

Abstract
The clinical management of locally recurrent or unresectable malignant melanoma continues to pose a significant challenge. These lesions are typically painful and currently available treatments, such as repeated intratumoral injections of interferon-alpha (IFN-α), are costly and inconvenient. Nanotechnology offers promise as a novel means of drug delivery. A capsule-like nanoporous miniature device (NMD) based on a biodegradable polymer, poly(polycaprolactone) (PCL) was developed for controlling the local delivery of immunological agents to the tumor microenvironment. The device consists of a nanoporous release gate, a fabricated drug reservoir loaded with IFN-α and a protective layer. To improve the biocompatibility of the device, a hydrophilic poly(ethylene glycol) monoacrylate was applied to the outside wall of the device via covalent bonding techniques. Microscopic visualization of the nanoporous gate from in vitro experiments exhibited good pore stability over a two-month period. In vitro experiments demonstrated a constant release rate of IFN-α from the NMD and showed that the release rate could be regulated by the gate area. The released IFN-α was biologically functional. Cytokine-containing supernatants from release experiments phosphorylated signal transducer and activator of transcription (STAT1) in peripheral blood mononuclear cells. Subcutaneous implantation of the NMDs was well tolerated and associated with an anti-tumor effect in a human xenograft model of melanoma. There was no evidence of a significant inflammatory response to the NMD or encapsulation of the NMD by fibrosis. These experiments show that the NMD can be fabricated and employed in vivo as a versatile drug delivery platform.
Keywords: Biodegradable nanoporous miniature device (NMD), PCL, Nanoporous membrane, Interferon-alpha release, Malignant melanoma
1. Introduction
Locally recurrent melanoma tumors are often unresectable due to their size and/or close proximity to critical structures. These lesions can disrupt normal organ function, are typically painful, and can present a cosmetic dilemma. These tumors provide further challenges to clinicians because radiation therapy is frequently unsuccessful, and chemotherapy induces regression in less than 20% of cases [1,2]. Spontaneous regressions of malignant melanoma lesions have been observed on rare occasions and careful histologic evaluations have revealed that this process is mediated by activated lymphocytes [3].
Local administration of cytokines can have beneficial effects in the setting of locally recurrent melanoma. Systemically administered interferon-alpha (IFN-α) is commonly used in the treatment of metastatic melanoma [4]. This cytokine is known to enhance the activity of immune effector cells as well as mediate anti-proliferative and pro-apoptotic effects against tumor cells [5]. The binding of IFN-α to its receptor results in the phosphorylation of Janus kinase 1 (Jak1) and tyrosine kinase 2 (Tyk2) on specific tyrosine residues. These phosphotyrosine residues provide docking sites for cytoplasmic transcription factors belonging to the signal transducer and activator of transcription (STAT) family of proteins. These STAT proteins (STAT1-α, STAT2) are phosphorylated by the Janus kinases and subsequently form high affinity DNA binding complexes that rapidly translocate to the cell nucleus to drive the expression of IFN-responsive genes [6]. The current study used IFN-α as an antitumor agent, as it is FDA-approved for the treatment of high-risk melanoma in patients following resection of involved lymph nodes and is also used in the setting of advanced disease [4]. While systemic administration of IFN-α can be accompanied by significant toxicities (50% of patients require drug holidays or dose reductions), local administration of IFN-α can have potent anti-tumor activity at low doses because of the high affinity receptors present on immune effector cells and tumor cells [7–9]. However, this approach is limited by the discomfort induced by repeated intratumoral injections and the need for frequent clinical visits.
Polymer-based drug delivery devices have gained more attention in recent years because of the stringent requirements of protein- and DNA-based therapy [10]. The potency of many protein- and DNA-based drugs is off-set by the fragility of these molecules and their short half-life in vivo. These drugs require an effective delivery device to realize their therapeutic potential. Delivering these drugs to the targeted tissues and cells in a safe, controlled, and patient-friendly manner remains a significant challenge [11]. Various techniques have been explored to tackle the drug-delivery challenges. Traditional fabrication protocols, such as phase separation and microemulsion, have been successfully used for the production of drug delivery carriers [12,13]. In the medical and pharmaceutical fields, drug-loaded chambers are capable of controlling the release rate and keeping the concentration within the therapeutic windows for prolonged periods of time [14]. When combined with relevant micro-/nanofabrication technologies, such devices can provide several advantages for protein delivery. These include high loading efficiency, good payload protection during fabrication, the ability to deliver multiple agents, and a pre-designed release profile.
