

Abstract
Objective
The aim of this work was to evaluate the role of Ubiquitin-Proteasome System (UPS) on mitochondrial-driven alpha-synuclein (aSN) clearance in in vitro, ex vivo and in vivo Parkinson disease (PD) cellular models.
Method
We used SH-SY5Y ndufa2 knock-down (KD) cells, PD cybrids and peripheral blood mononuclear cells (PBMC) from patients meeting the diagnostic criteria for PD. We quantified aSN aggregation, proteasome activity and protein ubiquitination levels. In PBMC of PD patients population we evaluated aSN levels in plasma and the influence of several demographic characteristics in the above mentioned determinations.
Results
We found that ubiquitin-independent proteasome activity was up-regulated in SH-SY5Y ndufa2 KD cells while a down regulation was observed in PD cybrids and PBMC. Moreover, we observed an increase in protein ubiquitination that correlates with a decrease in ubiquitin-dependent proteasome activity. Accordingly, proteasome inhibition prevented ubiquitin-dependent aSN clearance. Ubiquitin-independent proteasome activity was positively correlated with ubiquitination in PBMC.
We also report a negative correlation of chymotrypsin-like activity with age in control and late-onset PD groups. Total ubiquitin content is positively correlated with aSN oligomers levels, which leads to an age-dependent increase of aSN ubiquitination in LOPD. Moreover, aSN levels are increased in the plasma of PD patients.
Interpretation
aSN oligomers are ubiquitinated and we identified an ubiquitin-dependent clearance insufficiency with accumulation of both aSN and ubiquitin. However, SH-SY5Y ndufa2 KD cells showed a significant up-regulation of ubiquitin-independent proteasomal enzymatic activity that could mean a cell rescue attempt. Moreover, we identified that UPS function is age-dependent in PBMC.
Keywords: Parkinson’s disease, Ubiquitin-proteasome system, Mitochondria, Alpha-synuclein, Ubiquitin
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder, characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the presence of ubiquitylated alpha-synuclein (aSN)-containing intracytoplasmic inclusions called Lewy bodies (LBs) in surviving SNpc neurons (Forno, 1996, Wichmann and DeLong, 2003). Although most forms of PD are idiopathic (late-onset, LOPD) (de Lau and Breteler, 2006), mutations in at least 10 different genes encoding proteins including aSN, parkin (an ubiquitin E3 ligase), DJ-1, PINK1 (PTEN-induced kinase 1), LRRK2 (Leucine-rich repeat kinase 2), and UCLH-1 (ubiquitin carboxyl terminal esterase L1), have been described in familial forms of PD (early-onset, EOPD) (Hatano, et al., 2009). The identification of these genes linked to the disease allowed us to propose that mitochondria and quality control systems have central role in PD pathophysiology (Arduino, et al., 2010). Indeed, it was reported a decrease in proteasomal function in the SNpc of sporadic PD (McNaught and Jenner, 2001) and an accumulation of ubiquitin in LBs in the locus coeruleus of PD (Galloway, et al., 1988, Manetto, et al., 1988). Although, the role of UPS dysfunction on the initiation or progression of the neurodegenerative process in PD remains to be established. Moreover, mitochondria has a key role in PD etiophatogenesis (Cardoso, 2011), since it is implicated in most genetic forms of familial PD and its deficiency was observed in most PD patients’ tissues, including the brain (Esteves, et al., 2011, Schapira, et al., 1989). Since, UPS-dependent protein degradation requires covalent attachment of the polyubiquitin chain to the target protein in an ATP-dependent manner to be rapidly degraded by the 26S proteasome, which need ATP to assemble from the 19S and 20S subunits, mitochondrial-driven ATP depletion, could potentiate UPS dysfunction in PD.
Thus, in this work we propose to evaluate how mitochondrial dysfunction inhibits ubiquitin-dependent aSN clearance and potentiates aSN aggregation through the direct study of proteasome activity. We found that mitochondrial deregulation triggered an 26S impairment, which induced an increase in total protein ubiquitination and alpha-synuclein oligomers. Proteasomal inhibition failed to increase ubiquitinated aSN in mitochondrial deficient cell line models, although promoted an increase in aSN oligomers in ndufa2 KD cell line with increased ATP-independent proteasomal activity. Our findings, suggest that cells harboring a mitochondrial dysfunction have a proteasomal proteolytic impairment, strengthening the connection between ubiquitin-dependent aSN clearance insufficiency and mitochondrial function.
Material and Methods
Chemicals, antibodies and cell media
Uridine (Urd) and lactacystin (C15H24N2O7S) were from Sigma Chemical Co (St. Louis, MO, USA). The flurimetric substrates N-Succinyl-Leu-Leu-Val-Tyr-AMC (Suc-LLVYAMC) was from Bachem (Bubendorf,Switzerland); Boc-Leu-Ser-Thr-Arg-AMC (Boc-LSTA-AMC) and Z-Leu-Leu-Glu-AMC (Z-LLG-AMC) were obtained from Peptide Institute(Japan). For western blotting analysis the following antibodies were used and the working dilutions are given in brackets: mouse mAb anti-GAPDH [1:2,500] was from Chemicon International; rabbit pAb anti-ubiquitin [SC-9133, (1:200)] was from Santa Cruz Biotechnology; mouse mAb anti-aSN [clone LB509, (1:100)] was from Invitrogen Corporation (Camarillo, CA, USA); mouse mAb anti-alpha-tubulin [1:10,000] was from Sigma and rabbit pAb anti-NDUFA2 [1:1000] was a generous gift of Dr. Leo Nijtmans (Radboud University Nijmegen Medical Center, The Netherlands). Alkaline phosphatase-conjugated secondary antibodies [1:15,000] were from GE Healthcare UK Limited (Buckinghamshire, UK). For aSN immunoprecipitation (IP) the following antibody was used: mouse mAb anti-aSN antibody [211 sc-12767] from Santa Cruz Biotechnology; aSN levels and ubiquitin co-immunoprecipitation were quantified in the immunoprecipitated samples by western blotting analysis using the following antibodies, respectively: mouse mAb anti-aSN antibody [211 sc-12767, (1:200)] from Santa Cruz Biotechnology and rabbit pAb anti-ubiquitin [SC-9133, (1:200)] was from Santa Cruz Biotechnology.
SH-SY5Y human neuroblastoma cells were cultured in Dulbecco’s modified Eagle’s medium F12 (DMEM F12) medium supplemented with 10% nondialyzed fetal bovine serum (FBS), 1.2g/L NaHCO3, 10ml/L penicillin/ streptomycin. SH-SY5Y human neuroblastoma ndufa2 KD cells were cultured in DMEM F12 medium supplemented with 10% nondialyzed FBS, 1.2g/L NaHCO3, 10ml/L penicillin/ streptomycin, 100mM sodium pyruvate and 75mg/ml Uridine.
