when generating each map. The viSNE map remains consistent following the removal of each single marker (Figure 2A left and middle, Supplementary Figure 3). Remarkably, when removing the canonical markers for B cells, T cells and myeloid cells (CD19/CD20, CD3 and CD33, respectively), viSNE’s map remains consistent with the one constructed with all thirteen markers (Figure 2A, right). This demonstrates viSNE’s robustness. The more notable interpretation is that the expression levels of non-canonical markers, when analyzed together, contains the information needed to separate between immune subtypes. This speaks to a previously unappreciated level of organization where specialized immune subtypes have tightly coordinated surface marker expression beyond their canonical identifiers. The different subtypes reside in distinct well-separated shapes in high-dimensional space and the viSNE map takes advantage of this structure (see Supplementary Figure 4 and Supplementary Data 1).
Figure 2.
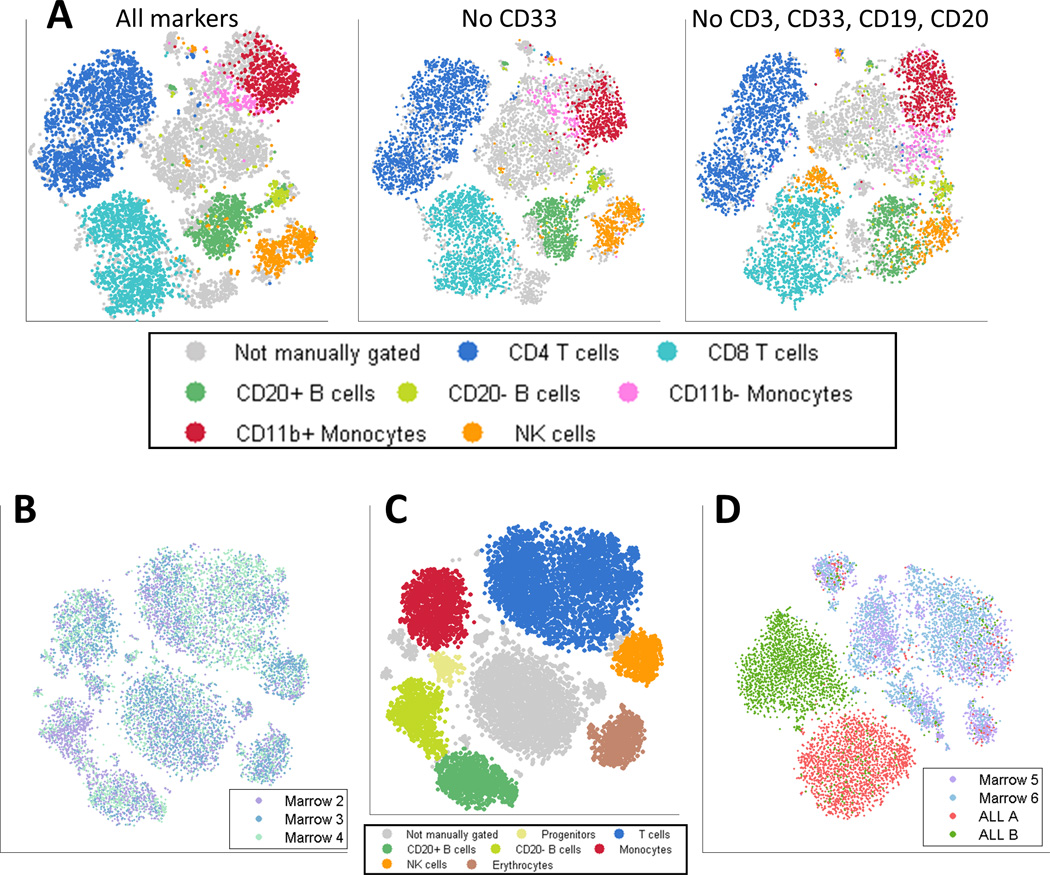
viSNE is robust: it does not require canonical markers and the viSNE map has a conserved structure across healthy samples and a distinct structure in each cancer sample. (A) Left: Same as 1B, viSNE map based on all 13 markers. Middle: viSNE map of the same cells, projected after removing CD33, results in a very similar map and still identifies monocytes. Right: viSNE map of same cells, removing CD33, CD3, CD19 and CD20. Despite removing four canonical markers, viSNE separates most major subtypes using the remaining nine channels. See Supplementary Figure 3 for additional marker subsets. (B) Samples from three healthy donors were mapped using viSNE. Each point represents a single cell, color coded by sample. The different samples overlap over all regions of the map. See Supplementary Figure 5 for a separate plot for each sample. (C) The same map as in 2B, color coded by subtypes, as identified by marker expression levels (Supplementary Figure 6). (D) Samples from two healthy donors and two ALL patients were mapped using viSNE. Each cell is colored based on sample. While the healthy samples overlap, the cancer samples are separate from both the healthy samples and each other.
viSNE highlights a consistent and reproducible map of healthy bone marrow
Having demonstrated viSNE’s robustness when applied to a single healthy bone marrow sample, we examined its robustness across multiple healthy individuals. Three healthy bone marrow samples were assayed by mass cytometry using a panel of 31 phenotypic surface markers (see Materials and Methods). The resulting viSNE map demonstrates both the consistency between healthy samples and viSNE’s ability to handle higher dimensionality. The cells are grouped into distinct subpopulations and cells from all three individuals overlap within each subpopulation (Figure 2B and Supplementary Figure 5). We used the Jenson-Shannon (JS) divergence to quantify the similarity between the viSNE maps (see Materials and Methods). The JS divergence between each pair of healthy individuals is 0.04, confirming that there is almost no divergence between the viSNE maps of healthy samples.
Using cyt to visualize the markers characterizing each subpopulation, we matched these to their known subtypes (Figure 2C, Supplementary Figure 6). Marrows 2–6 were measured using a panel of 29 antibodies optimized for the analysis of acute lymphoid leukemia (ALL, Supplemental Table 2). This resulted in the identification of a slightly different set of immune subpopulations: Recognition of erythrocyte and progenitor subsets was aided by the addition of CD61 and CD117 to the panel, while the CD4 and CD8 T cell populations could were merged because those markers were omitted.
