

Abstract
Disturbances in sodium concentration are common in the critically ill patient and associated with increased mortality. The key principle in treatment and prevention is that plasma [Na+] (P-[Na+]) is determined by external water and cation balances. P-[Na+] determines plasma tonicity. An important exception is hyperglycaemia, where P-[Na+] may be reduced despite plasma hypertonicity. The patient is first treated to secure airway, breathing and circulation to diminish secondary organ damage. Symptoms are critical when handling a patient with hyponatraemia. Severe symptoms are treated with 2 ml/kg 3% NaCl bolus infusions irrespective of the supposed duration of hyponatraemia. The goal is to reduce cerebral symptoms. The bolus therapy ensures an immediate and controllable rise in P-[Na+]. A maximum of three boluses are given (increases P-[Na+] about 6 mmol/l). In all patients with hyponatraemia, correction above 10 mmol/l/day must be avoided to reduce the risk of osmotic demyelination. Practical measures for handling a rapid rise in P-[Na+] are discussed. The risk of overcorrection is associated with the mechanisms that cause hyponatraemia. Traditional classifications according to volume status are notoriously difficult to handle in clinical practice. Moreover, multiple combined mechanisms are common. More than one mechanism must therefore be considered for safe and lasting correction. Hypernatraemia is less common than hyponatraemia, but implies that the patient is more ill and has a worse prognosis. A practical approach includes treatment of the underlying diseases and restoration of the distorted water and salt balances. Multiple combined mechanisms are common and must be searched for. Importantly, hypernatraemia is not only a matter of water deficit, and treatment of the critically ill patient with an accumulated fluid balance of 20 litres and corresponding weight gain should not comprise more water, but measures to invoke a negative cation balance. Reduction of hypernatraemia/hypertonicity is critical, but should not exceed 12 mmol/l/day in order to reduce the risk of rebounding brain oedema.
Introduction
Hyponatraemia and hypernatraemia are frequent in patients admitted to the ICU and may even be acquired during their stay [1-8]. These patients have a higher overall mortality, and even slight changes in plasma [Na+] (P-[Na+]) are associated with an increased risk of death [3,9].
Case story
A 58-year-old male is admitted somnolent (Glasgow Coma Scale = 11) to the emergency department. He has a newly discovered tonsil cancer, consumes too much alcohol, has arterial hypertension that is being treated with losartan/thiazid and is receiving nonsteroidal anti-inflammatory drugs (NSAIDs). He is spontaneously breathing with a patent airway. Mean arterial pressure is 100 mmHg, heart rate = 120 beats/minute. Arterial blood gas analysis is as follows: pH = 7.57, pCO2 = 3.4 kPa/ 26 mmHg, pO2 = 7.9 kPa/59 mmHg, Hgb = 7.3, lactate = 1.6 mmol/l, BE = 1.3, plasma [Glucose] (P-[Glc]) = 7.6 mmol/l, P-[Na+] = 90 mmol/l and P-[K+] = 3.5 mmol/l. Cerebral CT scanning shows no bleeding. Infusion of 0.9% NaCl is instituted and he is transferred to the ICU. After 6 hours (P-[Na+] = 95 mmol/l), the patient is more awake, but delirious. To achieve P-[Na+] = 120 mmol/l, the 0.9% NaCl infusions are increased. After 12 hours (P-[Na+] = 105 mmol/l), the patient is awake but confused, and phenobarbital and haloperidol are given. Respiratory insufficiency necessitates intubation after 24 hours (P-Na+ = 115 mmol/l). The patient is seen by an oncologist and diagnosed quite informally with syndrome of inappropriate antidiuretic hormone (SIADH). P-[Na+] rises progressively. On day 6, quadriplegia and coma (Glasgow Coma Scale = 3 without sedation) are observed (P-[Na+] = 144 mmol/l). A neurologist proposes a diagnosis of osmotic demyelinisation (OD). Over the next 2 days, P-[Na+] increases to 151 mmol/l and he dies from intractable cardiac arrest.
This case story illustrates common and important problems in managing the hyponatraemic patient. First, the initial 0.9 % NaCl approach is inadequate to ensure a rapid and controllable response [10]. Second, no measures to avoid overcorrection are taken, and he gets hypernatraemia despite being in the ICU [8]. Third, hyponatraemia often has multiple causes [11]. Fourth, nothing is done to identify the mechanisms of hyponatraemia [12].
This review takes a practical approach to the critically ill patient with dysnatraemia.
Plasma [Na+] is determined by water and electrolytes
Knowledge about what determines P-[Na+] is crucial for understanding the mechanisms behind dysnatraemia and how to correct/prevent these disorders. Within a population of heterogeneous patients, Edelman and colleagues [13] demonstrated that P-[Na+] is determined by exchangeable cations (eNa+ and eK+) and total body water (TBW) according to Equation 1:
| (1) |
where α and β are the coefficients from the linear regression.
Equation 1 is not readily useful at the bedside; however, it was recently demonstrated that it is valid in the individual and that changes in P-[Na+] can be determined by the external balances of water and cations (Na+ + K+) [14]. The simplified version (Equation 2) where α = 1 and β = 0 provides a good estimate of the P-[Na+] changes and is useful for planning fluid and electrolyte treatment in the individual patient [14-16]:
| (2) |
where [Na+]1 is the initial plasma concentration and [Na+]2 is the concentration that results from the change in the external balances of water (ΔTBW) and cations (Δ(Na+ + K+)).