NT2 Rho0 cells growth medium consisted of OPTIMEM supplemented with 10% heat inactivated FBS, 200 μg/ml sodium pyruvate, 150 μg/ml uridine and 10,000U/mL penicillin and 10μg/mL streptomycin. Cybrid cell lines selection medium consisted of DMEM, supplemented with 10% dialyzed FBS, 100 IU/ml penicillin, and 50 μg/ml streptomycin. Cybrid cell lines expansion medium consisted of OPTIMEM supplemented with 10% non-dialyzed FBS and 10,000U/mL penicillin and 10μg/mL streptomycin. Cybrid cell lines and NT2 cell line growth medium consisted of OPTIMEM medium supplemented with 10% heat inactivated FBS, 10,000 U/ml penicillin and 10μg/mL streptomycin.
Cell lines culture and treatments
SH-SY5Y human neuroblastoma cells (ATCC-CRL-2266) were obtained from ATCC (USA). The sequence for NDUFA2 siRNA was purchased from Invitrogen Online Ordering. The sequence was then cloned into lentiviral vector for siRNA pGreenPuro (System Biosciences) according to manufacturer’s instructions and further purified. The siRNA construct was packaged into pseudoviral particles tranduced into SH-SY5Y cells. Since infected cells stably express copGFP, as well as, the shRNA they were selected as green fluorescent protein (GFP) positive cells by FACS.
Human NT2 (Ntera2/D1) cells, a neuronally committed human teratocarcinoma cell line (Cardoso, et al., 2001, Pleasure and Lee, 1993, Sodja, et al., 2002) were purchased from Stratagene Cloning Systems (La Jolla, CA, USA).
SH-SY5Y NDUFA2 KD, wild-type SH-SY5Y and cybrids cell-lines were maintained at 37°C in a humidified incubator containing 95% air and 5% CO2.
Where indicated, 2μM of lactacystin was added in the culture medium 24 h after seeding the cells. For all experimental procedures, controls were performed in the absence of lactacystin. Incubations were performed for 6h for proteasome activity assay and for 12h to WB analysis and IP.
Generation of cybrid cell lines
Cybrid approach consists on the transfer of PD or healthy subjects’ platelet mitochondria to mtDNA-depleted recipient cells (NT2 Rho0 cells) generating hybrid cell lines (cybrids) (Esteves, et al., 2010). The resulting cybrid cell lines express the nuclear genes of the recipient Rho0 cell line and the mitochondrial genes of the platelet donor.
To generate cybrid cell lines for this study, we used a clonal stock of human teratocarcinoma cells containing no mtDNA (NT2- Rho0 cell line) created in NT2 cells by long-term exposure to 5 μg/mL ethidium bromide to deplete selectively mitochondrial DNA (mtDNA). Platelets (which contain mtDNA but not nDNA) from PD subjects are known to have reduced complex I activity relatively to control subjects’. We used platelet mitochondria to generate cybrid cell lines from both sPD and disease-free control subjects. Previously, platelets were isolated from the individual blood samples and then were fused with NT2-Rho0 cells by co-incubation in polyethylenoglycol (PEG) as previously described. The resulting cybrids were plated on T75 flasks, maintained for one week in Rho0 growth medium, and then switched to cybrids selection medium for 6 weeks. NT2 Rh0 cells lack intact mtDNA, do not possess a functional mitochondrial electron chain, and are auxotrophic for pyruvate and uridine. Maintaining cells in selection medium removes Rh0 cells that have not repopulated their mtDNA with platelet mtDNA. “Mock fusions”, in which NT2 Rho0 cells were not co-incubated with platelets, were performed in parallel with the proper fusions. During the selection period, all cells from the mock fusions died. After selection was complete, the cybrids were changed to cybrid expansion medium. Flasks were maintained in this medium at 37°C, 5% CO2 for 24 h prior to harvesting.
Human subjects
Subject participation was approved through the Institutional Review Board of the University Hospital of Coimbra. 26 PD patients, meeting diagnostic criteria (Hughes, et al., 1992), followed by the Movement Disorders Consulting of Neurology department of the University Hospital of Coimbra and 10 healthy, age-matched, volunteer individuals provided 10 ml blood samples after written informed consent, under the following exclusion criteria: Hepatic, Renal or Heart Failure, Severe Hypertension, Other Neurological Disease, Mini-Mental State Examination (MMSE) lower than 24, Cranial trauma in less than 6 months and anti-inflammatory, anti-neoplasic or immunosupressor drugs administration during the study. Blood was collected from the PD patients and from control individuals and drawn into a tube containing anticoagulant. PD patients samples were divided in two groups: LOPD group where age of onset was >50years and EOPD group where age of onset was <50years. For cybrids generation, 3 sPD and 2 age-matched control subjects underwent a 10 ml phlebotomy using tubes containing acid-citrate-dextrose, as an anticoagulant, to provide the platelets needed for cell fusions.
Isolation of PBMCs and Blood plasma
No later than 2h after drawing, 10ml of blood were carefully laid with Pasteur pipette over 8ml of histopaque (Sigma Aldrich, St. Louis, MO, USA) in a 50ml Falcon tube, avoiding mixing of blood and separation. The Falcon tube was centrifuged at 2,500rpm, 20min at 18°C in a swing-out rotor, without brake. After centrifugation, the mononuclear cells form a distinct band at the sample/medium interface and were removed without the upper layer of serum, using a Pasteur pipette. The harvested fraction was diluted in 45ml of phosphate-buffered saline (PBS) in a 50ml Falcon tube and centrifuged for 10min at 1,500rpm at 18°C. The supernatant was removed and the pellet resuspended in respective lysis buffer and further treated as cell culture extractions for fluorimetric proteasomal activity analysis and immunoblotting.
The blood plasma was collected after the first centrifugation into aliquots and centrifuged at 4,000rpm for 15minutes in order to sediment the platelets. Then, the plasma (supernatant) was collected and stored at −80°C and the platelets (pellet) were washed with 300μl of PBS. The centrifugation was repeated at 4,000rpm for 15min and the pellet was resuspended in 125μl of lysis buffer (0,25M Sacarose, 5mM Hepes, pH 7,4) and stored at −80°C. Plasma from human subjects’ blood samples was used in dot blot analysis.
Fluorimetric proteasomal activity analysis
To determine proteasome activity from the three individual cell models (SH-SY5Y ndufa2KD, PD Cybrids, PD patients’ PBMC and their respective control conditions) we used the method described by Domingues and colleagues (Domingues, et al., 2008) with modifications. Cellular extracts were incubated with proteasome activity buffer (0.5 mM EDTA and 50 mM Tris-HCl, pH 8) and 50 μM Suc-Leu-Leu-Val-Tyr-AMC, 100 μM Boc-Leu-Arg-Arg-AMC, or 400 μM Z-Leu-Leu-Glu-βNa, which were used as substrates to measure the chymotrypsin-like, trypsin-like, and peptidyl-glutamyl peptide hydrolytic-like (PGPH) proteolytic activities, respectively. ATP-dependent activity was measured supplementing the lysis buffer with 2 mM ATP. Data in the cell line models (SH-SY5Y ndufaKD, PD Cybrids and their respective control conditions) represents the difference between basal and post-6h lactacystin treatment for each condition. PBMC were not treated with lactacystin.