To further evaluate the robustness of viSNE’s map of healthy bone marrow, we applied viSNE to an additional bone marrow sample collected using conventional fluorescence-based flow cytometry. The resulting viSNE map is similar to the map generated by mass cytometry (Supplementary Figure 8), demonstrating not only consistency in the map between healthy samples, but also that viSNE is well-suited for the analysis of fluorescent based cytometry data.
The cellular subtypes comprising the human immune system are reproducibly represented by viSNE and the fidelity of this structure is maintained across multiple cytometry platforms, marker panels, and most importantly, individual patients. Furthermore, analysis of human bone marrow with viSNE reveals an additional layer of information: the inherent high dimensional structure of healthy human bone marrow and relationships between the cellular subtypes and their transitional states.
viSNE maps of cancer result in deformed shapes
Encouraged by the consistency and robustness of viSNE maps in healthy samples, we decided to use viSNE to examine cancer heterogeneity. We applied viSNE to a 29 marker panel measuring four bone marrow samples, two donated from healthy individuals and two from Acute Lymphoblastic Leukemia (ALL) patients (Figure 2D). Similarly to Figure 2B, the two healthy samples overlap and map together (JS divergence 0.04). In contrast, the two cancer samples occupy a completely separate region within the viSNE map (JS divergence 0.45), and each forms a distinct population separate from the other cancer sample (JS divergence 0.42). Some cells from the leukemia samples (~5%) overlap with the healthy samples. Inspection of these cells reveals marker combinations that correspond to healthy immune cells, supporting their placement with the other healthy cells.
To further investigate leukemia, viSNE was applied separately to each leukemia sample, providing more resolution and detail for each. Each sample mapped into a single large deformed shape (Figure 3A), along with a number of small subpopulations corresponding to healthy immune populations, indicating that the malignant cells were related to each other but sufficiently distinct from healthy cells. We applied viSNE to a second cancer type, acute myeloid leukemia (AML) and the viSNE maps of these cancer samples also resulted in a single large deformed shape (Figure 3B), in stark contrast to the separated and distinct subpopulations of healthy samples. We note that there is a considerable population structure within each cancer, as can be discerned by multiple peaks and saddle points in the contour map. Moreover, while the maps of healthy samples are comparable, each cancer forms a unique viSNE map: there is a consistent overlap of the normal, which is, in turn, consistently separated from the abnormal. Furthermore, the regions occupied by cancer cells had a distinguishable structure that could be observed in the expression patterns of the various markers.
Figure 3.
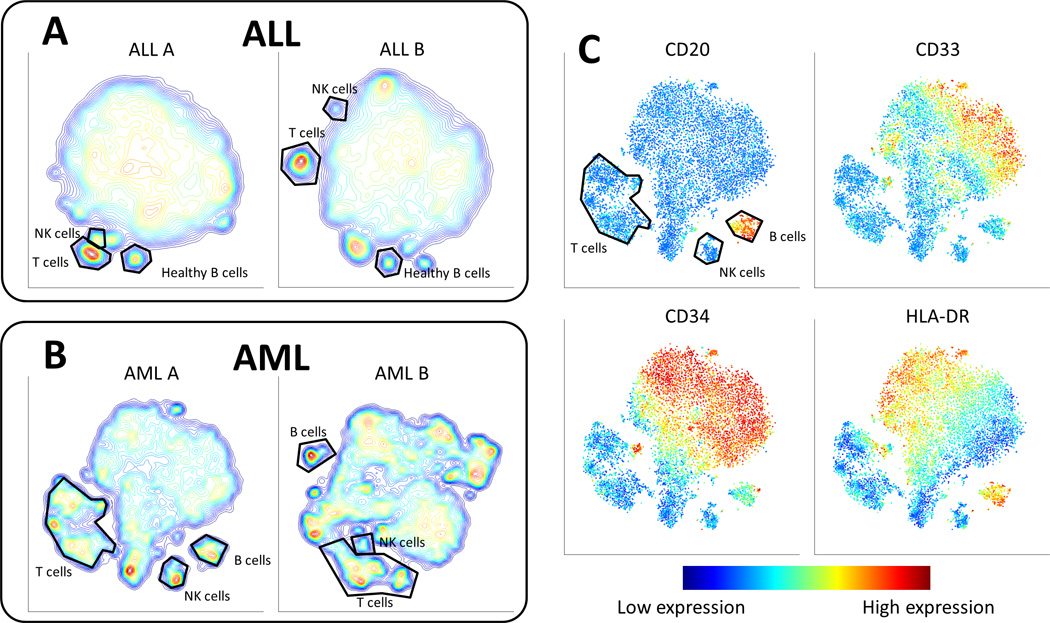
Cancer samples form contiguous but heterogeneous shapes. (A) Contour plots of the viSNE maps of two different ALL samples. The contours represent cell density in each region of the map. Each map has a single large population and a number of small separated subpopulations. These small populations are healthy immune subtypes as identified by their marker expression combination. Such healthy subtypes are highlighted in black. (B) Contour maps of two different AML samples. (C) viSNE map of a diagnosis bone marrow sample from AML patient 1. Cells are colored by marker expression levels. CD20 helps identify the healthy B cell subpopulation. The other markers form gradients on the viSNE map (blue to red) in different regions and directions.
Using viSNE to explore cancer heterogeneity
While healthy samples can be studied by biaxial gating based on known subtypes, exploring cancer heterogeneity in high dimensions can be a daunting task as cancer samples frequently involve uncharacteristic marker expression patterns and there are hundreds of possible biaxial combinations. In current clinical practice, hematopoietic malignancies are analyzed using at most four to eight markers simultaneously. The number of antigens examined can be further expanded using additional panels containing overlapping backbone markers [25]. Immunophenotyping results have typically been displayed using biaxial plots focused on key markers (Supplementary Figure 9). Combining mass cytometry with viSNE, we are able to visualize a single cell map of cancer heterogeneity that takes ~30 markers into account in a single viewpoint, revealing additional structure and sub-populations.
We demonstrate how viSNE can be used to comprehensively characterize a diagnostic AML bone marrow sample. Using cyt, we see that while the overall shape of cancer is deformed compared to the healthy bone marrow, many markers form clear structures. A number of markers form gradients, such as CD33, CD34 (hematopoietic progenitor cells) and HLA-DR (MHC class II, typically expressed on B cells and monocytes) shown in Figure 3C (see Supplementary Figure 10 for more examples).