Equation 2 is fundamental in understanding changes in P-[Na+]. It is, therefore, also fundamental in practical patient treatment. It is important, however, to keep in mind that the impending output side of the water and cation balances can only be guessed. Frequent measurement of output (especially diuresis) and P-[Na+] is necessary and input of fluids must be adjusted dynamically.
Equations 1 and 2 assume that plasma tonicity is determined by P-[Na+]. This is true in hypernatraemia and, by far, in the most common hyponatraemic conditions. Translocational hyponatraemia is an exception. In this condition osmotically active substances confined to the extracellular/plasma compartment cause a shift of water from the intracellular compartment. In this situation, the resulting hyponatraemia is hypertonic. The most frequent clinical condition is hyperglycaemia (P-[Na+] is reduced approximately 0.4 mmol/l per mmol/l increase in P-[Glc] (or a reduction of 2.4 meq/l per 100 mg/dl increase)) [17], but the condition can also be caused by mannitol. Pseudohyponatraemia is an unusual measurement flaw in patients with hyperlipaemia/hyperproteinaemia whose plasma tonicity is normal. In the rest of this article, 'hyponatraemia' refers to hyponatraemia where plasma tonicity is decreased.
Regulation of P-[Na+]
P-[Na+] is tightly regulated between 135 and 145 mmol/l primarily by water intake (thirst) and renal water excretion [15]. Of lesser quantitative importance, P-[Na+] is regulated by the kidney's regulation of cation excretion via the renin-angiotensin-aldosterone system. Thirst is stimulated when P-[Na+] increases a few percent [18]. It is also stimulated by a decrease in the effective circulating volume, which is the part of the extracellular volume (ECV) that effectively perfuses the tissue [19]. Arginine vasopressin (vasopressin, or antidiuretic hormone (ADH)) reduces renal water excretion. Vasopressin binds to the V2-receptor in the collecting duct. This promotes trafficking of aquaporin 2 to the apical membrane and passive water reabsorption to the hypertonic medullary interstitium [20]. Vasopressin secretion is stimulated when P-[Na+] increases [18]. Vasopressin release can also be stimulated non-osmotically by a reduced effective circulating volume, stress, pain, nausea, vomiting, various drugs and exercise.
Irrespective of vasopressin, the kidney's ability to excrete water is influenced by solute intake (protein/urea and cations) since the urine volume is the solute excretion divided by the urine osmolality:
A low solute intake reduces urine solute excretion and thereby urine volume despite maximally diluted urine. In contrast, a high protein/urea intake or generation will increase the urine volume. However, urea does not directly determine P-[Na+] (according to Equation 2). This is so because in the whole body perspective, urea eventually crosses cell membranes and therefore is an ineffective osmolyte that does not contribute to water flux between cells and extracellular volume [15].
Altogether, the renal effects influencing P-[Na+] are gathered in excretion of electrolyte-free water [21]:
where U[Na+] and U[K+] are urine Na+ and K+ concentrations. If electrolyte free water clearance (cH2Oe) is positive, the urine increases P-[Na+]; and if cH2Oe is negative, urine decreases P-[Na+].
Part l: hyponatraemia
In a patient with hyponatraemia, it must first be determined whether acute correction is required. This decision is made based on the patient's symptoms at a time when the mechanisms causing the hyponatraemia are rarely known. The discussion of hyponatraemia therefore begins with the symptoms and possible interventions (initial approach shown in Figure 1). Next, the mechanisms and final diagnostics are discussed.
Figure 1.

Initial approach to the hyponatraemic patient. P-[Na+], plasma [Na+].
Hyponatraemia with severe symptoms: airway, breathing, circulation and 3% NaCl bolus therapy
Severe hyponatraemia symptoms (Table 1) are believed to be caused by cerebral oedema. This corresponds well with cellular swelling as the extracellular tonicity decreases [22]. Intracranial pressure (ICP) rises when the brain cells swell in the rigid skull. An efflux of excitatory neurotransmitters (for example, glutamate) as a response to cell swelling [23] or decreased chloride conductance caused by the corresponding, low plasma [Cl-] might also, in part, explain the symptoms [24]. Because severe cerebral symptoms indicate ongoing brain damage and a substantial risk of incarceration, treatment should normally not be delayed by cerebral CT scanning as was the case in the case story [25].
Table 1.
Cerebral symptoms in hyponatraemia
| Severe symptoms |
| Decreased consciousness |
| Seizures |
| Muscle rigidity |
| Other symptoms |
| Nausea and vomiting |
| Headache |
| Bloating, 'puffiness' |
| Muscle weakness, cramps or spasms |
Crucially, secondary brain damage caused by hypoxia, hypercapnia and hypoperfusion should ardently be avoided with an ABC approach [10,26]. Next, immediate ICP reduction is best induced with one or more boluses of 2 ml/kg 3% NaCl (or a corresponding amount of more hypertonic NaCl) given intravenously/intraosseously [27-29]. The effect is immediate, and bolus doses may be repeated at 5-minute intervals. One 2 ml/kg 3% NaCl bolus causes a controllable rise in P-[Na+] of about 2 mmol/l (Example 1 in Box 1). Cerebral symptoms decrease when P-[Na+] increases by 4 to 6 mmol/l [27-29]. Infusion of 0.9% NaCl should not be used to acutely increase P-[Na+] as in the case story: such an infusion does not cause an immediate, controllable increase in P-[Na+], and 0.9% NaCl might worsen the hyponatraemia in SIADH (see SIADH section and Box 1). Vasopressin V2-receptor antagonists are not recommended: V2-receptor antagonists do not induce a controllable and fast increase in P-[Na+] and the induction/worsening of hypovolemia can be hazardous [30].