The nanoporous membrane is a key element to achieving a desirable release profile of biologic agents in the local tumor environment. Silicon- and alumina-based nanoporous membranes provide good mechanical stability. They have highly uniform and well-defined pore structures [15,16]. However, if implanted, they must be surgically removed after use. They may also stimulate a host fibrotic reaction that can significantly alter the tumor micro-environment. Inexpensive polymers such as PCL, on the other hand, can be designed to be fully biodegradable. A variety of low-cost techniques are available for the fabrication of polymeric micro/nanostructures, including porous membranes [17–20]. However, most porous PCL membranes prepared via those methods have the structure of microscale pores, and are unable to achieve a constant, zero-order, drug release rate. Recently, our group has published a series of papers investigating the preparation of nanoporous PCL membranes via the combination of thermally- and nonsolvent-induced phase separations [21–23]. The pore sizes of membranes were optimized and evaluated to achieve the constant release by controlling the preparation conditions (i.e. temperature, polymer concentration, and solvent composition). The surface treatment for the nanoporous membrane via the plasma technique was also studied. However, the release performance of the PCL-based nanoporous miniature device (NMD) has not been evaluated. Furthermore, animal studies are needed for clinical applications. We hypothesized that the zero order local release of high concentrations of immunotherapeutic agents from a locally implanted, biodegradable NMD would improve the ability of cytokines, such as IFN-α, to exert an antitumor effect on unresectable deposits of melanoma. We provided evidence that a biological molecule can be released from our NMD in a murine model, with minimal foreign body response.
2. Materials and methods
2.1. Reagents and cells
PCL (Mn ~80,000 Da) was purchased from Aldrich Chemicals (Milwaukee, WI). 1,4-Dioxane was obtained from Mallinckrodt Chemicals (Philipsburg, NJ). 2-Methoxyethanol (ACS reagent, ≥93%) was purchased from Sigma-Aldrich (St. Louis, MO). Polypropylene glycol 400 monoacrylate (PEG(400)MA) was purchased from Monomer-Polymer & Dajac Labs (Feasterville-Trevose, PA). Isopropanol was purchased from Fisher Scientific Inc. Immunoglobulin G (from bovine serum, reagent grade, ≥95%) and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO). All chemicals were used as received without further purification.
Recombinant human IFN-α-2b for in vitro and in vivo studies was purchased from Schering-Plough Inc. (Kenilworth, NJ). The A375 human melanoma cell line was obtained from the American Type Culture Collection (Manassas, VA). Peripheral blood mononuclear cells (PBMCs) were isolated from source leukocytes of healthy adult donors (American Red Cross, Columbus, OH) via density gradient centrifugation with Ficoll-Paque Plus (Amersham Pharmacia Biotech, Uppsala, Sweden) as previously described [24]. C57BL/6 mice were purchased from Taconic Farms Inc. (Germantown, NY). BALB/C-nu/nu mice where purchased from Charles River Laboratories (NCI).
2.2. Device design and fabrication
The fabrication protocol employed for the PCL-based NMDs used in this study has been described in detail elsewhere [25]. Briefly, the device consists of three parts: a drug reservoir for holding therapeutic agents, a nanoporous gate for controlling the drug release rate, and a protective layer for the nanoporous gate and reservoir. Fig. 1 outlines the fabrication protocol for the NMD. The drug reservoirs and nanoporous PCL membranes were made by using a hot embossing process and phase inversion, respectively. To achieve the constant release, the preparation condition for nanoporous membrane has been optimized: 25 wt.% of PCL in the solvent mixture (methoxyethanol/1,4-dioxane at the weight ratio of 13/3) and a 5 °C water bath [21].
Fig. 1.

Schematic of fabrication for a nanoporous PCL device.
In order to render the NMD more hydrophilic and thereby prevent fibrosis, hydrophilic PEG molecules were grafted onto the PCL device surface by using the plasma technique [23,26]. Briefly, the PCL porous membranes and PCL reservoirs were soaked in 10 wt.%. PEGMA (one type of PEG-based molecules) solution (20% water and 80% ethanol) for 2 h. After overnight drying, the pre-soaked samples were subjected to oxygen plasma for 20 s at a power of 25 W. After PEGMA-modification, the mixture of human recombinant IFN-α-2b and BSA was loaded into the modified reservoir at a ratio of 10:1 by weight. BSA was used as a stabilizer to prevent denaturation of the drug and maintain IFN-α activity. A sharp tweezer was used to pack the loose powder to improve the loading efficiency. Subsequently, the release gate was applied to the drug reservoir using a NuSil bioadhesive to form a laminated assembly. Finally, a 20 μm-thick PCL protection layer was attached to the top of the device to provide additional protection of the release gate from drug leakage and to regulate the release rate by adjusting the diffusion area. Here, R1 and R2 are the inner radius of the drug reservoir and the radius of the diffusion holes on the PCL protection layer, respectively.