One of the most dominant gradients is CD34 (Figure 3C, bottom left), a stem and progenitor marker in healthy hematopoiesis. We see that within the highly expressing CD34 cells (immature) there is a CD33 gradient (indicating differentiation into monocytes, figure 3C, top right) without the attenuation of CD34 that occurs during healthy development. These gradients suggest a derailed developmental program resulting in abnormal phenotypic states (combinations of phenotypic markers) that express progenitor markers (CD34) concurrently with differentiation markers (CD33). A possible hypothesis is that a progenitor-like state (associated with CD34) is enforced by oncogene activity, but the cells continue to differentiate aberrantly as indicated by the rise of CD33. The leukemia exhibits a heterogeneous spectrum of aberrant phenotypic states and a continuum of intermediate states between them. The single cell resolution of viSNE highlights cancer as a continuum of states, demarcated by gradients of marker expression, rather than distinct sub-populations.
viSNE elucidates progression between diagnosis and relapse cancer samples
Following the notion that the viSNE map might reflect aspects of cancer progression, we applied the algorithm to a matched pair of samples from a patient with AML: one sample taken before chemotherapy and another after relapse of the disease. Using viSNE on a merged dataset representing both samples, we mapped phenotypic states explored by the cancer and could clearly visualize a separation between the diagnosis and relapse samples (Figure 4A, Supplementary Figures 11 and 12). viSNE identifies phenotypes unique to the diagnosis sample (Figure 4A, bottom-middle lobe), which are presumably eliminated by chemotherapy, and new phenotypes that arise at relapse. Notably, the viSNE map identifies a region of shared phenotypes (Figure 4A, right side), which is occupied by both samples but considerably rarer at diagnosis. This potentially indicates enrichment of a rare drug-resistant clone that maintained a consistent phenotype from diagnosis to relapse.
Figure 4.
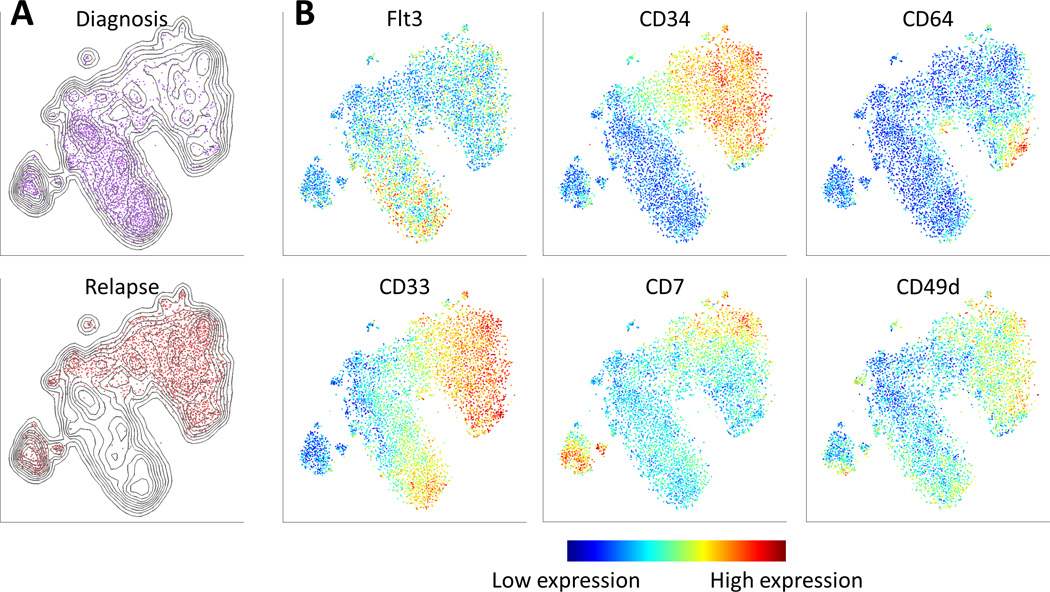
viSNE reveals the progression of cancer, from diagnosis to relapse. (A) Contour plots of the viSNE map combining diagnosis and relapse samples. The contours represent cell density in each region in the map. Points are cells from the diagnosis (top, purple) and relapse (bottom, red) samples. While some of the regions overlap, the two samples largely reside in different regions of the viSNE map. (B) The same map as in (A), cells from both samples are shown colored by marker expression levels, enabling the comparison of expression patterns before and after relapse. Flt3 is prevalent largely in the diagnosis sample. CD34 emerges in the relapse sample, as do CD64 and CD7. There is a CD33 gradient in both samples. The overlapping region has cells that express high levels of CD49d.
We note populations of healthy cells from both the diagnosis and relapse samples that overlap in the viSNE map (Figure 4A, left side), providing an internal technical control for the similarity of staining between samples. FLT3 expression is pervasive in the diagnosis sample, but diminished in the relapse sample. Genetic analysis of the diagnosis sample revealed an internal-tandem duplication of FLT3, a common mutation in AML [26], suggesting relapse derived from a clone lacking this mutation (FLT3 status at relapse was unavailable). The clone that reemerged at relapse had an altogether different and more immature phenotype, with cells expressing both high CD34 and CD33 throughout a large fraction of the sample (Figure 4B, Supplementary Figure 12). The relapse sample was highly heterogeneous: distinct regions expressed different markers from the myeloid lineage (CD64 and CD15) and lymphoid lineage (CD7). This heterogeneity and the marker expression profile of each cell are apparent when visualized using the colored viSNE map.
The viSNE map can further be used as a tool to characterize the structure of surface marker expression in AML bone marrow and identify subpopulations. When examining the relapse sample, we see distinct expression patterns of CD34, CD33, CD64 and CD7 (Figure 4B and Figure 5A). To allow further dissection of the sample using experimental tools such as DNA and RNA sequencing, we used the viSNE map to devise a gating scheme that is compatible with fluorescence-assisted cell sorting (FACS) and divides the sample into subpopulations based on these four markers. We classified each marker as either “on” (positive) or “off” (negative) according to a threshold that was chosen using the map (Figure 5A, black lines). These gates can be intersected as in Figure 5B, where we demonstrate an intersection of CD34+/− and CD33+/− (left of arrow) followed by further intersection of the top right quadrant by CD64+/− and CD7+/− (right of arrow). When examining the intersection of all eight groups (two for each marker), we identified six distinct subpopulations which are spread across the map (Figure 5C). The next step would be to physically separate the relapse sample into these subpopulations using FACS and characterize them via downstream experiments. Through the viSNE map we identified subpopulations of interest in the unknown landscape of cancer and defined a gating scheme for further experimental examination of these subpopulations.