Cerebral disease, hepatic encephalopathy and sedation can cause severe symptoms similar to those of hyponatraemia [31], but a slight P-[Na+] increase will not worsen these conditions [32,33]. However, the clinician must always consider other conditions if the patient fails to respond, and a maximum of three boluses should be given (complete awakening cannot be anticipated if the patient has suffered elevated ICP/seizures for hours [29]).
There is no set P-[Na+] level below which severe symptoms arise. Cerebral symptoms have been described at P-[Na+] levels between 125 and 130 mmol/l [34,35]. A MRI study in pigs demonstrated that cellular swelling in the brain oedema corresponds with the relative reduction in P-[Na+] [22]. This indicates that it is the relative reduction and its speed that are clinically interesting and not an arbitrarily defined absolute P-[Na+] value [36]. Hence, a P-[Na+] reduction from 160 to 128 mmol/linduces as much osmotic stress to the brain as a reduction from 113 to 90 mmol/l (illustrated in the case story). This also implies that patients with intracranial pathology (for example, intracranial bleeding, tumour or oedema) have an increased risk of cerebral deterioration if brain cells swell and that they may need correction at a higher P-[Na+] (for example, P-[Na+] = 135 mmol/l). Bolus treatment is therefore instituted based on the patient's symptoms and not on the basis of an arbitrarily defined P-[Na+] value.
Cerebral symptoms determine treatment, not the assumed time course
Treatment should be guided by the patient's symptoms and not by the assumed acute (<48 hours) or chronic nature of hyponatraemia. Classification of acute versus chronic hyponatraemia is based on brain-adaptive responses to hyponatraemia. Patients with days of hyponatraemia have fewer cerebral symptoms than patients with newly developed hyponatraemia [37]. This has been linked with cerebral volume regulation [38,39]. Adaption to sustained hyponatraemia by loss of organic osmolytes has also been proposed to increase the risk of OD (formerly known as central pontine myelinolysis) when correcting hyponatraemia [40-43]. OD is a devastating clinical condition with progressive quadriplegia, dysarthria, dysphagia and alterations of consciousness days after hyponatraemia correction, and it is proposed to be due to cell shrinkage and/or water diffusion differences in the brain [22,44]. Alcoholism, malnutrition, hypokalaemia, liver failure and malignant disease increase the risk of OD [45].
These observations lie at the root of the distinction between acute and chronic hyponatraemia in treatment protocols [46,47]. This distinction is arbitrary, though, and difficult to implement in practice. First, asymptomatic chronic hyponatraemic patients bear the highest risk of symptomatic hyponatraemia, that is, acute worsening of chronic hyponatraemia with looming brain oedema (for example, as in the case story) [48]. Second, it is difficult to determine the time frame of hyponatraemia in the comatose patient brought to the emergency department (for example, as in the case story). Finally, OD has been demonstrated in case reports after correction of acute (<48 h) hyponatraemia, so cautious correction is also important in these cases [42,49].
Measures to avoid P-[Na+] overcorrection
Avoiding overcorrection is pivotal to diminish the risk of OD (see the case story). No prospective studies have established an absolutely safe and definitive speed for correction of hyponatraemia. In retrospective studies, OD has been seen in patients corrected with more than 12 mmol/1 day and not in less rapidly corrected patients [41,43,50]. In a small clinical MRI study, OD lesions were seen in patients corrected with more than 10 mmol/l/day [42].
Because OD is more likely when the patients are corrected by more than 12 mmol/l/day, it seems reasonable, in the light of the brain's adaption mechanisms, not to correct at a faster pace, even though there is no absolutely safe rate. Conservative therapeutic goals for correction of 8 mmol/1 in 24 hours, 14 mmol/1 in 48 hours and 16 mmol/l in 72 hours have been proposed [51]. Therefore, only three 2 ml/kg 3% NaCl bolus doses should be given to patients with severe symptoms [51,52].
Brisk diuresis is the most common cause of over-correction even without sodium input [51,53]. Diuresis can be counteracted by administering water and, if necessary, using desmopressin [54]. Hypokalaemia correction can contribute to P-[Na+] overcorrection. If renal replacement therapy is necessary (for example, fluid overload or hyperkalaemia), P-[Na+] changes can be controlled by lowering the Na+ concentration in the replacement fluid to the desired P-[Na+] level by adding water (note that this will also reduce the concentration of all other components in the fluid) [55] or by reducing the blood flow [56].
Mechanisms and treatment of hyponatraemia without severe symptoms
The population of hyponatraemia patients in the ICU without severe symptoms comprises two groups: one initially with severe symptoms stabilized with bolus therapy and one initially not having severe symptoms. In these patients, treatment must be individualized and the underlying mechanisms identified for safe and lasting correction.