SDS-PAGE and Immunoblotting
To determine aSN oligomers and total protein ubiquitin conjugates, SH-SY5Y and cybrid cell lines were washed with PBS, scraped and lysed on ice in 1% Triton X-100 containing hypotonic lysis buffer (25mM HEPES, pH 7,5, 2mM MgCl2, 1mM EDTA and 1mM EGTA supplemented with 2mM DTT, 0,1mM PMSF and a 1:1000 dilution of a protease inhibitor cocktail). PBMC of PD patients and control individuals were resuspended in the same lysis buffer after separation. Cell suspensions were frozen three times in liquid nitrogen and centrifuged at 20,000g for 10min. The resulting supernatants were removed and stored at −80°C. Protein concentrations were determined by Bradford protein assay. For the analysis of ubiquitination levels and aSN aggregates in the three cell models, equal amounts of protein from Triton soluble fractions, were separated under reducing conditions on 7% or 10% SDS-PAGE gels, respectively. In the two cell line models 12h lactacystin treatment was performed to understand the influence of proteasome enzymatic inhibition in these protein levels. For the analysis of NDUFA2 protein a 10% SDS-PAGE gel was used.
After transfer to ImmobilonTM-P PVDF (polyvinylidene difluoride) membranes (Millipore, Billerica, MA, USA), the membranes were incubated for 1h in Tris-buffered saline (TBS) solution containing 0.1% Tween 20 and 5% nonfat milk or 5% bovine serum albumin (BSA) for aSN oligomers quantification, followed by an overnight incubation with the respective primary antibody at 4°C with gentle agitation. Membranes were further washed three times with TBS, 0.1% Tween and then incubated with the corresponding secondary antibody for 1h30min at room temperature. The membranes were washed again three times and bound antibodies detected using the enhanced chemifluorescence reagent ECF (Amersham Biosciences UK Limited, Buckinghamshire, UK) according to the manufacturer’s instructions. Blots were visualized using a VersaDoc imaging system (Bio-Rad, Hercules, CA, USA) and quantified using Quantity-One software (Bio-Rad, Hercules, CA, USA).
Immunoprecipitation assay
To determine aSN ubiquitination levels in SH-SY5Y and cybrid cell lines, cells were scraped and lysed on ice in lysis buffer [20 mM Tris-HCl (pH 7.0), 100 mM NaCl, 2 mM EDTA, 2 mM EGTA, supplemented with 0.1% SDS, 1% Triton X-100, 2 mM DTT, 0.1 mM PMSF and a 1:1000 dilution of a protease inhibitor cocktail)]. Cellular suspensions were centrifuged at 20,000 ×g, 10min at 4°C and whole lysates were assayed for protein concentration as described above. 500 μg of each sample were precleared with Protein A/G Sepharose beads (GE Healthcare Bio-Sciences, Uppsala, Sweden) for 1 h, 4°C, and then incubated with 2μg of primary antibody (mouse mAb anti-aSN antibody [211 sc-12767] from Santa Cruz Biotechnology), overnight at 4°C and with gentle agitation. Protein A/G Sepharose beads were then added to samples followed by 2 h incubation. The beads were spun down and supernatant was collected (INPUT). The beads were washed seven times in washing buffer [1% Triton X-100, 500 mM NaCl, 2 mM EDTA, 2 mM EGTA, 20 mM Tris-HCl (pH 7.0)]. The last supernatant was collected and 25 μl of 2x sample buffer was added. The samples were boiled at 95-100°C for 5 min to denature the protein and to separate it from the protein-A/G beads. The boiled proteins were centrifuged at 20,000 ×g for 5 min at room temperature and the supernatants collected. To evaluate aSN levels and ubiquitin co-precipitation, samples were separated by SDS–PAGE and subjected to Western blotting as aforementioned.
Dot Blot assay
To determine aSN levels in human subjects’ blood plasma a Dot Blot assay was done as previously described (Domingues, et al., 2008). Briefly, subjects’ serum was transferred to new tubes and kept at 4°C, until protein content was determined. Samples were then preserved at −80°C until assays were performed. PVDF membrane (Amersham Pharmacia Biotech) was placed on the top of the soaked sheets and equal amounts of protein in similar volume were put down in dots in specific zones. Once the dots were dried, nonspecific binding was blocked for 1 h at 4°C using 5% nonfat milk and 0.1% Tween 20 in Tris-buffered saline (TBS). Membranes were subjected to Western blotting as abovementioned.
Evaluation of mitochondrial respiratory chain NADH–Ubiquinone oxidoreductase and citrate synthase activities
The activities of mitochondrial NADH-Ubiquinone oxidoreductase (complex I: EC 1.6.99.3) and citrate synthase were determined as previously described (Esteves, et al., 2008).
MTT cell proliferation assay
Cell proliferation was determined by the colorimetric MTT assay previously described (Mosmann, 1983).
Data analysis
All data were expressed as mean ± SEM of at least two independent experiments and each experimental endpoint for each sample was run in duplicated. Experimental results were analyzed by Kolmogorov-smirnov normality test and depending on the result, p values were calculated by parametric or non-parametric distribution tests. One-way ANOVA or Kruskal-Wallis test, followed by a post hoc Bonferroni’s or Dunnet’s test, respectively, were used to compare multiple conditions. To compare two isolated conditions, unpaired t test or Mann-Whitney test were performed. Correlation studies were done using Pearson Correlation or Spearman Correlation test when appropriate. A p-value<0.05 was considered statistically significant.
Results
Mitochondrial-driven aSN oligomerization and accumulation in PD cellular models
As previously shown by our group, ETC CXI activity is reduced in platelets of PD patients and in PD Cybrids (Esteves, et al., 2008). We further characterized SH-SY5Y ndufa2 KD cells and observed that ETC CXI activity was reduced (p=0.0071) as compared to wild-type SH-SY5Y cells (Supplementary Figure 1A). This mitochondrial impairment was due to the decrease in ndufa2 gene expression in SH-SY5Y ndufa2 KD cells (p=0.0256) (Supplementary Figure 1B). Since, mitochondrial defects were associated with decreased ATP levels, increased free radical production, impaired mitochondrial calcium buffer, and aSN oligomerization (Esteves, et al., 2009, Esteves, et al., 2010, Esteves, et al., 2008), we further evaluated aSN conformational change in our in vitro, ex vivo and in vivo PD models harboring a CXI deficiency.
We observed that aSN oligomerization increased in SH-SY5Y ndufa2 KD when compared to respective parental cell-line (p=0.0007) (Figure 1A). Moreover, as it was previously shown by our group (Esteves, et al., 2010), there was an increased aSN oligomerization in PD cybrids (p=0.0485) (Figure 1B). In PBMC of PD patients we can just observe a tendency to an increased aSN oligomerization, probably due to high variability of human samples (Figure 1C). An important factor for aSN accumulation and aggregation is whether the protein degradation pathway is functioning properly. Since, aSN is known to be a target of proteasome degradation in the cytosol (Bennett, et al., 1999), we treated our in vitro and ex vivo models with lactacystin, a proteasome inhibitor, and observed an increase in aSN oligomerization in both in control and SHSH5Y ndufa2 KD and CT cybrids cells (Figure 1A and 1B). The concentration of lactacystin used did not reduce cell viability (Supplementary Figure 2).