Figure 5.
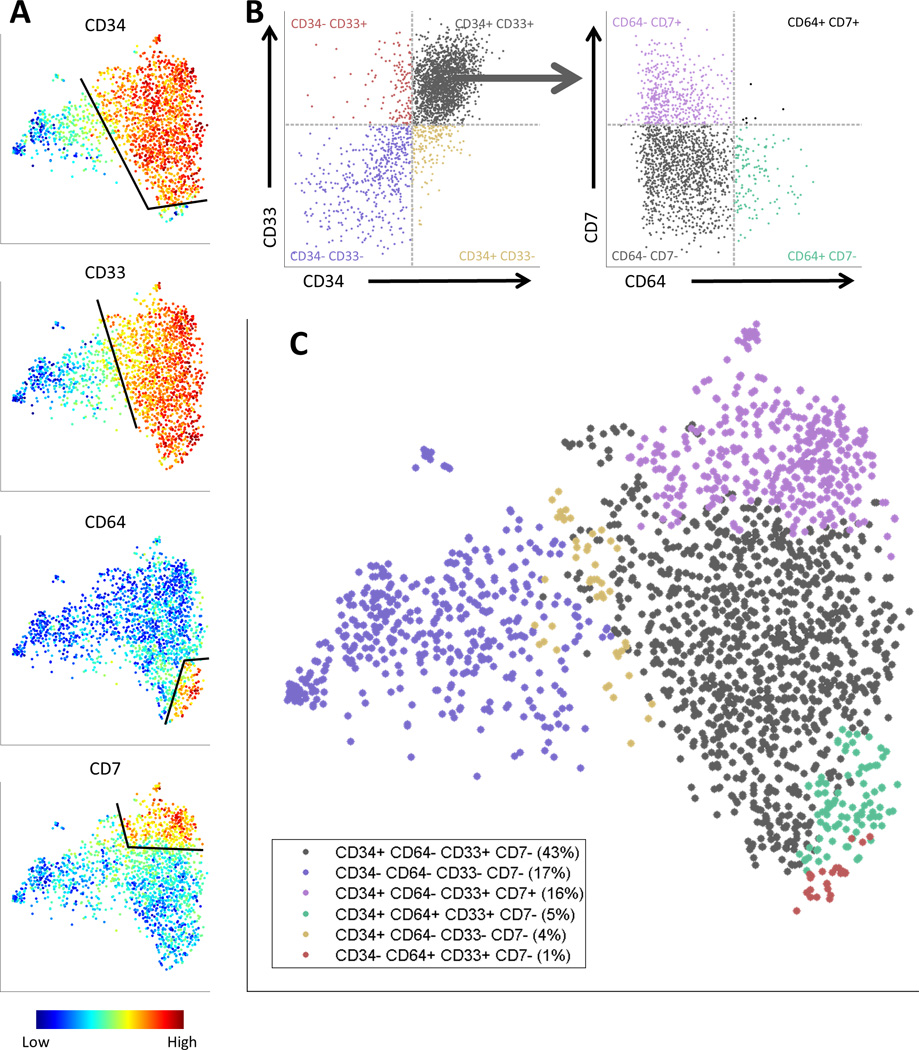
A gating scheme for fluorescence-activated cell sorting (FACS) of the AML relapse sample based on the viSNE map. (A) The viSNE map, colored by (from top to bottom) CD34, CD33, CD64 and CD7. For each marker, cells were separated into two subpopulations: “on” (positive) and “off” (negative), based on an expression threshold for the marker (black lines). (B) Left: Biaxial plot of CD34 versus CD33, demonstrating the intersection of each of the CD34+/− subpopulations with each of the CD33+/− subpopulations. Each point is a cell, the X-axis is CD34 expression level and the Y-axis is CD33 expression level. The 4 quadrants correspond to different CD34+/− and CD33+/− combinations; the cells are colored and labeled by the quadrants. Right: Biaxial plot of CD64 versus CD7, demonstrating the intersection of the CD34+ CD33+ subpopulation with each of the four CD64+/− CD7+/− subpopulations. The X-axis is CD64 expression and the Y-axis is CD7 expression. As in the left side, the cells are colored and labeled by the quadrants. (D) The six subpopulation gating scheme projected onto the viSNE map. Cells are colored by their respective subpopulation from B. The relapse sample can now be sorted into these subpopulations via fluorescence-activated cell sorting (FACS) and further studied through downstream experiments such as DNA and RNA sequencing.
viSNE aids detection of minimal residual disease in acute lymphoblastic leukemia
Minimal residual disease (MRD) is increasingly used to gauge the early response to therapy in many types of leukemia and some solid tumors. The ability to detect small numbers of cancerous cells as identified by an aberrant phenotypic “fingerprint” by flow cytometry has been recently used to risk-stratify patients and direct treatment decisions. The presence of MRD has been shown to be associated with risk of relapse [27, 28]. There are two competing manual methods for assessing MRD by flow cytometry. The first method involves identifying aberrant antigen expression (leukemia-associated immunophenotype, or LAIP) on the leukemia cells at diagnosis, and then looking for that same phenotype after chemotherapy [29]. The second method involves identifying leukemic cells based on a ‘different-from-normal’ compared to a historical bank of healthy bone marrow samples [30]. While the prognostic value of MRD measurement has been validated in several clinical trials, both of these approaches rely on an expert pathologist to identify abnormal cells by manually inspecting biaxial plots.
The detection of MRD indicates a need for intensified therapy that unfortunately carries an increased risk of toxicity. Consequently, accurate detection of rare malignant populations is paramount in correctly assigning risk to an individual patient. Current practice of MRD detection is limited by a number of factors. Manual inspection of the immuno-phenotype of a given MRD sample relies upon the ability to identify abnormal cells that are sufficiently distinct from normal bone marrow. Additionally, detection of a phenotype, which relies on the diagnostic phenotype, may fail to detect remaining malignant populations which were not detected at diagnosis, due to their rarity or the modulation of surface marker expression during therapy. Thus, a tool which automatically identifies abnormal cellular phenotypes allows more clear identification and evaluation of possible remaining cancer cells.