Traditional classifications according to volume status are of little use in clinical practice since hypo- and normovolaemia cannot be reliably separated [57,58], and multiple combined causes are common so a simple scheme cannot be used (see the case story) [11,59]. Mechanisms causing hyponatraemia are often revealed by the patient's treatment response [60]. Importantly, the initial mechanisms causing hyponatraemia may be evanescent and a rapid P-[Na+] increase may occur. In all patients with hyponatraemia spot urine for urine osmolality and U-[Na+] determination should be sampled as fast as possible and preferably before treatment.
The mechanisms causing hyponatraemia are discussed in the following sections and summarized in Figure 2.
Figure 2.
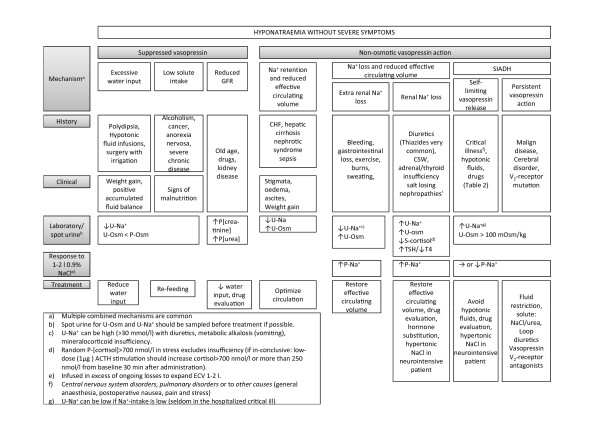
Hyponatraemia without severe symptoms: diagnostics and treatment. ACTH, adrenocorticotropic hormone; CHF, congestive heart failure; CSW, cerebral salt wasting; ECV, extracellular volume; GFR, glomerular filtration rate; SIADH, syndrome of inappropriate antidiuretic hormone; TSH, thyroid-stimulating hormone; U-[Na+], urine [Na+]; U-Osm, urine osmolality.
Hyponatraemia despite suppressed vasopressin
Young, normally functioning kidneys have an enormous capacity to excrete water (1 l/h), so excessive water input (polydipsia, infusion of hypotonic fluids or absorption of irrigant solutions) must exceed 1 l/h to produce hyponatraemia. Much less water ingestion can produce hyponatraemia when the kidney's ability to excrete water is reduced. In beer potomania and severe malnutrition (for example, due to chronic disease like cancer or anorexia nervosa), a low solute intake (protein and cations) decreases the kidney's ability to excrete water as pure water cannot be excreted [15,61]. This is a possible contributing mechanism in the case story. The kidney's diluting capacity is also reduced independently of vasopressin [62] with decreased delivery of fluid to the distal nephron. A reduction in the glomerular filtration rate with increasing age, various drugs (Table 2) and various disease states (for example, reduced effective circulating volume) will therefore render the patient more vulnerable to water ingestion. Treatment should address these mechanisms: reduce water intake (avoid hypotonic fluids), improve nutrition and restore kidney function.
Table 2.
Drugs associated with hyponatraemia
| Groups | SIADH mechanism | Other mechanisms |
|---|---|---|
| Diuretics | Thiazides Indapamide |
Thiazides Indapamide |
| Antidepressant agents | SSRI TCA (mirtazapine) SNRI (venlafaxine/duloxetin) |
|
| Antipsychotic agents | Phenothiazine Butyrophenones |
|
| Anti-seizure drugs | Carbamazapine Oxcarbazepine Valproate Nicotine Clofibrate |
|
| Antineoplastic agents | Cyclophosphamide Vincristine Ifosfamide |
Cisplatin |
| V2-receptor agonist | Desmopressin Vasopressin Oxytocin(?) |
|
| Miscellaneous | NSAIDs Voriconazole Methylenedioxymethyl- amphetamine (ecstacy) |
NSAIDs |
| Hypotonic fluids |
NSAID, nonsteroidal anti-inflammatory drug; SIADH, syndrome of inappropriate antidiuretic hormone; SNRI, serotonin-norepenephrine reuptake inhibitor; SSRI, selective serotonin-reuptake inhibitor; TCA, tricyclic antidepressant.
Thiazide-induced hyponatraemia
Thiazide-induced hyponatraemia (TIH) is common and may have contributed to the hyponatraemia in the case story [50,63]. The mechanisms by which some individuals (females more than males) develop TIH are not clear [63]. In contrast to loop diuretics, thiazides do not reduce the medullary concentration gradient in the kidneys necessary for concentrating the urine, but reduce NaCl transport in the diluting segment of the nephron. Desalination is a necessary consequence of thiazides [64], but whether this results in overt hyponatraemia is dependent on other partly unknown factors. A preexisting urine dilution defect (old age, medications like NSAIDs) may be worsened by thiazides independently of the vasopressin levels [65]. One study proposes increased thirst as a mechanism for TIH [66]. However, a small decrease in effective circulating volume with non-osmotically stimulated thirst/vasopressin-secretion may also contribute. Following TIH, a substantial risk of overcorrection looms.