2.3. Scanning electron microscopy (SEM) for membrane morphology
To visually examine the surface morphology of the nanoporous PCL membranes, a Hitachi Model S-4300 SEM was used to analyze the pore structure. The air-dried samples were loaded on the surface of an aluminum SEM specimen holder and sputter coated with gold for 40 s (Pelco Model 3 Sputter Coater) before observation. A working distance of about 8–10 mm, an accelerating voltage of 10 KV, and a chamber pressure of 10−8 Torr were found to be suitable for obtaining high-resolution images. The magnification used in this study varied from 2000× to 120,000× depending on the pore structure.
Changes of the pore size and morphology of PCL membranes in buffer solution were determined by SEM observation. First, PCL membranes were immersed in 5 ml of 10 wt.% of human serum albumin buffer solution. Samples were then incubated at 37 °C, at various time intervals the samples were taken out of the buffer solution and dried in a vacuum oven for 24 h at room temperature. The dried membranes were then imaged by SEM.
2.4. In vitro release performance
Diffusion studies of IFN-α through the PCL membrane were conducted using a PCL membrane that had been cut in a circular pattern (1.4 cm diameter) and then bonded to the bottom of the Transwell insert by a bioadhesive (NuSil Technology, Carpinteria, CA) to separate the two chambers (the effective diffusion area was 1.0 cm2). After the adhesive was completely solidified (around 24 h), 0.3 ml of a 1.0 mg/ml IFN-α solution was pipetted into the donor chamber and allowed to diffuse into the receptor chamber which contained 6.0 ml of PBS solution (pH 7.4) with 0.2% sodium azide. These diffusion experiments were carried out at 37 °C and subjected to constant shaking at 200 rpm. At various time intervals, the donor chamber was put into a new receptor chamber containing 6 ml fresh buffer. Samples containing the released IFN-α were analyzed using the VeriKine™ Human IFN-α ELISA Kit (PBL InterferonSource, NJ) per the manufacturer’s recommendations. ELISA plates were analyzed with a TECAN GENios plate reader using a 450 nm absorbance filter. And the concentrations of IgG molecules and BSA molecules were measured with a Bio-Rad protein assay using the microplate assay protocol at 595 nm.
To evaluate the effect of diffusion area on release profiles, NMDs (n=3) loaded with 1.5 mg of IFN-α were placed into a 6-well microtiter plate filled with 6 ml of PBS solution and placed on a 37 °C rotating platform. The plate was shaken at a rate of 200 rpm. Samples were withdrawn from each well and the devices were transferred into new microtiter plates containing 6 ml of fresh PBS daily. In vitro release was monitored for 23 days. Samples were stored at −80 °C until further analysis. The concentration of IFN-α in the buffer solution was obtained from a calibration curve, and the amount of IFN-α release at time t (Mt) was calculated from accumulating the total IFN-α release up to that time. The fractional drug release, Mt/M0, could then be calculated. Here M0 is the amount of initially loaded IFN-α.
2.5. Cell stimulation assays and flow cytometric assays
To confirm the biologic activity of IFN-α released from the NMDs during the in vitro release experiments, we employed a previously described intracellular flow cytometric assay [9] that can detect the phosphorylated form of the STAT1 protein (p-STAT1), a downstream modulator of IFN-α activity. Peripheral blood mononuclear cells (PBMCs) were stimulated at 37 °C, 5% CO2 for 15 min using the supernatants obtained from Day 5 of the in vitro studies of IFN-α-loaded NMDs. PBMCs treated with PBS or directly treated with human IFN-α, served as negative and positive controls, respectively. After stimulation cells were stained for intracellular p-STAT1. Analyses were performed as previously described using a Becton Dickinson FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ) equipped with a 488-nm air-cooled argon laser and a 633-nm helium-neon laser, using at least 10,000 PBMCs gated in the lymphocyte region as determined by light scatter properties. [9].