Based on the striking contrast of leukemic bone marrow from normal marrow, we sought to test whether viSNE can aid manual MRD detection. We spiked metal-barcoded [31] cells from an ALL patient into a healthy bone marrow sample, creating a synthetic in-vitro sample with 0.25% MRD. A single healthy bone marrow sample served as a guide for interpretation (similar to the “different-from-normal” approach). We used a biased subsampling method to enrich for unique non-control subpopulations and generated a viSNE map using eight markers (CD3, CD7, CD10, CD15, CD20, CD34, CD38, CD45) to emulate a MRD scenario using fluorescence-based flow cytometry (see Materials and Methods). The algorithm was blinded to the metal-barcoded channel.
A good MRD candidate region is a distinct region of the viSNE map that contains cells from the MRD sample, but no cells from the healthy control. Cells from both samples were well-mixed across most of the viSNE map, except for one suspect region that was composed almost entirely from cells from the MRD sample (Figure 6A, black arrow). We compared marker expression levels in the suspect region to the rest of the sample (Figure 6B) and found that the suspect cells strongly expressed CD10 and CD34, exhibited below-average expression of CD45 and also expressed CD15, a phenotypic combination often seen in B precursor ALL. Taken together, the combination of these surface markers and the absence of similar cells in the healthy control suggest that these were leukemic cells. Upon removing the blind these cells proved to be positive for our metal-barcoding channel (Figure 6C), confirming that these were indeed the spiked ALL cells.
Figure 6.
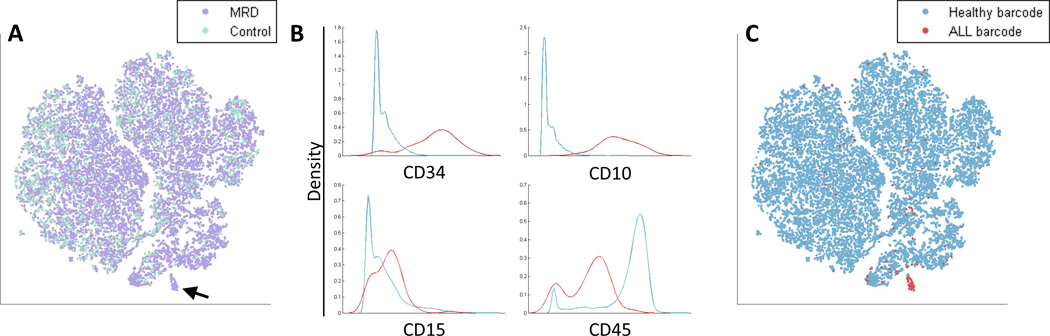
Identification of Minimal Residual Disease (MRD). (A) An in vitro synthetic MRD sample was created by spiking a healthy sample with ALL cells (see Methods). The viSNE map of the MRD sample (purple) and a healthy control sample (cyan) includes a suspect region (marked by an arrow) that is almost entirely cells from the MRD sample (purple). (B) Marker expression of cells in the suspect region (red) and the non-suspect region (cyan). Marker distributions are plotted, x-axis represents marker expression level and y-axis represents density of cells. The suspect region is CD34+ CD10+ CD15+ CD45−, a possible fingerprint for ALL. (C) The viSNE map from 6A, color coded by healthy barcode (cyan) and ALL barcode (red). The suspect region is indeed almost entirely composed of ALL cells. The viSNE algorithm was blinded to this barcode, yet managed to separate this small sub-population from the healthy cells. ALL cells outside of the suspect region have marker expression levels conforming to healthy cells. See Supplementary Figure 13 for an additional example.
We repeated this analysis with a different set of markers and achieved similar results (Supplementary Figure 13). As a control, we repeated the same procedure with an “MRD” sample that only included healthy cells. The two healthy samples were well mixed across the entire viSNE map and there was no region that contained only MRD cells (Supplementary Figure 14), demonstrating that the subsampling method and viSNE do not spuriously create suspect regions. While only an in-vitro example, this demonstrates viSNE’s success in identifying a minuscule cancer subpopulation in the data, suggesting that viSNE can be effectively used for MRD detection by exploiting the subtle high-dimensional differences between cancer populations and healthy cells.
Discussion
We developed viSNE, a tool for the visualization of high-dimensional single-cell data that projects each cell onto a two-dimensional map while preserving the separation between immune subtypes. This mapping takes advantage of the inherent structure of the data: different subtypes reside in separate regions in high-dimensional space. Conventional analysis of cytometry data, which views only two dimensions at a time, ignores the higher-order structure and complex relationships between markers in the data. While viSNE plots resemble conventional biaxial plots, their utility comes from combining and representing information from all dimensions simultaneously.
The viSNE map is consistent across multiple healthy individuals, while cancer samples occupy regions distinct from healthy cells and from each other. We illustrated how viSNE can be utilized to characterize heterogeneity within cancer samples, mark disease progression from diagnosis to relapse, and identify rare cancer populations lurking among predominantly healthy cells. Finally, viSNE is a flexible method that can be applied to various single-cell datasets, including flow cytometry.
viSNE has a number of advantageous features for the analysis of single-cell data. It is sensitive to small subsets (Supplementary Figure 15), which allows us to subsample uniformly, preserving the original frequencies of cell populations. Second, viSNE takes into account similarities between all pairs of cells, providing information about both nearby and distant cells. Third, viSNE plots each individual cell, showing the diversity within populations and maintaining single-cell resolution. This single cell resolution is a distinguishing feature relative to SPADE [18], it contributes to viSNE’s increased robustness over SPADE (Supplementary Figure 16) and is critical for viSNE’s success in the MRD scenario. We applied SPADE to the same “in-vitro” MRD sample and the ALL cells were indistinguishable from healthy cells in the resulting SPADE tree (Supplementary figure 17), rendering SPADE inappropriate for MRD.
viSNE belongs to the class of nonlinear dimensionality reduction (NLDR) algorithms: unlike principal component analysis (PCA), NLDR methods do not assume linear relationships between parameters. Immune subsets are nonlinear and hence PCA, unlike viSNE, fails to separate between them (Supplementary Figures 18, 19). We evaluated several other NLDR algorithms [32] and among these only viSNE resulted in consistent maps that separated immune subtypes across multiple subsamples of the same data (Supplementary Figures 19). The other methods might be confounded by the noise inherent in biological systems and measurement technologies or by the complex geometry in high-dimensional hematopoietic space.