Figure 1. aSN aggregation in PD cellular models.

(A) Densitometry analysis of triton-soluble aSN oligomers in SH-SY5Y ndufa2 KD cells and representative WB. N=3, ***p<0.001. (B) Densitometry analysis of triton-soluble aSN oligomers in PD Cybrids and representative WB. N=3, *p<0.05 (C) Densitometry analysis of triton-soluble aSN oligomers in PD patients PBMC and representative WB.
UPS reply to aSN buildup in PD cellular models
Quality control systems have a central role in PD pathophysiology. Indeed, proteasomal dysfunction in the substantia nigra in sporadic PD has been reported (McNaught and Jenner, 2001). Therefore, aSN aggregation could be the consequence of proteasomal impairment or instead can directly induce proteasome dysfunction (Chen, et al., 2006), which could lead to a toxic vicious cycle.
In order to delineate a relationship between mitochondrial deficits, proteasomal dysfunction and aSN aggregation we evaluated UPS function in our cellular PD models. Although ndufa2 KD cells have a deficient ETC, ATP-dependent chymotrypsin-like activity was found unchanged and surprisingly ATP-independent chymotrypsin-like activity increased in ndufa2 KD cells (p=0.0207). Despite of this, the levels of total protein ubiquitination and ubiquitin monomer were increased (Figure 2). Lactacystin, by inhibiting ATP-dependent and independent proteasome activity, induced an increase in the accumulation of both ubiquitinated species and ubiquitin monomer (Figure 2B, C, D). Our results in this in vitro model may indicate that despite chymotrypsin-like site is the rate limiting site (Chen, et al., 2006), other proteasome-like activities may be impaired and responsible for the decrease in the degradation of ubiquitinated proteins processed via the proteasome.
Figure 2. UPS function in SH-SY5Y ndufa2 KD cells.

(A) Proteasome 26S and 20S chymotryosin-like activity. N=4, *p<0.05. (B) Densitometry analysis of total ubiquitinated protein content. N=3, p=0,0534 (C) Densitometry analysis of ubiquitin monomer. N=3, *p<0.05. (D) Representative WB of ubiquitinated proteins in SH-SY5Y control and ndufa2 KD cell lines under basal and lactacystin-treated conditions.
In PD cybrids, we observed an increase in protein ubiquitination levels (p=0.0044), as it was previously showed by our group (Esteves, et al., 2010) (Figure 3B, C). These results were due to a decrease in ATP-dependent trypsin-like activity in PD cybrids (p=0.0438). Interestingly, in our ex vivo model, we could not observe an up-regulation of ATP-independent (20S) proteasomal function, like previously described for ndufa2 KD cells. Lactacystin treatment promoted a non significant accumulation of ubiquitinated species (Figure 3B, C).
Figure 3. UPS function in PD Cybrids.

(A) Proteasome 26S and 20S chymotryosin-like; trypsin-like and PGPH activities. N=4, *P<0.05. (B) Densitometry analysis of total ubiquitinated protein content. N=3, **p<0.01. (C) Representative WB of ubiquitinated proteins in CT and PD cybrid cell lines under basal and lactacystin-treated conditions.
Regarding the study of PBMC of PD patients we observed a significant difference between means (p=0.0362). Interestingly, we detected an increase of proteasome activities in younger groups, in both CT and PD cells, versus older individuals’, being this effect greater for ATP-dependent chymotrypsin-like activity (Figure 4A).
Figure 4. UPS function in PBMC of LOPD and EOPD patients.

(A) Proteasome 26S and 20S chymotrypsin-like activity. *p<0,05 (B) Densitometry analysis of total ubiquitinated protein content and representative WB. (C) Ubiquitination influence on proteasome activity in LOPD patients: 26S chymotrypsin-like activity do not correlated with ubiquitinated protein content. N=13, Pearson r=0.2840, p=0.3470, r2=0.08066; 20S chymotrypsin-like activity has a significant positive correlation with ubiquitinated protein content. N=13, Pearson r=0.6486, *P<0,05, r2=0,4207.
Regardless of great variability, we can see an increase in the mean of ubiquitination levels in both disease groups when compared with their counterparts, similarly to what we observed in our other PD cellular models. There were also increased protein ubiquitination levels in younger individuals group (Figure 4B). Ubiquitination levels have a significant positive correlation with 20S chymotrypsin-like activity in LOPD group (p=0,0165) (Figure 4C). This positive correlation between ATP-independent proteasome activity and ubiquitination, allow us to hypothesize that UPS-dependent protein degradation is impaired in PBMC of PD patients.
To further correlate UPS impairment with aSN aggregation and accumulation we decided to determine the levels of ubiquitinated aSN. In ndufa2 KD cells we observed an increase in the levels of ubiquitinated aSN as compared to parental cells (p=0.0157) (Figure 5A). Interestingly, lactacystin potentiated the accumulation of ubiquinated aSN in parental cells (p=0.0325), but failed to do so in ndufa2 KD cells, which indicates a previous impairment of UPS.
Figure 5. Ubiquitinated aSN in PD cell-line models.

(A) Densitometry analysis of the ratio between ubiquitinated aSN and aSN after aSN IP in SH-SY5Y ndufa2 KD; and representative WB. SH-SY5Y ndufa2 KD cells show an increased amount of ubiquitinated aSN compared to parental cell-line. Lactacystin induced an increase in aSN ubiquitination in parental cells. N=2 *p<0.05. (B) Densitometry analysis of ratio between ubiquitinated aSN and aSN after aSN IP in PD Cybrids and representative WB. PD cybrids show an increase in the amount of ubiquitinated aSN compared to CT cybrids. Lactacystin promote the accumulation of ubiquitinated aSN in CT cells. N=2, p<0.05. p<0,05. (C) aSN oligomers levels have a positive correlation with total ubiquitination in PBMC of PD patients. N=11, Pearson r=0.6924, *p<0.05, r2=0.04795.
We observed identical findings in PD cybrids, where the levels of ubiquitinated aSN were increased in PD cells (p=0.0044) (Figure 5B). Moreover, similarly to what we observed in ndufa2 KD cells, lactacystin was able to promote the accumulation of ubiquitinated aSN only in CT cells (p=0.0277). Again, PD cybrids with dysfunctional mitochondria also have an impaired UPS that may precede aSN ubiquitination, since lactacystin failed to potentiate the build-up of these aSN species.