2.6. In vivo IFN-α release and biocompatibility of NMDs
For in vivo studies, a human xenograft model of melanoma was used [27]. The animal protocol was approved by the OSU ULAR and employed under the supervision of a licensed veterinarian. A375 human melanoma cells were thawed and expanded by standard cell culture techniques. Cells were cultured in complete medium (DMEM containing 10% fetal bovine serum (FBS), 1% sodium bicarbonate, 1% sodium pyruvate, and 1% antibiotic/antimycotic solution). Female BALB/C-nu/nu severe combined immunodeficiency (SCID) mice 4–6 weeks of age were injected subcutaneously (right flank) with 2×106 A375 cells on Day 0. On Day 23 when the tumors were established (approx. 55 mm3) the mice were randomly separated into two treatment groups. One group was treated with an IFN-α-loaded NMD (n=4) and the other was treated with a PBS-loaded NMD (n=4). For implantation of the NMD, the mice were anesthetized with isoflurane and a transverse nuchal incision was made to from a subcutaneous pocket that was extended along the animal’s right flank. The NMD was placed into the subcutaneous pocket so that it was in close proximity but did not touch the tumor. Bidimensional tumor measurments were obtained weekly using calipers. Tumor volume was calculated using the formula: (width2×length)/2. Mice were euthanized on Day 37 by CO2 inhalation (due to tumor size) and exsanguinated via cardiac puncture. At the time of sacrifice, the mice underwent a final tumor measurement. Melanoma tumors were harvested from each of the mice. These samples were fixed in formalin and embedded in paraffin. After sectioning and staining with H&E, they were analyzed by an expert pathologist.
To assess the biocompatibility of PEGMA-modified NMDs, immunocompetent C57BL/6 were implanted with PEGMA-modified NMDs or unmodified NMDs loaded with either PBS or IFN-α. The NMDs were implanted subcutaneously as described. On Day 30 the mice were euthanized and the tissue surrounding the implanted devices was harvested and subjected to H&E staining. The specimens were examined by an expert pathologist for inflammatory response.
2.7. Statistical analysis
Statistic analysis was performed using the Student’s t-test with significance set at p<0.05. All data reported are means±SD, unless otherwise noted.
3. Results and discussion
3.1. Fabrication and characterization of nanoporous release gate
Porous membranes play an important role in a variety of drug delivery devices and phase inversion is a widely used method for membrane fabrication. The basic principle of the phase inversion process is the bidirectional diffusion of solvent and non-solvent, which allows the formation of a nanoporous structure within the cast membrane. The structural elements, such as pore size and pore size distribution can be controlled by a number of experimental variables, including the solvent type, the ratio of solvents to non-solvents, the polymeric composition, the evaporation time, and the temperature of the coagulation bath, among others. By tuning the PCL concentration, it was possible to adjust the pore size over a range of 20 to 60 nm (measured by ASAP 2010, Micromeretics). Fig. 2 shows the pore structure of the PCL nanoporous membrane as imaged by SEM. As can be seen from the membrane cross-section, the PCL membrane with a thickness of 50 μm has an asymmetric structure consisting of a thin and very dense layer associated with a thick and porous support layer. The thick support layer has micro-sized open cells (approx. 1–2 microns) and contributes to the mechanical stability of the membranes. The thin layer determines the diffusion rate of molecules through the membrane.
Fig. 2.

SEM images of membrane formed with 25 wt.% PCL solution observed from side view.
3.2. In vitro stability of nanoporous membrane
When biodegradable nanoporous membranes are used as release gates in controlled drug release, the morphological stability of the nanoporous structure in a buffer solution is essential. Therefore, we investigated the morphological changes of PCL membranes following immersion in a liquid medium designed to mimic tissue fluid. Fig. 3 compares the porous structure of PCL membranes after being immersed in a PBS buffer solution containing 10 wt.% of human serum albumin over time. As can be seen from the SEM images, there was no discernable deformation of the membrane or the structure of the pores that had been immersed in a PBS buffer solution for 8 weeks at 37 °C as compared to membranes that were immersed for only a few minutes. The stability of the membrane ensures that the release gate will maintain its desired physical characteristics and allow continued passage of drug molecules at a pre-specified release rate.
Fig. 3.

SEM images of PCL membrane after immersed in PBS buffers with 10 wt.% of human serum albumin for (A) 0 and (B) 8 weeks.
3.3. In vitro release studies
The polymer concentration of the casting solution, the solvent composition, and temperature are tunable parameters that permit control to be exerted over the porous characteristics of the nanoporous membrane. To achieve the constant release, the preparation conditions for nanoporous membrane have been determined to be: 25 wt.% of PCL in the solvent mixture (methoxyethanol/1,4-dioxane at the weight ratio of 13/3) and a 5 °C water bath [21]. The pore size on the controlled side of the PCL membranes is around 20–30 nm.