Optimize effective circulating volume in hypervolaemic conditions
In conditions with increased ECV/plasma volume (congestive heart failure, cirrhosis, nephrotic syndrome and sepsis), hyponatraemia is caused by non-osmotic stimulation of vasopressin and thirst due to a reduced effective circulating volume. Stigmata and the patient's history may help the diagnosis. Optimizing the haemodynamics is the cornerstone of treatment in this situation. The underlying conditions can occasionally be improved, such as by angiotensin-converting-enzyme (ACE) inhibitors in heart failure [67] and the use of spironolactone in advanced liver disease [68]. P-[Na+] is corrected with water restriction (avoid hypotonic fluids) and loop diuretics [68]. Treatment with V2-receptor antagonists seems rational, but a randomized trial has shown no survival benefits [69], and overcorrection may occur [70]. Hypertonic saline infusion (improves effective circulating volume) together with loop diuretics has been shown to be effective sometimes in refractory heart failure and ascites [33].
Restore effective circulating volume in conditions with hypovolaemia
In hypovolaemia conditions, loss of total body solutes (Na+ and K+) results in a reduced effective circulating volume, non-osmotic vasopressin secretion and thirst. In this situation, hypotonic fluid ingestion/infusion causes hyponatraemia. Differentiating these conditions from SIADH can be challenging because the reduced effective circulating volume (and ECV) is difficult to determine clinically [57,58].
Extra-renal causes of solute loss are diagnosed by the patient's history (bleeding, gastrointestinal loss, exercise [71] and sweating) and by U-[Na+] <30 mmol/l in spot urine; however, exceptions exist (concurrent use of diuretics, compensating renal loss of NaHCO3 in metabolic alkalosis (vomiting) or mineralocorticoid insufficiency).
The solute loss can also have renal causes. TIH has previously been discussed as a potential cause. Other renal mechanisms are osmotic diuresis, salt-losing nephropathy including mineralocorticoid deficiency, and cerebral salt wasting (CSW).
CSW is a condition with reduced effective circulating volume and unexplained/inappropriate natriuresis. The mechanisms leading to it are not clear [72,73]. Diagnostic difficulties are rooted in the problem of determining the effective circulating volume and in the fact that the neurointensive patient (for example, traumatic brain injury, subarachnoid haemorrhage) receives large amounts of normal and hypertonic saline to avoid hypovolaemia and hyponatraemia [74]. This sodium loading induces natriuresis in the normal kidney [75,76]. Inadequate release of brain natriuretic peptide has been proposed as a contributing mechanism in CSW [77].
Renal causes of hyponatraemia result in high U-[Na+] levels and may have characteristics similar to those of SIADH. Responses to infusion of 1 to 2 l 0.9% NaCl can help clarify the diagnosis. Increased P-[Na+] favours ECV/Na+ deficit: sodium, and to a lesser extent water, is retained, which increases P-[Na+]. Additionally, normal saline restores the effective circulating volume and reduces the non-osmotic stimulus of vasopressin secretion. Unchanged or decreased P-[Na+] makes SIADH likely: the patient with SIADH is normally in sodium balance (output is reflected in input) and the infused Na+ is excreted. However, the urine is concentrated and urine cations are generally higher than the 0.9% NaCl (308 mOsm) resulting in retention of water and a decrease in P-[Na+] (cH2Oe is negative) (Example 2 in Box 1) [15]. Infusions of 0.9% NaCl must increase ECV by 1 to 2 l and the infusion speed must therefore exceed ongoing losses [78].
Hyponatraemia induced by diuretics may entail a fractional excretion of uric acid (FE-UA = (U-[uric acid] × P-[creatinine])/(U-[creatinine] × P-[uric acid])) below 8% [79]. However, CSW and SIADH can both have an FE-UA >12%, and it may be necessary to distinguish between the two conditions in terms of their response to water restriction or a vasopressin V2-receptor antagonist (FE-UA and P-[Na+] is normalized in SIADH) [80]. However, this approach is not recommended in the neurointensive patient because it involves a substantial risk of worsening the hypovolaemia [74]. A practical approach is to control P-[Na+] with 0.9% NaCl and hypertonic NaCl in the neurointensive patient [81].
Once the diagnosis is established, the underlying disease should be treated and the balances, including potassium and water deficit, should be restored. Abolition of the non-osmotic stimulus of vasopressin secretion involves a substantial risk of overcorrection caused by brisk diuresis.
Syndrome of inappropriate antidiuretic hormone
Non-osmotic vasopressin secretion and abnormal thirst are present in SIADH despite a normal effective circulating volume [78,82-85]. In the critically ill patient, the mechanisms of inappropriate vasopressin secretion and thirst are heterogeneous. This may be due to various drugs (Table 2), malignant disease, central nervous system disorders (infection, bleeding, thrombosis, space-occupying disorders, psychosis and generalised disorders), pulmonary disorders (infection, asthma, respirator treatment) or other causes (general anaesthesia, postoperative nausea, pain and stress) [78,86]. These causes of SIADH may be divided into self-limiting mechanisms with an inherent risk of overcorrection when the vasopressin stimuli are abolished, and persistent conditions, for example, a paraneoplastic phenomenon that, in the absence of vasopressin V2-receptor antagonist treatment, will rarely be overcorrected.
