

The bone marrow failure (BMF) syndromes are a collection of heterogeneous disorders characterized by failure to produce blood cells of one or more lineage. Aplastic anemia (AA) is a rare, life threatening disorder defined by pancytopenia and a hypocellular bone marrow. Approximately 75% of cases are considered idiopathic (IAA), whereby no family history is observed and the cause remains unexplained.1 In some cases, however, AA patients have been found to harbor mutations in genes encoding the RNA or reverse transcriptase component of telomerase (TERC and TERT, respectively).2,3 Here we performed targeted resequencing of 52 genes associated with 11 distinct BMF syndromes in 45 patients with AA (known to be normal for TERC) that was seemingly idiopathic in nature (patients had no extra-hematopoietic features, family history or stigmata of known inherited BMF syndromes and normal chromosomal breakage scores). Through this systematic approach we sought to verify whether a subset of patients had occult genetic variants causing or predisposing to disease. DNA samples were prepared using a TruSeq Custom Amplicon Kit and sequenced on a MiSeq Benchtop Sequencer (Illumina). The success of the method was confirmed by 3 positive control samples. A patient with AA, previously identified with the TERC variant r.95G>C, was included in the screen and this variant was successfully called by the on-system program MiSeq reporter. Furthermore, we identified disease-causing variants in 2 previously genetically uncharacterized BMF syndrome patients. In a Fanconi anemia (FA) patient, a homozygous frameshift deletion (c.1158delC, p.Ser387ProfsX16) was found in FANCG, which has not been described before. FANCG is a component of the FA nuclear complex involved in DNA repair.4 In a patient with Diamond-Blackfan anemia, we identified a heterozygous frameshift mutation in RPL5 (c.172_173delAG, p.Asp59TyrfsX53) which has been reported previously.5 Both changes were confirmed by Sanger sequencing.
Screening of 45 IAA patients resulted in 8398 variant calls which were filtered using Ingenuity® Variant Analysis™ (Ingenuity Systems; www.ingenuity.com/variants) on the assumption that any disease causing or predisposing variant would not be seen at a high frequency in the general population. Variants were selected that were rare (minor allele frequency of 0.0001 or less), had passed the Illumina filter, had a quality score of Q80 or above and resulted in a missense, stop gain/loss, insertion/deletion or splice site change. All of these variant calls were then examined manually using the Integrated Genomics Viewer (Broad Institute) and variant calls that were clear sequence artefacts were ignored. Twenty rare variants remained and 19, from 12 different genes, were validated by Sanger sequencing (Table 1).
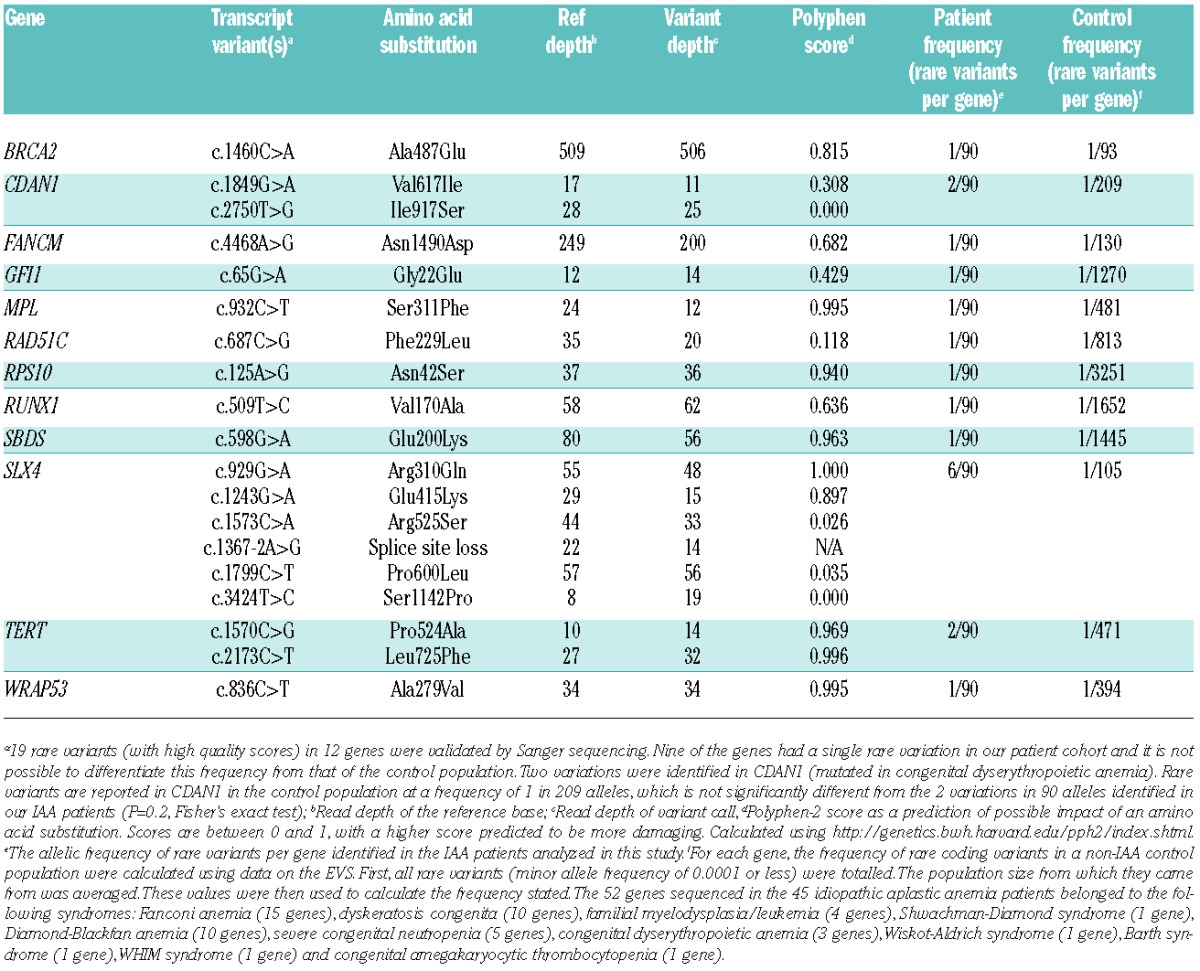
Table 1.