Despite its extensive utility, as with all dimensionality reduction tools viSNE is inherently limited: low-dimensional mapping cannot represent all of the information in high-dimensional space. Therefore, viSNE only captures the most dominant structures. One way to gain more detail is to run viSNE on a well-defined subset of the data; for example, instead of analyzing several cancer samples together (Figure 2D), running viSNE on each sample separately (Figure 3A,B). Alternatively, it is possible to limit the mapping to only a subset of parameters of interest. Another consequence of dimensionality reduction is the “crowding problem”, which typically limits the number of cells we can map to 30,000. This limits applications such as gating, since a subsampling of the cells is required, meaning only cells in the subsample can be classified. An effective solution for gating is combining viSNE with a clustering algorithm. We clustered the cells using FLOCK [19], a state-of-the-art clustering tool for cytometry data, and labeled the viSNE map according to this clustering (Supplementary Figure 20). While FLOCK separated the immune subtypes, it splits each subtype into multiple clusters. The viSNE map helps interpret these clusters and their relationships.
viSNE is an unsupervised algorithm and does not require prior knowledge of the system (for example, manual gating of subtypes) and is thus suitable for navigating less explored systems such as cancer. The structure in healthy samples is formed through an orderly program of hematopoietic development, whereas cancer’s derailed developmental program leads to loss of normal order and structure. The true value of viSNE is that the abnormal structure and heterogeneity unique to the cancer can be explored: viSNE literally becomes the map to further investigate regions of the cancer and the expression patterns of various markers. viSNE helps characterize the plethora of abnormal phenotypes unique to each cancer, enabled by its ability to take all markers into account simultaneously, rather scanning through hundreds of biaxial plots, two markers at a time (see Figure 4B versus Supplementary Figure 9).
A characteristic feature that repeated across multiple cancer maps was the emergence of distinct gradients of marker expression levels that resemble developmental progression in healthy cells (Figure 1D). Comparing gradients in AML diagnosis and relapse samples (Figure 4B) supports the notion that the cells first gain CD34 and subsequently the cells acquire lineage-specific markers without attenuation of CD34. There is a large diversity in the lineage-specific markers expressed. In different regions of the map, the cancer is exploring markers of both the myeloid lineage (CD33, CD64, CD15) and the lymphoid lineage (CD7). This process appears to be development gone awry. After identifying unexpected populations using viSNE, one can design a sorting strategy for physical isolation and downstream experiments and characterization of these populations (Figure 5).
We have examined the utility of viSNE in the analysis of mass cytometry data measuring surface marker expression. However, mass cytometry is also capable of integrating the surface marker panel with a panel of functional markers that probe signaling, cell cycle and metabolism, under many experimental perturbations (such as cytokines and drugs) [3]. Such experiments result in a huge explosion of data that cannot be analyzed well with existing methods. viSNE offers a powerful approach to characterize such data, revealing the internal computational processes of cells. viSNE’s ability to distinguish rare subsets which comprise only a tiny fraction of the population (Figure 6 and Supplementary Figure 15) could be advantageous toward the identification and characterization of rare drug resistant subpopulations. Mass cytometry combined with viSNE has enormous potential to shed new light on signaling, both in healthy systems and in cancer.
We have demonstrated viSNE’s capability to analyze mass cytometry and flow cytometry data. viSNE provides a unique perspective on questions involving the high-dimensional nature of biological systems. The emerging trend in biological research revolves around dozens of dimensions in tens of thousands of cells: mass cytometry will soon measure a hundred epitopes; high-resolution microscopy promises to examine more than fifty features [9, 10]; and sequencing technologies are quickly approaching the resolution needed for accurate single-cell DNA and RNA sequencing [11–13]. Making sense of these data is a daunting challenge that requires powerful computational approaches. viSNE’s visualization is a crucial asset whose importance will only increase as mass cytometry and other technologies expand in their capabilities.
Materials and Methods
The t-SNE algorithm
The t-Distributed Stochastic Neighbor Embedding (t-SNE) algorithm maps points from high-dimensional space to low-dimensional space by minimizing the difference in all pairwise similarities between points in high- and low-dimensional spaces [22]. The axes of the low-dimensional spaces are given in arbitrary units. Briefly, the algorithm proceeds as follows: First, the pairwise distance matrix is calculated in high-dimensional space. Next, the distance matrix is transformed to a similarity matrix using a varying Gaussian kernel, so that the similarity between points X_i and X_j represents the joint probability that X_i will choose X_j as its neighbor or vice versa (based on their Euclidean distance and local density). Then, a random low-dimensional mapping is rendered and pairwise similarities are computed for points in the low-dimensional map. However, the low-dimensional similarities are computed using Student’s t-distribution rather than a Gaussian distribution. Finally, gradient descent is used to minimize the Kullback-Leibler divergence between the two probability distributions, leading to the final low-dimensional map.
The optimization step may be interpreted as a set of springs. Each pair of points Y_i and Y_j is connected by a spring which repels or attracts the points from each other depending on whether the similarity between the points in the projection is lower or greater than the similarity in the high-dimensional space. The gradient reduces each point’s springs into a single force. The heavy tailed t-distribution helps alleviate the “crowding problem” by exerting more force when pushing distant points further apart. See Supplementary Methods and [22] for full technical details.
The viSNE implementation
viSNE is a fast, distributed implementation of the t-SNE algorithm, improved and tailored for the analysis of single-cell data. In viSNE, each cell is represented by a point in high-dimensional space, each coordinate representing one measured parameter (e.g, the protein expression level). Our implementation then maps these points into two or three dimensions. viSNE also includes cyt, a tool for the interactive visualization of the resulting maps (see Supplementary Methods for more details).
The robustness and accuracy of t-SNE derives from the computation of all pairwise similarities. But, the similarity matrix comes at a heavy computational price, limiting the original implementation to 10,000 points. Our distributed implementation relies on the fact that each of t-SNE’s computations are local and do not require the entire matrix. The technical computational limit of viSNE is 100,000 points. However, beyond 30,000 the limit is not computational, but rather the “crowding problem” {Van der Maaten, 2008 #22}[22]: the volume in high-dimensional space grows polynomial with the number of dimensions, and as a result a two-dimensional map cannot accommodate a large number of points while conserving the high-dimensional distances between them. Instead, distant points collapse onto nearby areas of the map, creating one large, dense region, with no separation between populations. To solve this, viSNE subsamples cells uniformly at random and maps the sampled population. The algorithm is robust to such subsampling (Supplementary Figure 2) and even after subsampling, we still detect rare subpopulations that constitute a mere 0.2% of the population (Supplementary Figure 14).
viSNE is publicly available for download at http://www.c2b2.columbia.edu/danapeerlab/html/index.html.