When hyponatraemia persists, SIADH may be diagnosed by conventional criteria: plasma hypo-osmolality (P-Osm <275 mOsm/kg) with a not maximally diluted urine osmolality (typically U-Osm >100 mOsm/kg), high U[Na+] (>30 mmol/l), normal effective circulating volume and normal renal, thyroid and adrenal function [78,86-88].
Other hyponatraemia mechanisms are likely to co-exist in the critically ill patient (for example, thiazide therapy, low solute intake, renal impairment), and it is important to determine and correct these causes. Urine investigation (U-Osm and U-Na+) is important, although it is not always performed [12]. U-Na+ can be low in SIADH with a low salt intake. Failure to increase P-[Na+] with 1 to 2 l 0.9% NaCl intravenously is a practical way of tracing SIADH without inviting the risk of circulatory collapse (Example 2 in Box 1). Plasma uric acid below 238 μmol/l (<4 mg/dl) and FE-UA above 12% also likely suggest SIADH [79].
When a reduced effective circulating volume has been ruled out and persistent SIADH is a likely diagnosis, the cornerstone is investigation and treatment of its underlying causes. In the critically ill patient, the input side of Equation 2 is controlled and hyponatraemia may be corrected with water restriction and avoidance of hypotonic fluids. Loop diuretics and increased solute intake (for example, 0.5 to 1 g/kg/day urea in the gastric tube [89]) can be used to increase water excretion. Compliance problems with urea administration in the critically ill patient seem small, and urea (as ineffective osmolytes) has been shown to reduce the risk of OD in experimental studies [90]. If SIADH persists, vasopressin V2-receptor antagonists may be the most effective option in terms of correcting P-[Na+]. However, lack of studies in the critically ill population with persistent SIADH weakens this approach [91].
Exclude adrenal and thyroid insufficiency
In adrenal insufficiency with glucocorticoid deficiency, the hyponatraemia mechanisms include an increased vasopressin/thirst response [92] and a decreased effective circulating volume [93]. In the critically ill patient with suspected adrenal insufficiency, a random serum cortisol and plasma adrenocorticotropic hormone is obtained followed by 100 mg hydrocortisone intravenously. A serum cortisol level above 700 nmol/l (25 μg/dl) virtually excludes adrenal insufficiency [94,95]. If this is inconclusive, low-dose adrenocorticotropic hormone stimulation should be performed after cessation of hydrocortisone (details in Figure 2).
Hypothyroidism is occasionally associated with hyponatraemia. The mechanisms are unclear. Fluid retention, impaired cardiac and renal function are likely [96]. Patients should be screened with plasma thyroid-stimulating hormone and T4 [95].
Part 2: hypernatraemia
Why did the man in the case story develop hypernatraemia while staying in the ICU and how could it have been prevented? Hypernatraemia is less common than hyponatraemia, but the patient is generally more ill and has a higher mortality [3]. The initial approach is ABC management followed by treatment of underlying diseases and restoration of the distorted physiology. The discussion of hypernatraemia therefore addresses first the mechanisms, then treatment. The discussion of the mechanisms addresses situations of water and solute loss, pure water loss and increased total body solutes. Multiple combined causes are common. Mechanisms and treatment are summarized in Figure 3.
Figure 3.
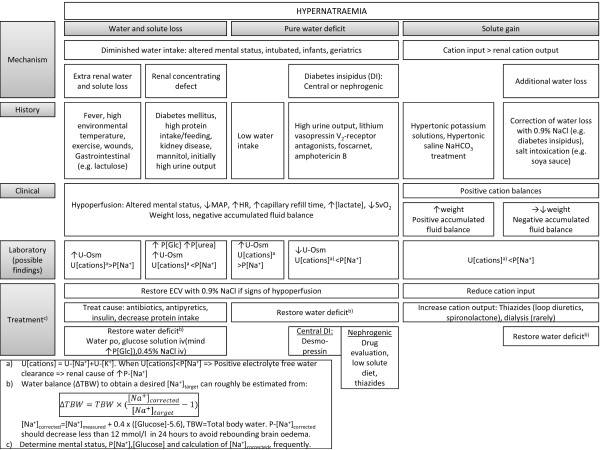
Hypernatraemia: diagnostics and treatment. ECV, extracellular volume; HR, heart rate; MAP, mean arterial pressure; P, plasma; P-[Glc], plasma [Glucose]; SvO2, central venous oxygen saturation; U, urine; U-Osm, urine osmolality.
Hypernatraemia with water and solute loss
This condition arises by a negative water balance exceeding a concomitant negative cation balance (Equation 2). The resulting hypovolaemia is the most common condition in hospitalised hypernatraemic patients [97].
Diminished water intake is a pivotal mechanism of increased P-[Na+] [97]. Individuals at risk of insufficient drinking often have an altered mental status (critical illness, sedation, neurological impairment) or they are intubated patients, infants [98] or geriatric patients [99].
An enhanced water above salt loss contributes to hypernatraemia. Fever is a common contributing factor [21,97,100]. A high loss through the skin can result from a high temperature in the environment, from exercise or wounds (for example, burns). Gastrointestinal loss of hypotonic fluid is increased by diarrhoea (for example, infection, lactulose).