In PBMC of PD patients we evaluated the interplay between UPS and aSN accumulation by determining a statistically significant positive correlation between aSN and total ubiquitin content (p=0,0182) (Figure 5C)
Since we observed in PBMC of PD patients a tendency for aSN accumulation we quantified the levels of aSN in the plasma by dot blot. We observed an increase in aSN secretion in the both groups, although we only obtained statistical significance in the EOPD group compared to the respective age-matched control group (p=0,0111) (Figure 6). Our data allow us to hypothesize that proteasomal dysfunction, a key feature of PD pathogenesis, occurs downstream mitochondrial dysfunction and could be due to mitochondrial-dependent ATP depletion and/or oxidative stress.
Figure 6. aSN quantification in the plasma of PD patients.

Densitometry analysis of aSN levels in the plasma of PD patients and representative dot blot. There is an increase in the amount of aSN in the plasma of PD patients, which is significative in EOPD. N=4-10. *P<0,05.
Correlation studies in PBMC PD model
Due to high variability observed in the previous results with PBMC model, some correlation studies were performed in order to better understand the influence of some demographic characteristics of patients’ population (Table 1).
Table 1.
Demographic characteristics of patient population
| Gender | Duration of L- DOPA treatment |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Condition group |
N | ♂ | ♀ | Age | Age of diagnostic |
Duration of disease |
UPDRS III |
MMSE | |
| CT (LOPD) |
6 | 2 | 4 | 65,17±3,31 | |||||
| LOPD | 14 | 9 | 5 | 74,295±7,39 | 64,64±10,2 | 9,64±7,75 | 7,27±6,10 | 45±9,06 | 26,08± 2,29 |
| CT (EOPD) |
4 | 2 | 2 | 54,75±3,86 | |||||
| EOPD | 6 | 2 | 4 | 58,83±3,19 | 47,17±1,47 | 11,67±2,42 | 10,5±4,32 | 44,5± 27,58 |
26,83± 0,41 |
We previously have shown that younger individuals seem to have higher chymotrypsin-like activities (20S and 26S), herein we observed a negative correlation between age of individuals and 26S and 20S chymotrypsin-like activities, in both control individuals (p=0.0307) (Figure 7A) and LOPD patients (p=0.0067) (Figure 7B, C). 20S chymotrypsin-like activity in EOPD patients had a positive correlation tendency (Figure 7C). The other demographic features were accessed but there were neither significant correlations nor strong associations (data not shown).
Figure 7. Correlation between Age and Chymotrypsin-like activity in PBMC.
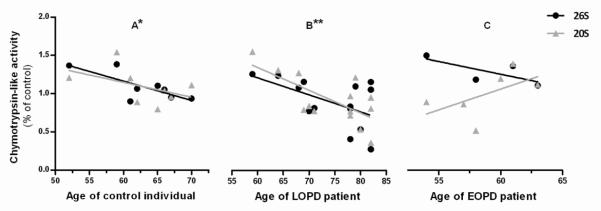
(A) In control individuals 26S chymotrypsin-like activity has a significant negative correlation with age in control indivuduals. N=8, Pearson r=−0,754, *p<0,05, r2=0,5686 and 20S chymotrypsin-like activity isnegatively correlated with age. N=8, Pearson r=−0,468, p=0,2422, r2=0,219 (B) In LOPD patients 26S chymotrypsin-like activity is negatively correlated with age. N=14, Pearson r=−0,5275, p=0,0526, r2=0,2783; 20S chymotrypsin-like activity has a significant negative correlation with age. N=14, Pearson r = −0,6864, **p<0,01, r2=0,4712 (C) In EOPD patients 26S chymotrypsin-like activity is negatively correlated with age. N=4, Pearson r=−0,7365, p=0,2635, r2=0,5424 and 20S chymotrypsin-like activity is positively correlated with age. N=6, Pearson r=0,5648, p=0,2429, r2=0,3190.
Concerning to ubiquitination levels, age at the time of participation in the study is again the independent variable from demographic characteristics that has stronger impact. Although there is no influence of this variable in the amount of ubiquitinated species and aSN oligomers on healthy individuals, there is a negative correlation between age and ubiquitination levels in LOPD group of patients (p=0,0114) (Figure 8A). aSN/ubiquitin ratio is not correlated with duration of disease but is positively correlated with age, with an exponential nonlinear fit (p=0,0041) (Figure 8C and D). Interestingly, ubiquitination has a positive correlation tendency with duration of disease (Figure 8B). Even though, aSN oligomers levels remain unchangeable, just with a very small positive slope in the linear regression, both depending on age or duration of disease. The other demographic features were accessed but there were neither significant correlations nor strong associations (data not shown).
Figure 8. Correlations studies between demographic characteristics and Ubiquitination or aSN oligomers.

(A) In LOPD patients ubiquitination has a significantly negative correlation with age. N=13, Pearson r=−0,6748, *p<0,05, r2=0,4553; aSN has a very low and weak positive correlation with age. N=13, Pearson r=0,2511, p=0,4079, r2=0,06306 (B) In LOPD patients ubiquitination has a positive correlation with duration of disease. N=10, Pearson r=0,6058, p=0,0634, r2=0,3670; aSN has a very low and weak positive correlation with duration of the disease. N=10, Pearson r=0,2214, p=0,5387, r2=0,04902 (C) In LOPD patients aSN/ubiquitin ratio has a positive correlation with age. N=13, Pearson r=0,736, **p<0,01, r2=0,5417 (D) In LOPD patients aSN/ubiquitin ratio has a very weak negative correlation with duration of disease. N =10, Pearson r=0,4997, p=0,1414, r2=0,2497.
Discussion
The most direct evidence indicating a key role of mitochondria in PD pathogenesis comes from studies in idiopathic PD patients where a deficiency of ETC CX I activity was observed in platelets, PBMC, skeletal muscle and brain (Esteves, et al., 2011, Schapira, et al., 1989). Moreover, CXI inhibitors induce parkinsonism in humans, non-human primates and rodents (Langston, et al., 1983). Remarkably, mitochondria are also implicated in most genetic forms of familial PD (Cardoso, 2011). Our study directly addresses the consequences of defects on mitochondrial function on the UPS and aSN oligomerization in PD. We used different PD cellular models with dysfunctional mitochondria, such as, cells that were genetically depleted of CXI subunit ndufa2 (in vitro model), cells that carry mtDNA from PD patients, PD cybrids (ex vivo model) and PBMC of PD patients (in vivo model) to validate our results.