Rare variants identified in patients with IAA and their frequency per gene compared to a control population.
Two patients from our cohort presented with novel heterozygous TERT variants (Table 1). In one patient, the variant c.1570C>G, p.Pro524Ala was found in the RNA-interacting domain. In another patient, we identified c.2173C>T, p.Leu725Phe, which occurs in the reverse transcriptase domain necessary for catalytic activity. The telomere lengths of the 45 AA patients in the study were quantified by monochrome multiplex quantitative PCR6 and were compared to 124 healthy controls aged 0–49 years (Figure 1A). Although this group of IAA patients did not have shorter telomeres than the age-matched controls, the 2 patients with the TERT variants stand out, with telomere lengths below the 5th percentile. The effect of these variants on telomerase activity was measured using a TRAPeze RT Telomerase Detection Kit (Millipore). Both mutations had a negative impact resulting in less than 22% of wild-type telomerase activity (Figure 1B). This finding demonstrates that pathogenic TERT variants can be successfully identified through a relatively large resequencing experiment.
Figure 1.
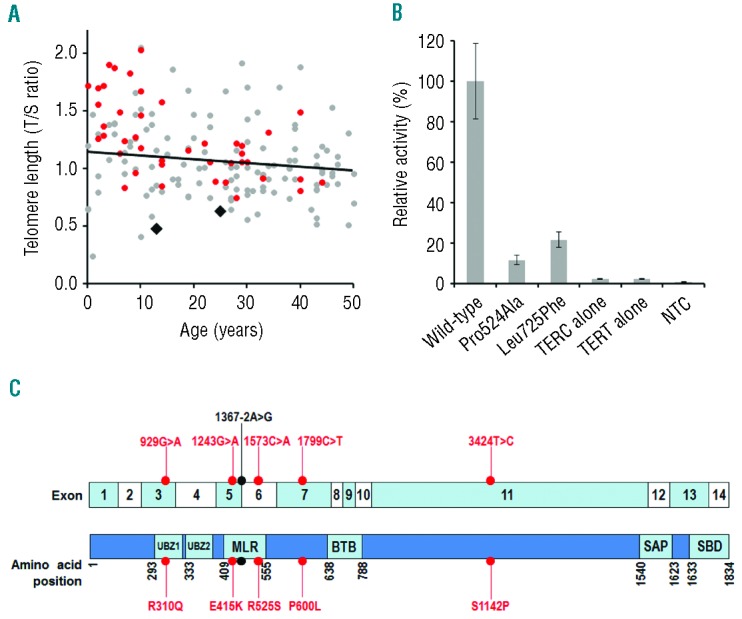
The impact of variants identified in TERT and the location of variants identified in SLX4. (A) Telomere lengths were measured in the 45 IAA patients studied and in 124 normal, age-matched controls by quantitative PCR and expressed as a T/S ratio. The 2 IAA patients with TERT variants had telomeres below the 5th percentile for their age group. Gray circles: controls; red circles: IAA patients; black diamonds: IAA patients with TERT variants. (B) The telomerase repeat amplification protocol showed that the two TERT variants reduced telomerase activity. Wild-type TERC and TERT were expressed in WI38 cells as a positive control to give 100% activity, and the variants were compared to this. NTC: no template control. Bars show standard error of mean of duplicate readings from 2 independent experiments. (C) Schematic illustration of SLX4 with exon positions and functional domains highlighted. Variants in IAA patients are indicated. Red dots: missense variants; black dot: splice site variant; UBZ1/UBZ2: ubiquitin-binding zinc finger domains 1 and 2; MLR: MUS312-MEI9 interaction-like region; BTB: broad-complex, tramtrack, and bric à brac; SAP: SAF-A/B, Acinus and PIAS; SBD: SLX1 binding domain.