A renal concentrating defect frequently contributes to hypernatraemia. Several mechanisms are responsible for this. Loop diuretics contribute, especially in the critically ill patient [21]. Osmotic diuresis (U-Osm >300 mOsm with hypernatraemia) can be induced by hyperglycaemia. Osmotic urea diuresis is seen with excessive protein nutrition and protein wasting and diagnosed by increased electrolyte-free water excretion [101]. Mannitol also induces osmotic diuresis. A renal concentrating defect is seen with kidney insufficiency. Rare causes are hypercalcaemia and potassium depletion.
The patient is treated by restoring the ECV and water deficit (Example 3 in Box 1).
Hypernatraemia with pure water loss
This condition develops when the water balance is negative and the cation balance is normal. The reduction in ECV is far less than with a concomitant salt loss. Diminished water intake is obligatory [102]. Individuals with reduced water intake (infants, old, debilitated patients) will have appropriate, maximally concentrated urine. Patients with a functionally decreased vasopressin response to hypertonicity (central or nephrogenic diabetes insipidus) only develop hypernatraemia when water intake is restricted (for example, critical illness). Diabetes insipidus occasionally develops in the critically ill patient (for example, by traumatic brain injury and late-phase septic shock) and U-Osm is inappropriately low. In rare situations, the set point for osmolality is directly increased due to cerebral disease resulting in essential hypernatraemia and reduced thirst at a given sodium concentration.
Treatment is restoration of the water deficit (Example 3 in Box 1). Water loss in central diabetes insipidus can be reduced with desmopressin. Treatment of nephrogenic diabetes insipidus is challenging and may comprise drug evaluation, correction of hypercalcaemia or hypokalaemia and low-solute diet to decrease urine volume and thiazides.
Hypernatraemia with increased total body solute
Hypernatraemia with increased ECV develops when the cation balance is positive, which is frequently observed in ICU-acquired hypernatraemia [6,21]. Increased input of cations is mandatory. This is seen in conjunction with correction of water loss with 0.9% NaCl. Also, correction of hypokalaemia with hypertonic potassium-containing solutions (for example, 0.9% NaCl added 40 mmol KCl) may contribute to hypernatraemia. Hypernatraemia can be induced therapeutically with hypertonic saline to reduce ICP [103] or as a side effect seen with NaHCO3 treatment. In healthy individuals, an increased salt load is excreted in the urine [75,76]. Natriuresis can be diminished in the critically ill patient with a reduced effective circulating volume and a low glomerular filtration rate [104]. A diagnosis of solute overload is made from the history, water- and cation balances and, if available, weight changes. Salt intoxication outside the hospital is rare, but may arise by suicide attempts with soya sauce and by inappropriate administration of solute in nursery homes [105].
Treatment consists of creating a negative cation balance by reducing the cation input and increasing the cation output with diuretics or, rarely, dialysis.
Symptoms and correction of hypernatraemia
Hypernatraemic patients are generally critically ill and it may be difficult to determine whether cerebral symptoms stem from hypernatraemia (decreased level of consciousness, irritability, hyper-reflexia, spasticity and seizures) or the underlying disease.
The first principle in treatment is ABC. Circulatory collapse/hypoperfusion is treated with infusion of 0.9% NaCl according to haemodynamic parameters (Figure 3) [106]. This should be paralleled by investigation and treatment of the underlying mechanisms (for example, insulin, antibiotics and antipyretics). Clinical evaluation of volume status is notoriously imprecise, and the only sign may be cerebral symptoms in the elderly/infant [98,99]. Next, total body solute cannot be measured in a clinical setting. Simple measures can be helpful (for example, history, weight, accumulated fluid/electrolyte balances and electrolyte-free water clearance to diagnose the mechanisms behind the hypernatraemia (Figure 3)). Finally, the water/cation balance is restored.
Too rapid P-[Na+] correction can provoke seizures, probably from cerebral oedema [107-110]. This has been linked with the brain cells' adaption to the hypertonic state by accumulation of solute [108]. The relative reduction in tonicity results in cellular swelling. However, failure to correct the hypernatraemia is associated with higher mortality [111,112]. No optimal correction ratehas been determined, but it has been suggested that it should not exceed 0.5 mmol/l/h [107]. A practical approach is to decrease P-[Na+] 5 to 6 mmol/l the first hours (1 mmol/l/h) and then slow down the correction rate so the total correction is 12 mmol/l in 24 hours (0.5 mmol/l/h) [97,98]. The change in tonicity is important. The measured P-[Na+] must therefore be corrected for any hyperglycaemia.
In hypernatraemia with water loss, the water correction rate can roughly be estimated from:
where [Na+]corrected is the measured P-[Na+] corrected for any hyperglycaemia and Target [Na+] is the desired P-[Na+] (Example 3 in Box 1). However, frequent measurements of P-[Na+], P-[K+] and P-[Glucose] and calculation of [Na+]corrected are necessary to avoid overcorrection.
In severe cases with renal failure, renal replacement therapy should be instituted. Here, the Na+ content in the replacement fluid/dialysate must be increased to the desired P-[Na+] by NaCl addition to avoid overly rapid correction [55].