Protein aggregation has also a role in both familial and sporadic PD pathogenic process (Esteves, et al., 2011). Indeed, the presence LBs, composed of aSN, parkin, ubiquitin, synphilin-1, tubulin and other cytoskeletal proteins, in surviving SNpc neurons, is a PD neuropathological feature (Esteves, et al., 2011). We previously showed that a mitochondrial dysfunction induces aSN oligomerization, via ATP depletion–driven microtubule depolymerization and via ROS increase–driven protein oxidation (Esteves, et al., 2009, Esteves, et al., 2010). Moreover, it is known that aSN aggregation process involves degradative mechanisms, such as, autophagy and UPS (Arduino, et al., 2011). Our results show that aSN soluble oligomers build-up in mitochondrial-deficient PD cells, and that proteasomal inhibition potentiated this effect in controls of both in vitro and ex vivo models, but failed to do so in PD cybrids. Evidences exist that aSN is primarily degraded by the proteasome, although its mutant forms can compromise proteasomal function, leading to further accumulation of misfolded aSN and other proteins (Esteves, et al., 2011, Webb, et al., 2003). Moreover, the existence of EOPD forms caused by mutations in genes that codify proteins of the proteasome pathway, the co-localization of proteasome subunits in LBs (Ii, et al., 1997), the presence of ubiquitinated proteins in LBs and proteasomal dysfunction in the SN of LOPD (McNaught and Jenner, 2001) indicate an UPS involvement in PD. Data from the literature shows that transgenic mouse models for aSN have defective proteasome function (Chen, et al., 2006). Moreover, mutant aSN expression significantly reduced proteasomal activities such as, chymotrypsin-like, trypsin-like and PGPH. In these cells, lactacystin induced increase sensitivity to aSN-induced toxicity (Tanaka, et al., 2001). In order to disclosure if mitochondrial dysfunction induces aSN aggregation due to proteasomal impairment, we evaluated UPS function in our PD models. We observed an increase in total protein ubiquitination levels in all PD cellular models, but no decrease in chymotrypin-like activity. Although chymotrypsin-like site is thought to be the rate limiting proteasomal catalytic activity and its impairment would lead to the accumulation of both ubiquitinated and non-ubiquitinated proteins, the inhibition of trypsin-like and/or PGPH proteasomal activities may also decrease protein degradation via the proteasome (Davies, 2001). We observed a decrease in trypsin-like activity in PD cybrids, both dependent and independent of ATP. Moreover, PGPH activity was also decrease in these cells in an ATP-independent manner. Reduced activity of 20S element is likely to contribute to an increase in the quantity of oxidized damaged proteins in the cell (Davies, 2001). Highly oxidative intracellular environment due to mitochondrial dysfunction, increases DA metabolism and can compromise the integrity of vulnerable DAergic neurons, thus contribute to neuronal degeneration (Cardoso, et al., 2009, Ciechanover and Brundin, 2003, Goldberg, 2003). This is a point of intersection between mitochondria and UPS function, since mitochondrial dysfunction, producing excessive ROS, may induce protein oxidation, which affects proteasomal activity. Additionally, ATP reduction may compromise protein degradation by the UPS in ATP-dependent processes, like ubiquitin tagging by ubiquitin ligases E3 and the assemble of 26S subunits, 19S and 20S (Goldberg, 2003). Indeed, it was reported that 26S proteasome is more sensitive to oxidative stress than 20S proteasome (Reinheckel, et al., 2000). Under our conditions of elevated oxidative stress and ATP decrease, only 20S proteasome would be able to degrade oxidized proteins in an ubiquitin-independent way. In fact, it was shown that levels of 20S proteasome subunits were elevated upon H2O2 stimulation (Godon, et al., 1998). Accordingly, we observed an increase of ATP-independent proteasome enzymatic activity in ndufa2 KD cells followed by increased levels of total ubiquitin content, and a positive correlation between total ubiquitination content and 20S chymotrypsin-like activity in LOPD. These results indicate that an UPS dysfunction may up-regulate an ATP-independent proteasome activation, that is not enough to avoid aSN oligomerization as we can observe aSN accumulation in ndufa2 KD.
Hence, these studies suggest that UPS impairment can occur as a consequence of mitochondrial dysfunction in PD. Accordingly with our results showing that chymotrypsin-like activity decreases with age in PBMC of both control individuals and LOPD patients, other groups demonstrated an UPS loss of function with aging, reflected on the decrease in the expression of proteasome subunits, activity and response to oxidative stress (Bulteau, et al., 2000, Keller, et al., 2000). Furthermore, we showed an age-dependent decrease of total ubiquitin content as well as exponential increase of aSN/ubiquitin ratio in LOPD patients, consistent with an UPS dysfunction. A recent study showed a decrease in E2 levels and proteasomal activity but no alterations in total ubiquitin and E1 levels in PBMC of PD patients (Ullrich, et al., 2010).
UPS dysfunction triggered by an impaired mitochondria correlates positively with an increase in the accumulation of ubiquitinated aSN in ndufa2 KD and PD cybrids, being also suggestive in PBMC of LOPD. aSN oligomers formation could be probably explained by an insufficient clearance due to increased formation or by deficits in protein tagging and/or ubiquitin recognition that are ATP-dependent processes. Moreover, when we treated ndufa2 KD and PD cybrids with proteasome catalytic activity inhibitor, lactacystin, we did not observed an increase in the levels of aSN ubiquitinated species, as observed in the parental and CT cybrid cells. Despite this, an increase in total protein ubiquitination occurs in ndufa2 KD cells. Our hypothesis is compatible with a direct effect of dysfunctional mitochondria on UPS ability to degrade misfolded aSN. Our results can be correlated with a predominant ATP-independent degradation pathway in ndufa2 KD cells, since 20S proteasome inhibition increased aSN aggregation in these cells, but failed to boost aSN ubiquitination. In PD cybrids, a decrease in ATP levels (Esteves, et al., 2008) and increased ROS production (Esteves, et al., 2009) induces an UPS dysfunction that potentiates aSN oligomerization. Further inhibition of proteasomal proteolysis (20S plus 26S) did not increase total ubiquitination, aSN ubiquitination or aggregation, which indicates that UPS deregulation is an upstream event. Our previous study (Esteves, et al., 2008) showed that PD cybrids have increased levels of oxidatively modified aSN, which is known to potentiate its oligomerization process (Ono and Yamada, 2006). Considering our and others studies, there are several lines of evidence that suggest a cross-talk between mitochondria and UPS in PD (Branco, et al., 2010). Some authors claim that mitochondrial compromise is the primary event followed by proteasome impairment and consequent aSN aggregation. However, it was reported that proteasome inhibition leads to the accumulation of polyubiquitinated proteins in the mitochondria, which activates mitochondrial apoptosis in dopaminergic neuronal cells (Sun, et al., 2009). Recently, it was shown that the accumulation of monoubiquitinated forms of aSN protein promoted subsequent aggregation (Rott, et al., 2008). Soluble misfolded monomers and dimers can be recognized and degraded by the UPS but macroautophagy is the only mechanism available to clear the more insoluble and highly ordered aggregates (oligomer or fibrils). Indeed, our group also showed that macroautophagy is impaired in PD cybrid and PBMC cells due to mitochondrial-mediated intracellular traffic deficits (Arduíno, et al., in press).
Considering that aSN oligomerizes due to an increase in oxidative stress (aSN oxidized) and due to an inefficient degradation, by the UPS (aSN monoubiquitinated and poliubiquitinated) or by macroautophagy (aSN oligomers), its secretion through cell membranes to extracellular space can be a protective strategy. Our results showed a tendency to increased levels of aSN in plasma of patients, mainly in those suffering from EOPD. This probably represents a cellular mechanism to avoid soluble oligomeric aSN toxicity and it could be of great interest if we can understand that this is an early process in aging and disease progression.