Fig. 4 compares the accumulative diffusion profiles of IFN-a, BSA and IgG through the PCL membranes (the effective diffusion area of 1.0 mm2). The diffusion rate of the molecules through membranes depends on their solubility (initial concentration), the molecular size, the hydrophilic properties of the molecules and membranes, the interaction between the molecules and diffusion path, etc. In this study, the modified nanoporous membranes have the hydrophilic diffusion path, facilitating the hydrophilic molecules to pass through. We also used similar initial concentrations for three molecules. Therefore, the determined parameter for diffusion rate here is the size of the molecules. The molecular weights of IFN-α, BSA and IgG are 19.2 KD, 65.0 KD, and 150 KD, respectively. Among these reagents, IFN-α has the smallest size and the highest diffusion rate (7%). As the molecule size increased, the diffusion rate decreased. For BSA, less than 1.5% was diffused through the membrane after 192 h. BSA acts as a stabilizer in this study. The low diffusion rate allowed BSA to be held inside of the reservoir to maintain IFN-α activity. IgG molecule is an immune molecule and has the largest size of the three molecules. From Fig. 4 we can see the accumulative percentage of IgG was close to zero showing that most of IgG molecules were blocked from release. This demonstrates that the PCL nanoporous membrane is selective, allowing the diffusion of small molecules and blocking the large molecules.
Fig. 4.

Comparison of diffusion of IFN-α, BSA and IgG release through the PCL nanoporous. membranes.
In addition to the pore size of the release gate, the gate diffusion area also affects the rate of drug release. The release characteristics of NMDs were investigated using IFN-α as a model molecule. Fig. 5 compares the release profiles of IFN-α-loaded devices with different diffusion areas. Here, R1 is equal to 1.8 mm and R2 is 1.8 mm for a diffusion area of 10.2 mm2. R2 is corresponding reduced to 1.2 mm for a diffusion area of 4.5 mm2 and 0.8 mm for a diffusion area of 2.1 mm2. And 1 Unit is equal to 1.42 pg/ml. All release gates had the same pore size. The pore size on the controlled side for this type membrane is around 20–30 nm. As can be seen, IFN-α was released in a burst pattern after one day and was then released very rapidly when the diffusion area was large. When the diffusion area was decreased it took time for the buffer solution to diffuse into the PCL membrane and dissolve the IFN-α. Therefore, there is no IFN-α released the first day. The starting time of the burst drug release depends on the diffusion area. The larger the diffusion area, the faster the IFN-α molecules inside dissolved and the earlier the IFN-α molecules were released. When the release area was decreased, nearly constant IFN-α release profiles were achieved for many days. The area under the release curves are nearly the same, indicating that all devices were initially loaded with the same amount of IFN-α. Moreover, the IFN-α remained bioactive for up to 23 days. When the diffusion area was decreased from 10.2 to 4.5 mm2 (a ratio of around 2.27), the ratio of maximum release concentration (the maximum release concentration when the diffusion area is 10.2 mm2 to the maximum release concentration when the area is 4.5 mm2) was around 2.20. Similarly, when the diffusion area was decreased from 10.2 to 2.1 mm2 (a ratio of around 4.85), the ratio of maximum release concentration was 4.71, implying a linear relationship between the release rate and the diffusion area. By adjusting the diffusion area and the reservoir size of NMDs, one can pre-design the release rate and the release time period. We also can design the device dimensions to control the amount of drug loaded and adjust the release time period. Drug delivery carriers can be fabricated by using traditional protocols. For example, the double emulsion and emulsification-solvent diffusion have been utilized for producing nanoparticles loaded with hydrophilic proteins. However, the fabrication conditions may damage the bioactivity of proteins through aggregation, chemical inactivation, or denaturation. Our protocol to make the NMD doesn’t use the organic solvent and can provide good payload protection during fabrication.
Fig. 5.

IFN-α release from the PCL nanoporous devices with different diffusion areas. Data were derived from n=3 NMDs for each diffusion area.
3.4. Determination of functional activity of IFN-α release in vitro
To evaluate whether the released IFN-α from the NMDs was biologically active, we utilized phosphorylated STAT1 (p-STAT1) in PBMCs as a marker of IFN-α activity. Supernatants from day 5 of the in vitro IFN-α release experiment (n=6) were used to stimulate PBMCs at 37 °C, 5% CO2 for 15 min. PBMCs were then stained for intracellular p-STAT1, a transcription factor that is activated by IFN-α and critical for its anti-tumor activity, and evaluated by flow cytometric analysis. Our data confirmed that immunologically active IFN-α can diffuse through the NMD membrane, where the levels of p-STAT1 were significantly higher in PBMCs incubated with the supernatants from NMDs loaded with IFN-α than those loaded with PBS (Fig. 6).
Fig. 6.