Mass cytometry data
Fresh, Ficoll-enriched human bone marrow was obtained from healthy donors from AllCells, Inc. (Emeryville, CA). Leukemia samples were obtained under IRB-approved protocols at St. Jude Children’s Research Hospital, Memphis, TN (Pediatric acute myeloid and lymphoblastic leukemia) or at Princess Margaret Hospital, Toronto, ON (adult acute myeloid leukemia). All samples were deidentified.
Samples were processed as previously described in Bendall et al [21]. Briefly, cells were used fresh prior to mass cytometry experiments, or frozen in FCS with 10% DMSO, thawed and re-suspended in 90% RPMI with 10% FCS (supplemented with 20 U/mL sodium heparin (Sigma) and 0.025U/mL benzonase (Sigma) in the case of frozen samples), 1X L-glutamine and 1X penicillin/streptomycin (Invitrogen).
Cells were fixed with formaldehyde (PFA; Electron Microscopy Sciences, Hatfield, PA) added directly to growth media at a final concentration of 1.6% for 10 minutes at room temperature. Cells were then centrifuged at 500g for 5 minutes and washed once with staining media (PBS with 0.5% BSA, 0.02% sodium azide) to remove residual PFA, and blocked with Purified Human Fc Receptor Binding Inhibitor (eBioscience Inc., San Diego, CA) following manufacturer’s instructions. Surface marker antibodies were added yielding 50 or 100 uL final reaction volumes and stained at room temperature for 30min (Supplementary Table 1). Following staining, cells were washed 2 more times with cell staining media then permeabilized with 4°C methanol for at 10 min at 4°C, then optionally stored at −80°C for later use. Cells were then washed twice in cell staining media to remove remaining methanol, and then stained with surface and phospho-specific antibodies in 50 or 100 µL for 30 min at room temperature. Cells were washed once in cell staining media, then stained with 1 mL of 1:5000 191/193Ir DNA intercalator(2) (www.dvssciences.com; DVS Sciences, Richmond Hill, Ontario, Canada) diluted in PBS with 1.6% PFA for 20 mins at room temperature. Cells were then washed once with cell staining media and then finally with water alone before running on the CyTOF.
Processing of mass cytometry data
Data was transformed using hyperbolic arcsin with a cofactor of five. Single cells were gated based on cell length and DNA content (in order to avoid debris and doublets) as described in Bendall et al. [21]. The manual classification of Marrow1 was taken from [21], where the complete gating strategy can be found.
viSNE analysis
Generating viSNE maps included the following steps (exact details can be found in Supplementary Table 2). First, between 6,000 and 12,000 cells were uniformly subsampled from the data. After subsampling, viSNE was run for 500 iterations to project the data into 2D. Unlike t-SNE, PCA was not used as a preprocessing step. All runs used an identical random seed and the default t-SNE parameters (perplexity = 30, momentum = 0.5 for initial 250 iterations, momentum = 0.8 for remaining iterations, epsilon = 500, lie factor = 4 for initial 100 iterations, lie factor = 1 for remaining iterations). viSNE maps were visualized using cyt, which was also used to generate figures (color coding by subtype (as in 1B), by marker expression levels (as in 1D) and in plotting expression level densities (as in 2B)).
Quantifying similarity between viSNE maps
We use the Jensen-Shannon (JS) divergence (see Supplementary Methods) to quantify the similarity between viSNE maps. Each map is converted into a probability distribution. We define the similarity between two maps as the JS divergence between their respective distributions.
A gating scheme for fluorescence-activated cell sorting
The viSNE map has been utilized in devising a gating scheme for fluorescence-activated cell sorting (FACS) of the AML relapse sample (Figure 5). Due to the limits of flow cytometry, the gating scheme can only employ a limited number of channels and use hard thresholds. Through manual inspection of the viSNE map we identified four markers that lead to distinct subpopulations which could be of interest for downstream analysis: CD34, CD64, CD33 and CD7 (Figure 4). For each marker we defined a threshold for a binary negative/positive gate. The four binary gates were combined to create a total of sixteen composite gates covering all negative/positive combinations. Only 6 composite gates had more than 20 cells. The cells residing in each of these 6 composite gates are color coded on the viSNE map of Figure 5.
Subsampling of in-vitro MRD experiment
The MRD experiment involved two samples: the MRD sample, which is composed of an in-vitro mix of 99.5% healthy bone marrow cells and 0.5% barcoded ALL sample, and the control sample, which is a 100% healthy bone marrow cells (taken from a donor other than the MRD sample’s donor). To capture a higher proportion of ALL cells for the viSNE map, we devised the following subsampling procedure. The cells from the MRD sample and from the control sample were combined computationally and clustered using the Louvain algorithm [33]. Next, the clusters were weighted by the proportion of MRD sample cells in them; the higher the proportion of MRD sample cells, the higher the weight. Finally, 10,000 cells were chosen one at a time in a two-step process: one of the clusters was chosen randomly (biased by cluster weight) and a cell was uniformly chosen from that cluster. Note, the subsampling procedure is blind to the ALL barcode; it can only access the mass cytometry measurement and the source of the sample (MRD or control). Following the subsampling, viSNE was run as described above.
Additional algorithms
Isomap, LLE, KernelPCA and LLTSA were run using the Matlab Toolbox for Dimensionality Reduction [32]. FLOCK was compiled from the code available in the ImmPort FLOCK SourceForge page (http://sourceforge.net/projects/immportflock/). SPADE was run using the implementation available in Cytobank [34].