Conclusions
Based on our data we propose that an impairment of mitochondrial function leads to the depletion of ATP levels and to an increase in the production of ROS. These mitochondrial induced perturbation lead to a decrease in 26S proteasomal function and to an increase in aSN oxidation. Moreover, aSN oxidation and partially ubiquitinated soluble aSN promotes aSN oligomerization. aSN oligomers may themselves potentiate 20S proteasomal inhibition and increase mitochondrial deficits in what is usually called a toxic feed-back loop. Under these conditions, macroautophagy is activated to degrade aSN oligomeric toxic species (aggrephagy) and dysfunctional mitochondria (mitophagy). In a PD context, and due to an impaired microtubular network, autophagic clearance is also inhibited, which potentiates the neurodegenerative process.
Our findings strongly support that UPS is involved in the age dependent mechanism of disease progression, so recognition of mitochondrial and UPS interplay may open a new window to PD therapeutics.
Supplementary Material
Supplementary Figure 1. Mitochondrial characterization of NDUFA KD cells. (A) ETC CXI activity in SH-SY5Y ndufa2 KD. There is a reduction in CXI activity in SHSY5Y ndufa2 KD cells. N=2, **p<0,01. (B) WB and respective densitometry of protein ndufa2 in SH-SY5Y ndufa2 KD cells. SH-SY5Y ndufa2 KD cells show reduced amount of ndufa2 protein as expected. N=3, *p<0.05
Supplementary Figure 2. Effect of lactacystin on cell proliferation. (A) MTT reduction ability of SH-SY5Y and ndufa2 KD cells cells; (B) MTT reduction ability of cybrid cells; Lactacystin concentration used did not affect viability in both cell-line models. N=3.
Highlights.
Mitochondria dysfunction trigger Ubiquitin-Protesome System impairment in PD models
PD models show increased total protein ubiquitination and alpha-synuclein levels
Ubiquitinated aSN is increased, particularly in the in vitro and ex vivo models
Proteasome inhibition fail to increase ubiquitinated aSN in these cell-line models
ATP-independent proteasomal activity is increased in the in vitro PD model
Acknowledgements
The authors would like to acknowledge the Neurology internist Fradique Moreira, MD, who contributed with a great help to this work and to Isabel Nunes, PhD, for cell culture support. Diogo Martins-Branco, A. Raquel Esteves and Daniela M. Arduino were supported by Fundação para a Ciência e a Tecnologia, Portugal (BII, Pos-Doc and PhD grants, respectively). This work was supported by PTDC/SAU-NEU/102710/2008, FCT and by GAPI of Faculdade de Medicina da Universidade de Coimbra, Portugal. Russell H Swerdlow is supported by P30AG035982 to the KU ADC.
Abbreviations
- ANOVA
Analysis of variance
- aSN
Alpha-synuclein
- ATP
Adenosine-5′-triphosphate
- BSA
Bovine serum albumin
- CT
Control
- CXI
Complex I
- DA
Dopamine
- DMEM F12
Dulbecco’s Modified Eagle Medium Nutrient Mixture F-12
- DTT
Dithiothreitol
- EDTA
Ethylenediamine tetraacetic acid
- EGTA
Ethylene glycol tetraacetic acid
- EOPD
Early on-set Parkinson’s Disease
- ETC
Mitochondrial Electron Transport Chain
- FACS
Fluorescence-activated cell sorting
- FBS
Fetal Bovine Serum
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GFP
Green fluorescent protein
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IP
Immunoprecipitation
- KD
Knock-down
- LBs
Lewy Bodies
- LOPD
Late on-set Parkinson’s Disease
- LRRK2
Leucine-rich repeat kinase 2
- MD
Mitochondrial Disorder
- MMSE
Mini-Mental State Examination
- mtDNA
Mitochondrial DNA
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- nDNA
nuclear DNA
- NADH
Nicotinamide adenine dinucleotide
- PBMC
Peripheral Blood Mononuclear Cells
- PBS
Phosphate-Buffered Saline
- PD
Parkinson’s Disease
- PEG
Polyethylenoglycol
- PGPH
Peptidyl-glutamyl peptide hydrolytic
- PINK1
PTEN-induced kinase 1
- PMSF
Phenylmethanesulfonylfluoride
- PVDF
Polyvinylidene difluoride
- ROS
Reactive oxygen species
- SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEM
Standard error of the mean
- SN
Substantia Nigra
- SNpc
Substantia Nigra pars compacta
- TBS
Tris-buffered Saline
- UCLH-1
Ubiquitin carboxyl terminal esterase L1
- UPDRS
Unified Parkinson’s Disease Rating Scale
- UPS
Ubiquitin-Proteasome System
- WB
Western Blot
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Potential Conflicts of Interest Nothing to report.
References
- 1.Arduino DM, Esteves AR, Cardoso SM. Mitochondrial fusion/fission, transport and autophagy in Parkinson’s disease: when mitochondria get nasty. Parkinsons Dis. 2011;2011:767230. doi: 10.4061/2011/767230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arduíno DM, Esteves AR, Cortes L, Silva DFF, Patel B, Grazina MM, Swerdlow RH, Oliveira CR, Cardoso SM. Mitochondrial Metabolism in Parkinson’s Disease Impairs Quality Control Autophagy by Hampering Microtubule-Dependent Traffic. Human Molecular Genetics. doi: 10.1093/hmg/dds309. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arduino DM, Esteves AR, Oliveira CR, Cardoso SM. Mitochondrial metabolism modulation: a new therapeutic approach for Parkinson’s disease. CNS Neurol Disord Drug Targets. 2010;9:105–119. doi: 10.2174/187152710790966687. [DOI] [PubMed] [Google Scholar]
- 4.Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of alpha-synuclein by proteasome. J Biol Chem. 1999;274:33855–33858. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- 5.Branco DM, Arduino DM, Esteves AR, Silva DF, Cardoso SM, Oliveira CR. Cross-talk between mitochondria and proteasome in Parkinson’s disease pathogenesis. Front Aging Neurosci. 2010;2:17. doi: 10.3389/fnagi.2010.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bulteau AL, Petropoulos I, Friguet B. Age-related alterations of proteasome structure and function in aging epidermis. Exp Gerontol. 2000;35:767–777. doi: 10.1016/s0531-5565(00)00136-4. [DOI] [PubMed] [Google Scholar]
- 7.Cardoso SM. The mitochondrial cascade hypothesis for Parkinson’s disease. Curr Pharm Des. 2011;17:3390–3397. doi: 10.2174/138161211798072508. [DOI] [PubMed] [Google Scholar]
- 8.Cardoso SM, Esteves AR, Arduíno DM, Domingues AF, Oliveira CR. The crucial role of mitochondria in Parkinson’s disease. Recent Res. Devel. Neurosci. 2009;3:43–84. [Google Scholar]
- 9.Cardoso SM, Santos S, Swerdlow RH, Oliveira CR. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. Faseb J. 2001;15:1439–1441. doi: 10.1096/fj.00-0561fje. [DOI] [PubMed] [Google Scholar]