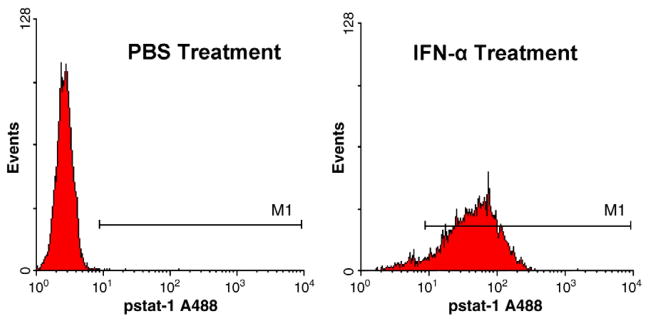
The biological activity of the released IFN-α on PBMCs assessed via flow cytometric analysis for phosphorylated signal transducer and activator of transcription 1 (p-STAT1). Histograms are representative of n=6 device supernatants.
3.5. Confirmation of IFN-α release in vivo and anti-tumor activity
For the in vivo evaluation of IFN-α release from NMDs (with a pore size on the controlled side around 20–30 nm, and a diffusion area of 2.1 mm2), a human xenograft model of melanoma was used. Tumor growth was significantly inhibited in mice that received IFN-α-loaded NMDs as compared to those that received PBS (0.1 wt.% BSA)-loaded NMDs (p<0.01) (Fig. 7). Examination of the tumors revealed that the IFN-treated mice had tumors with smaller areas of coagulation necrosis (17% versus 22.67%) and fewer mean mitotic figures (7.33 versus 11.67) when compared to PBS-treated mouse tumors. These findings represent a less advanced disease process in the IFN treated group. The controlled, local delivery of IFN-α from NMDs and the observed anti-tumor effect may therefore be useful clinically.
Fig. 7.

Comparison of A375 melanoma tumor growth in mice bearing locally implanted NMDs loaded with IFN-α or PBS. Surgical implantation occurred on Day 0. A statistically significant inhibition in tumor growth was observed in mice treated with IFN-loaded NMDs starting at Day 9, this effect continued until sacrifice on day 14 (p<0.01). The graph depicts a representative experiment of n=2.
3.6. Inflammatory response to NMD implantation in vivo
The foreign body response remains a major consideration in the development of implantable drug delivery devices. PEG-based molecules have widely been used to make implanted devices more biocompatible, as biofouling or host encapsulation have arisen as a common barrier for numerous implant and in vivo drug delivery platforms. They have also been used to coat the surface of nanoparticles such as liposomes and polyplexes for drug and gene delivery. PEG-based molecules are nontoxic, non-antigenic, non-immunogenic and FDA-approved for internal consumption [28]. These molecules are significantly effective at reducing protein adsorption and cell adhesion because of their rapid mobility, steric stabilization effect and minimal interfacial energy with water [29–31]. We implanted NMDs (the pore size on the controlled side is around 20–30 nm, and the diffusion area is 2.1 mm2) with and without PEGMA modification into immunocompetent mice. After 30 days, the tissues surrounding the NMD were evaluated for an inflammatory response. Gross examination revealed no obvious fibrous capsule or other reaction to the PEGMA-modified devices. Any evidence of unwanted foreign body response had been replaced over time by a benign connective tissue layer surrounding the devices. Pathologic analysis of H&E sections revealed no significant inflammatory response at the site of capsule implantation for the PEGMA modified devices (Fig. 8). However, for the devices without PEGMA grafting, there was significant inflammation and cell-infiltration. PEGMA modification appears to inhibit the inflammatory reaction to the foreign body.
Fig. 8.

Microscopic images of the inflammatory reaction after implantation of PCL-unmodified compared to PCL-PEG modified NMDs in immune competent mice at Day 30.
We also fabricated PLGA-based NMD for in vivo testing (data not shown here). The degradation time of PLGA was reported to be two months. However, we found the devices disappeared at the implanted site of mice after 30 days. This demonstrated that the actual degradation of the NMDs at the implanted site is faster than that reported. To achieve 6-month delivery, we used PCL with 2-year degradation time for device preparation. As shown by the SEM images of PCL membrane (Fig. 3), the device did not degrade after 8 weeks.
Ultimately, we plan to use the NMDs to deliver other biologically active compounds and therapeutics in a clinical setting. Since proteins may degrade at room temperature, each protein needs to be loaded and tested individually using this drug delivery platform. Other potential stable therapeutic agents that warrant future investigation include oligonucleotides and DNA vaccines. Moreover, besides the NMD application for melanoma treatment, potential targets include other skin cancers, hepatocellular carcinoma, renal cell carcinoma and pancreatic cancer as well as metastatic deposits from other cancers in the liver. Many of these cancers use cytokines as part of their treatment regimen, as well there are many new cytokines that are being explored for the treatment of cancer such as IL-12, IL-29 and IL-15.