Notably, 6 of the 45 IAA patients presented with rare heterozygous variants in the FA gene SLX4, 5 of which are novel and one (c.1367-2A>G) that has been reported in a homozygous state in a family with FA (Table 1).7 SLX4 acts as a scaffold, binding proteins involved in DNA damage repair. These include the nuclease XPF, involved in DNA interstrand crosslink repair,8 and the DNA mismatch repair complex MSH2/MSH3.9 The integrity of SLX4 is necessary for their binding and activity and loss of these interactions results in impaired DNA repair pathways.10 The SLX4 ubiquitin-binding zinc finger domains 1 and 2 (UBZ1 and UBZ2) and the MUS312-MEI9 interaction-like region (MLR) act as the binding sites for MSH2/MSH3 and XPF, respectively.10 Five variants identified in our patients clustered between exons 3 to 7, which encode these domains (Figure 1C). Three impact on the MLR domain and another lies in the UBZ1 domain. When SLX4 lacking a UBZ1 domain is ectopically expressed, cells exhibit an increased sensitivity to the DNA cross-linking agent mitomycin C.11 The importance of the UBZ1 and MLR domains in SLX4 is, therefore, well established, and it is possible that the variants we have identified here could impact on their function.
Data on the Exome Variant Server (EVS, http://evs.gs.washington.edu/EVS) were used to indicate the frequency of a rare SLX4 variant in a control population (likewise with a minor allele frequency of 0.0001 or less). When considering both European and African populations together, the frequency is one in 105 alleles. The increased frequency of 6 in 90 alleles that we observe in our IAA patient cohort is of borderline significance (P=0.05; Fisher’s exact test). When considering only exons 3 to 7, the frequency of rare variants on the EVS is one in 461 alleles, which is significantly different to the 5 variants in 90 alleles seen in our cohort of AA patients (P<0.01; Fisher’s exact test).
In summary, we have successfully utilized a method of targeted resequencing to analyze 52 genes responsible for inherited BMF syndromes in patients with IAA. This study is the first to investigate a gene panel of this scale using this technology and report on the frequency of rare variants in these genes in an IAA patient cohort. Intriguingly, we find an increased incidence of novel variants in the FA gene SLX4. These observations suggest heterozygous variations in SLX4 may be a predisposing factor for the development of AA, as proposed previously.12 Functional studies may provide additional information on whether all or only some of these SLX4 variants contribute to the pathophysiology of IAA. It is also notable that in 50 out of 52 bone marrow failure genes, resequencing has not detected variants that can be presumed to predispose to IAA.
Footnotes
The online version of this article has a Supplementary Appendix.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Young N. The etiology of acquired aplastic anemia. Rev Clin Exp Hematol. 2000;4(3):236–59 [Google Scholar]
- 2.Vulliamy TJ, Marrone A, Dokal I, Mason PJ. Association between aplastic anaemia and mutations in telomerase RNA. Lancet. 2002;359:2168–70 [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Engl J Med. 2005; 352:1413–24 [DOI] [PubMed] [Google Scholar]
- 4.de Winter J, Waisfisz Q, Rooimans M, van Berkel C, Bosnoyan-Collins L, Alon N, et al. The Fanconi anaemia group G gene FANCG is identical with XRCC9. Nat Genet. 1998;20:281–3 [DOI] [PubMed] [Google Scholar]
- 5.Gazda HT, Sheen MR, Vlachos A, Choesmel V, O’Donohue MF, Schneider H, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am J Hum Genet. 2008;83:769–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cawthon R. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009; 37(3):e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuster B, Knies K, Stoepker C, Velleuer E, Friedl R, Gottwald-Mühlhauser B, et al. Whole exome sequencing reveals uncommon mutations in the recently identified Fanconi anemia gene SLX4/FANCP. Hum Mut. 2013;34(1):93–6 [DOI] [PubMed] [Google Scholar]
- 8.Kuraoka I, Kobertz W, Ariza R, Biggerstaff M, Essigmann J, Wood R. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J Biol Chem. 2000;275(34):26632–6 [DOI] [PubMed] [Google Scholar]
- 9.Svendsen J, Smogorzewska M, Sowa B, O’Connell S, Gygi S, Elledge S, et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138(1):63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim Y, Spitz G, Veturi U, Lack F, Auerbach A, Smogorzewska A. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood. 2013;121(1):54–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto K, Kobayashia S, Tsuda M, Kurumizaka H, Takata M, Jiricnye J, et al. Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. PNAS. 2011; 108(16):6492–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hays L. Multifunctionality of the FA pathway. Blood. 2013;121(1):54. [DOI] [PubMed] [Google Scholar]