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Nir Friedman, Dr. Itsik Pe’er and Oren Litvin for valuable comments. The authors would also like to thank Dr. Mark Minden (Princess Margaret Hospital), Dr. Charles Mullighan, Dr. Jim Downing and Dr. Ina Radtke (St. Jude Children’s Hospital) for generously providing leukemia samples for mass cytometry analysis. This research was supported by the National Science Foundation CAREER award through grant number MCB-1149728, National Institutes of Health Roadmap Initiative, NIH Director's New Innovator Award Program through grant number 1-DP2-OD002414-01 and National Centers for Biomedical Computing Grant 1U54CA121852-01A1. E.D.A. is a Howard Hughes Medical Institute International Student Research Fellow. K.L.D. is supported by Alex's Lemonade Fund Young Investigator Award and St. Baldrick's Foundation Scholar Award. S.C.B. is supported by the Damon Runyon Cancer Research Foundation Fellowship (DRG-2017-09). G.P.N. is supported by the Rachford and Carlota A. Harris Endowed Professorship and grants from U19 AI057229, P01 CA034233, HHSN272200700038C, 1R01CA130826, CIRM DR1-01477 and RB2-01592, NCI RFA CA 09-011, NHLBIHV-10-05(2), European Commission HEALTH.2010.1.2-1, and the Bill and Melinda Gates Foundation (GF12141-137101). D.P. holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund and Packard Fellowship for Science and Engineering.
References
- 1.Bendall SC, et al. A deep profiler's guide to cytometry. Trends Immunol. 2012;33(7):323–332. doi: 10.1016/j.it.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petilla Interneuron Nomenclature, G et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci. 2008;9(7):557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irish JM, et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118(2):217–228. doi: 10.1016/j.cell.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 4.Sachs K, et al. Causal protein-signaling networks derived from multiparameter single-cell data. Science. 2005;308(5721):523–529. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- 5.Majeti R, Park CY, Weissman IL. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1(6):635–645. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tarnok A, Ulrich H, Bocsi J. Phenotypes of stem cells from diverse origin. Cytometry A. 2010;77(1):6–10. doi: 10.1002/cyto.a.20844. [DOI] [PubMed] [Google Scholar]
- 7.O'Brien CA, Kreso A, Dick JE. Cancer stem cells in solid tumors: an overview. Semin Radiat Oncol. 2009;19(2):71–77. doi: 10.1016/j.semradonc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Bandura DR, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal Chem. 2009;81(16):6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- 9.Cornett DS, et al. MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat Methods. 2007;4(10):828–833. doi: 10.1038/nmeth1094. [DOI] [PubMed] [Google Scholar]
- 10.Mercer J, et al. RNAi Screening Reveals Proteasome- and Cullin3-Dependent Stages in Vaccinia Virus Infection. Cell Rep. 2012;2(4):1036–1047. doi: 10.1016/j.celrep.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet. 2011;12(2):87–98. doi: 10.1038/nrg2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Gerstein MM. Snyder, RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalerba P, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol. 2011;29(12):1120–1127. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lubeck E, Cai L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat Methods. 2012;9(7):743–748. doi: 10.1038/nmeth.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benoist C, Hacohen N. Immunology. Flow cytometry, amped up. Science. 2011;332(6030):677–678. doi: 10.1126/science.1206351. [DOI] [PubMed] [Google Scholar]
- 16.Bendall SC, Nolan GP. From single cells to deep phenotypes in cancer. Nat Biotechnol. 2012;30(7):639–647. doi: 10.1038/nbt.2283. [DOI] [PubMed] [Google Scholar]
- 17.Herzenberg LA, et al. Interpreting flow cytometry data: a guide for the perplexed. Nat Immunol. 2006;7(7):681–685. doi: 10.1038/ni0706-681. [DOI] [PubMed] [Google Scholar]
- 18.Qiu P, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotechnol. 2011;29(10):886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian Y, et al. Elucidation of seventeen human peripheral blood B-cell subsets and quantification of the tetanus response using a density-based method for the automated identification of cell populations in multidimensional flow cytometry data. Cytometry B Clin Cytom. 2010;78(Suppl 1):S69–S82. doi: 10.1002/cyto.b.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pyne S, et al. Automated high-dimensional flow cytometric data analysis. Proc Natl Acad Sci U S A. 2009;106(21):8519–8524. doi: 10.1073/pnas.0903028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van der Maaten L, Hinton G. Visualizing data using t-SNE. Journal of Machine Learning Research. 2008;9(2579–2605):85. [Google Scholar]
- 23.Kondo M, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 24.van Lochem EG, et al. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: reference patterns for age-related changes and disease-induced shifts. Cytometry B Clin Cytom. 2004;60(1):1–13. doi: 10.1002/cyto.b.20008. [DOI] [PubMed] [Google Scholar]
- 25.van Dongen JJ, Orfao C, EuroFlow A. EuroFlow: Resetting leukemia and lymphoma immunophenotyping. Basis for companion diagnostics and personalized medicine. Leukemia. 2012;26(9):1899–1907. doi: 10.1038/leu.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wakita S, et al. Mutations of the epigenetics modifying gene (DNMT3a, TET2, IDH1/2) at diagnosis may induce FLT3-ITD at relapse in de novo acute myeloid leukemia. Leukemia. 2012 doi: 10.1038/leu.2012.317. [DOI] [PubMed] [Google Scholar]
- 27.Campana D. Status of minimal residual disease testing in childhood haematological malignancies. Br J Haematol. 2008;143(4):481–489. doi: 10.1111/j.1365-2141.2008.07350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borowitz MJ, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood. 2008;111(12):5477–5485. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ossenkoppele GJ, van de Loosdrecht AA, Schuurhuis GJ. Review of the relevance of aberrant antigen expression by flow cytometry in myeloid neoplasms. Br J Haematol. 2011;153(4):421–436. doi: 10.1111/j.1365-2141.2011.08595.x. [DOI] [PubMed] [Google Scholar]
- 30.Loken MR, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children's Oncology Group. Blood. 2012;120(8):1581–1588. doi: 10.1182/blood-2012-02-408336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30(9):858–867. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van der Maaten L, Postma E, Van Den Herik H. Dimensionality reduction: A comparative review. Journal of Machine Learning Research. 2009;10:1–41. [Google Scholar]
- 33.Blondel VD, et al. Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment. 2008;2008(10):P10008. [Google Scholar]
- 34.Kotecha N, Krutzik PO, Irish JM. Web-based analysis and publication of flow cytometry experiments. Curr Protoc Cytom. 2010;Chapter 10(Unit10):17. doi: 10.1002/0471142956.cy1017s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.