4. Conclusion
In this study, we have described the fabrication and characterization of PCL-based NMDs for controlled drug release in vitro and in vivo at a local site. Although composed of biodegradable materials, the nanopore stability was confirmed over the desirable drug delivery period for up to 2 months. In vitro studies demonstrated constant release of IFN-α, a prototypical molecule used for immunotherapy of advanced melanomas. The release of immunologically activity IFN-α was confirmed by flow cytometric analysis for activated signaling molecules (p-STAT1) in PBMCs. The NMDs were studied in vivo using a SCID mouse model bearing A375 human melanoma. Mice implanted with IFN-loaded devices showed decreased tumor growth compared to those implanted with PBS-loaded devices. Implantation of the NMDs was easily accomplished and well tolerated by the animals. No significant inflammatory response or biofouling was observed.
Acknowledgments
This work is funded by the National Science Foundation sponsored Nanoscale Science and Engineering Center for Affordable Nanoengineering of Polymeric Biomedical Devices (NSEC-CANPBD). This work was also funded by the National Institutes of Health grants K24 CA093670 (WEC), T32 CA090223 (WEC), and P30 CA016058.
References
- 1.McMasters KM, Sondak VK, Lotze MT, Ross MI. Recent advances in melanoma staging and therapy. Ann Surg Oncol. 1999;6:467–475. doi: 10.1007/s10434-999-0467-8. [DOI] [PubMed] [Google Scholar]
- 2.Belardelli F, Ferrantini M, Proietti E, Kirkwood JM. Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev. 2002;13:119–134. doi: 10.1016/s1359-6101(01)00022-3. [DOI] [PubMed] [Google Scholar]
- 3.Atkins MB. Immunotherapy and experimental approaches for metastatic melanoma. Hematol Oncol Clin N Am. 1998;12:877–902. doi: 10.1016/s0889-8588(05)70029-0. [DOI] [PubMed] [Google Scholar]
- 4.Crosby T, Fish R, Coles B, Mason M. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database Syst Rev. 2009;4:1–9. doi: 10.1002/14651858.CD001215. [DOI] [PubMed] [Google Scholar]
- 5.Chung ES, Sabel MS, Sondak VK. Current state of treatment for primary cutaneous melanoma. Clin Exp Med. 2004;4(2):65–77. doi: 10.1007/s10238-004-0040-5. [DOI] [PubMed] [Google Scholar]
- 6.Haque SJ, Williams BR. Signal transduction in the interferon system. Semin Oncol. 1998;25:14–22. [PubMed] [Google Scholar]
- 7.von Wussow P, Block B, Hartmann F, Deicher H. Intralesional interferon-alpha therapy in advanced malignant melanoma. Cancer. 1988;61(6):1071–1074. doi: 10.1002/1097-0142(19880315)61:6<1071::aid-cncr2820610603>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 8.Ikic D, Spaventi S, Padovan I, Kusic Z, Cajkovac V, Ivankovic D, Dakovic N, Nola P. Local interferon therapy for melanoma patients. Int J Dermatol. 1995;34(12):872–874. doi: 10.1111/j.1365-4362.1995.tb04429.x. [DOI] [PubMed] [Google Scholar]
- 9.Lesinski GB, Kondadasula SV, Crespin T, Kendra K, Walker M, Carson WE. Multiparametric flow cytometric analysis of interpatient variation in STAT1 phosphorylation following IFN-alpha immunotherapy reveals dose-dependent signal transduction. JNCI J Natl Cancer Inst. 2004;96(17):1142–1331. doi: 10.1093/jnci/djh252. [DOI] [PubMed] [Google Scholar]
- 10.Duncan R, Ringsdorf H, Satchi-Fainaro R. Polymer therapeutics: polymers as drugs, drug and protein conjugates and gene delivery system: past, present, and future opportunities. Adv Polym Sci. 2006;192:1–8. doi: 10.1080/10611860600833856. [DOI] [PubMed] [Google Scholar]
- 11.Lee LJ. Polymer nanoengineering for biomedical applications. Ann Biomed Eng. 2006;34:75–88. doi: 10.1007/s10439-005-9011-6. [DOI] [PubMed] [Google Scholar]
- 12.Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 13.Langer R. Biomaterials in drug delivery and tissue engineering: one laboratory’s experience. Acc Chem Res. 2000;33(2):94–101. doi: 10.1021/ar9800993. [DOI] [PubMed] [Google Scholar]
- 14.LaVan DA, McGuire T, Langer R. Small-scale systems for in vivo drug delivery. Nat Biotechnol. 2003;21:1184–1191. doi: 10.1038/nbt876. [DOI] [PubMed] [Google Scholar]