- 10.Chen L, Thiruchelvam MJ, Madura K, Richfield EK. Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol Dis. 2006;23:120–126. doi: 10.1016/j.nbd.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 12.Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 13.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 14.Domingues AF, Arduino DM, Esteves AR, Swerdlow RH, Oliveira CR, Cardoso SM. Mitochondria and ubiquitin-proteasomal system interplay: relevance to Parkinson’s disease. Free Radic Biol Med. 2008;45:820–825. doi: 10.1016/j.freeradbiomed.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Esteves AR, Arduino DM, Silva DF, Oliveira CR, Cardoso SM. Mitochondrial Dysfunction: The Road to Alpha-Synuclein Oligomerization in PD. Parkinsons Dis. 2011;2011:693761. doi: 10.4061/2011/693761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteves AR, Arduino DM, Swerdlow RH, Oliveira CR, Cardoso SM. Oxidative stress involvement in alpha-synuclein oligomerization in Parkinson’s disease cybrids. Antioxid Redox Signal. 2009;11:439–448. doi: 10.1089/ars.2008.2247. [DOI] [PubMed] [Google Scholar]
- 17.Esteves AR, Arduino DM, Swerdlow RH, Oliveira CR, Cardoso SM. Dysfunctional mitochondria uphold calpain activation: contribution to Parkinson’s disease pathology. Neurobiol Dis. 2010;37:723–730. doi: 10.1016/j.nbd.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Esteves AR, Arduino DM, Swerdlow RH, Oliveira CR, Cardoso SM. Microtubule depolymerization potentiates alpha-synuclein oligomerization. Front Aging Neurosci. 2010;1:5. doi: 10.3389/neuro.24.005.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esteves AR, Domingues AF, Ferreira IL, Januario C, Swerdlow RH, Oliveira CR, Cardoso SM. Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion. 2008;8:219–228. doi: 10.1016/j.mito.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Galloway PG, Grundke-Iqbal I, Iqbal K, Perry G. Lewy bodies contain epitopes both shared and distinct from Alzheimer neurofibrillary tangles. J Neuropathol Exp Neurol. 1988;47:654–663. doi: 10.1097/00005072-198811000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Godon C, Lagniel G, Lee J, Buhler JM, Kieffer S, Perrot M, Boucherie H, Toledano MB, Labarre J. The H2O2 stimulon in Saccharomyces cerevisiae. J Biol Chem. 1998;273:22480–22489. doi: 10.1074/jbc.273.35.22480. [DOI] [PubMed] [Google Scholar]
- 23.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 24.Hatano T, Kubo S, Sato S, Hattori N. Pathogenesis of familial Parkinson’s disease: new insights based on monogenic forms of Parkinson’s disease. J Neurochem. 2009;111:1075–1093. doi: 10.1111/j.1471-4159.2009.06403.x. [DOI] [PubMed] [Google Scholar]
- 25.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ii K, Ito H, Tanaka K, Hirano A. Immunocytochemical co-localization of the proteasome in ubiquitinated structures in neurodegenerative diseases and the elderly. J Neuropathol Exp Neurol. 1997;56:125–131. doi: 10.1097/00005072-199702000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Keller JN, Hanni KB, Markesbery WR. Possible involvement of proteasome inhibition in aging: implications for oxidative stress. Mech Ageing Dev. 2000;113:61–70. doi: 10.1016/s0047-6374(99)00101-3. [DOI] [PubMed] [Google Scholar]
- 28.Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 29.Manetto V, Perry G, Tabaton M, Mulvihill P, Fried VA, Smith HT, Gambetti P, Autilio-Gambetti L. Ubiquitin is associated with abnormal cytoplasmic filaments characteristic of neurodegenerative diseases. Proc Natl Acad Sci U S A. 1988;85:4501–4505. doi: 10.1073/pnas.85.12.4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McNaught KS, Jenner P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci Lett. 2001;297:191–194. doi: 10.1016/s0304-3940(00)01701-8. [DOI] [PubMed] [Google Scholar]
- 31.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 32.Ono K, Yamada M. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J Neurochem. 2006;97:105–115. doi: 10.1111/j.1471-4159.2006.03707.x. [DOI] [PubMed] [Google Scholar]
- 33.Pleasure SJ, Lee VM. NTera 2 cells: a human cell line which displays characteristics expected of a human committed neuronal progenitor cell. J Neurosci Res. 1993;35:585–602. doi: 10.1002/jnr.490350603. [DOI] [PubMed] [Google Scholar]
- 34.Reinheckel T, Ullrich O, Sitte N, Grune T. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Arch Biochem Biophys. 2000;377:65–68. doi: 10.1006/abbi.2000.1717. [DOI] [PubMed] [Google Scholar]
- 35.Rott R, Szargel R, Haskin J, Shani V, Shainskaya A, Manov I, Liani E, Avraham E, Engelender S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem. 2008;283:3316–3328. doi: 10.1074/jbc.M704809200. [DOI] [PubMed] [Google Scholar]
- 36.Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet. 1989;1:1269. doi: 10.1016/s0140-6736(89)92366-0. [DOI] [PubMed] [Google Scholar]
- 37.Sodja C, Fang H, Dasgupta T, Ribecco M, Walker PR, Sikorska M. Identification of functional dopamine receptors in human teratocarcinoma NT2 cells. Brain Res Mol Brain Res. 2002;99:83–91. doi: 10.1016/s0169-328x(01)00324-2. [DOI] [PubMed] [Google Scholar]
- 38.Sun F, Kanthasamy A, Anantharam V, Kanthasamy AG. Mitochondrial accumulation of polyubiquitinated proteins and differential regulation of apoptosis by polyubiquitination sites Lys-48 and -63. J Cell Mol Med. 2009;13:1632–1643. doi: 10.1111/j.1582-4934.2009.00775.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, V LD, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- 40.Ullrich C, Mlekusch R, Kuschnig A, Marksteiner J, Humpel C. Ubiquitin enzymes, ubiquitin and proteasome activity in blood mononuclear cells of MCI, Alzheimer and Parkinson patients. Curr Alzheimer Res. 2010;7:549–555. doi: 10.2174/156720510792231766. [DOI] [PubMed] [Google Scholar]
- 41.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 42.Wichmann T, DeLong MR. Functional neuroanatomy of the basal ganglia in Parkinson’s disease. Adv Neurol. 2003;91:9–18. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Mitochondrial characterization of NDUFA KD cells. (A) ETC CXI activity in SH-SY5Y ndufa2 KD. There is a reduction in CXI activity in SHSY5Y ndufa2 KD cells. N=2, **p<0,01. (B) WB and respective densitometry of protein ndufa2 in SH-SY5Y ndufa2 KD cells. SH-SY5Y ndufa2 KD cells show reduced amount of ndufa2 protein as expected. N=3, *p<0.05
Supplementary Figure 2. Effect of lactacystin on cell proliferation. (A) MTT reduction ability of SH-SY5Y and ndufa2 KD cells cells; (B) MTT reduction ability of cybrid cells; Lactacystin concentration used did not affect viability in both cell-line models. N=3.