- 15.Martin F, Walczak R, Boiarski A, Cohen M, West T, Cosentino C, Ferrari M. Tailoring width of microfabricated nanochannels to solute size can be used to control diffusion kinetics. J Control Release. 2005;102:123–133. doi: 10.1016/j.jconrel.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 16.Desai TA, Hansford D, Ferrari M. Nanopore technology for biomedical applications. Biomed Microdevices. 1999;2:11–40. [Google Scholar]
- 17.Lin WJ, Lu CH. Characterization and permeation of microporous poly(ε-caprolactone) films. J Membr Sci. 2002;198:109–118. [Google Scholar]
- 18.Tiaw KS, Teoh SH, Chen R, Hong MH. Processing methods of ultrathin poly (ε-caprolactone) films for tissue engineering applications. Biomacromolecules. 2007;8:807–816. doi: 10.1021/bm060832a. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka T, Tsuchiya T, Takahashi H, Taniguchi M, Lloyd DR. Microfiltration membrane of polymer blend of poly(L-lactic acid) and poly(ε-caprolactone) Desalination. 2006;1993:367–374. [Google Scholar]
- 20.Chiang HI, Perrie Y, Coombes AGA. Delivery of the antibiotic gentamicin sulphate from precipitation cast matrices of polycaprolactone. J Control Release. 2006;110:414–421. doi: 10.1016/j.jconrel.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 21.Yen C, He H, Lee LJ, Ho W. Synthesis and characterization of polycaprolactone nanoporous membranes for biomedical applications. J Membr Sci. 2009;343:180–188. [Google Scholar]
- 22.Yen C, He H, Fei Z, Zhang X, Lee LJ, Winston Ho WS. Surface modification of nanoporous poly(caprolactone) membrane with poly(ethylene glycol) to prevent biofouling: part i. Effects of plasma power and treatment time. Int J Polym Mater. 2010;59(11):923–942. [Google Scholar]
- 23.Yen C, He H, Fei Z, Zhang X, Lee LJ, Winston Ho WS. Surface modification of nanoporous poly(caprolactone) membrane with poly(ethylene glycol) to prevent biofouling: part ii. Effects of graft density and chain length. Int J Polym Mater. 2010;59(11):943–957. [Google Scholar]
- 24.Parihar R, Dierksheide J, Hu Y, Carson WE. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J Clin Invest. 2002;110:983–992. doi: 10.1172/JCI15950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, He Test H. A biodegradable, immunoprotective, dual nanoporous capsule for cell based therapies. Biomaterials. 2008;29(31):4253–4259. doi: 10.1016/j.biomaterials.2008.07.032. [DOI] [PubMed] [Google Scholar]
- 26.Carlisle ES, Mariappan MR, Nelson KD, Thomes BE, Timmons RB, Constantinescu A. Enhancing hepatocyte adhesion by pulsed plasma deposition and polyethylene glycol coupling. Tissue Eng. 2000;6:45–52. doi: 10.1089/107632700320883. [DOI] [PubMed] [Google Scholar]
- 27.Lesinski GB, Raig ET, Guenterberg K, Brown L, Go MR, Shah NN, Lewis A, Quimper M, Hade E, Young G, Chaudhury AR, Ladner KJ, Guttridge DC, Bouchard P, Carson WE. IFN-alpha and bortezomib overcome Bcl-2 and Mcl-1 overexpression in melanoma cells by stimulating the extrinsic pathway of apoptosis. Cancer Res. 2008;68:8351–8360. doi: 10.1158/0008-5472.CAN-08-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harris JM. Biotechnical and Biomedical Applications. Plenum Press; New York: 1992. Poly(ethylene glycol) Chemistry. [Google Scholar]
- 29.Faucheux N, Schweiss R, Lutzow K, Werner C, Groth T. Self-assembled monolayers with different terminating groups as model substrates for cell adhesion studies. Biomaterials. 2004;25:2721–2730. doi: 10.1016/j.biomaterials.2003.09.069. [DOI] [PubMed] [Google Scholar]
- 30.Nie FQ, Xu ZK, Huang XJ, Ye P, Wu J. Acrylonitirle-based copolymer membranes containing reactive group: surface modification by the immobilization of poly(ethylene glycol) for improving antifouling property and biocompatibility. Langmuir. 2003;19:9889–9895. [Google Scholar]
- 31.Goddard JM, Hotchkiss JH. Polymer surface modification for the attachment of bioactive compounds. Prog Polym Sci. 2007;32:698–725. [Google Scholar]