Conclusion
Dysnatraemia is common in the critically ill patient and is associated with increased mortality. The case story illustrates common and important treatment problems in the hyponatraemic patient. The key principle in treatment and prevention is that P-[Na+] is determined by external water and cation balances. First, the patient should be treated according to an ABC approach to diminish secondary organ damage. Next, symptoms are critical when handling a patient with hyponatraemia. Severe symptoms are treated with 2 ml/kg 3% NaCl bolus infusions irrespective of the proposed time course. The goal is to reduce cerebral symptoms. The bolus therapy gives an immediate, controllable rise in P-[Na+]. A maximum of three boluses are given. In hyponatraemic patients, any correction exceeding 10 mmol/l/day must be avoided to reduce the risk of OD. Reduced vasopressin action and brisk diuresis are the most common mechanism and they must be counteracted by increasing water input and, if necessary, by desmopressin. The risk of overcorrection is associated with the mechanisms causing hyponatraemia. Traditional classifications according to volume status are notoriously difficult to apply in clinical practice. Moreover, multiple mechanisms are common and may easily be mixed. More than one mechanism must therefore be investigated for safe correction.
First and foremost, the patient in the case story develops hypernatraemia in the ICU because the problem is not addressed. Pivotal is treatment of the underlying diseases and restoration of the distorted water and salt balances based on knowledge of what determines P-[Na+]. Multiple combined mechanisms are common and must be identified. Importantly, hypernatraemia is not only a matter of water deficit, and treatment of a critically ill patient with an accumulated fluid balance of 20 litres and corresponding weight gain is not more water, but a negative cation balance. Reduction of P-[Na+]/plasma tonicity in hypernatraemia is important, but should not exceed 12 mmol/l/day to reduce the risk of rebounding brain oedema.
Abbreviations
ABC: airway, breathing and circulation; cH2Oe: electrolyte free water clearance; CSW: cerebral salt wasting; CT: computed tomography; ECV: extracellular volume; FE-UA: fractional excretion of uric acid; ICP: intracranial pressure; MRI: magnetic resonance imaging; NSAID: nonsteroidal anti-inflammatory drug; OD: osmotic demyelination; P-[Glc]: plasma [Glucose]; P-[Na+]: plasma [Na+]; SIADH: syndrome of inappropriate antidiuretic hormone; TBW: total body water; TIH: thiazide-induced hyponatraemia; U-[Na+]: urine [Na+]; U-Osm: urine osmolality; Vasopressin: arginine vasopressin.
Competing interests
COS has participated in a Nordic Hyponatremia Network meeting sponsored by Otsuka Pharma Scandinavia AB and has lectured twice for the company without receiving a fee.
Authors' contributions
COS and TR have equally participated in the development of the idea and COS has drafted the manuscript. Both authors read and approved the final manuscript.
Box 1. Calculations
Example 1: Treatment with 2 ml/kg 3% NaCl
A 70 kg female with severe cerebral symptoms and P-[Na+]1 = 118 mmol/l is given a 2 ml/kg bolus of 3% NaCl ([Na+] = 513 mmol/l). According to Equation 2, the resulting P-[Na+]2 can be calculated.
TBW: estimated to 35 l (TBW is 50% of the total weight)
Conclusion: One bolus of 2 ml/kg 3% NaCl results in an immediate, controllable rise i P-[Na+] of about 2 mmol/l.
Example 2: Treatment of SIADH with 0.9% NaCl
A 70 kg male with P-[Na+]1 = 112 mmol/l is given 1 litre of 0.9% NaCl and has a diuresis of 750 ml with U-[Na+] = 154 mmol/l and U-[K+] = 80 mmol/l over the next 12 h.
According to Equation 2, the resulting P-[Na+]2 can be calculated.
TBW: estimated to 42 l (TBW is 60% of the total weight)
.
Conclusion: 0.9% NaCl worsens his hyponatraemia and SIADH is likely.
This is also illustrated by a negative electrolyte free water clearance cH2Oe:
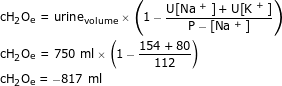 |
Example 3: Estimated water deficit in hypernatraemia
An 80 kg male is admitted with P-[Na+] = 158 mmol/l and P-[Glucose] = 32 mmol/l (577 mg/dl with a correction factor of 2.4 meq/l per 100 mg/dl increase in P-[Glucose]). What is his estimated water deficit?
First, P-[Na+] is corrected for hyperglycaemia:
From Equation 2:
With Δ(Na+ + K+) = 0 and P-[Na+]2 = 140 mmol/l:
With P-[Na+]corrected = 169 mmol/l and TBW 60% of weight:
So, he has an estimated water deficit of about 10 l. There is concomitant, reduced total body sodium because sodium is lost with the osmotic diuresis.
Exclude adrenal and thyroid insufficiency
Contributor Information
Christian Overgaard-Steensen, Email: ketamin@dadlnet.dk.
Troels Ring, Email: tring@gvdnet.dk.
References
- Funk GC, Lindner G, Druml W, Metnitz B, Schwarz C, Bauer P, Metnitz PG. Incidence and prognosis of dysnatremias present on ICU admission. Intensive Care Med. 2010;17:304–311. doi: 10.1007/s00134-009-1692-0. [DOI] [PubMed] [Google Scholar]
- Lindner G, Funk GC, Schwarz C, Kneidinger N, Kaider A, Schneeweiss B, Kramer L, Druml W. Hypernatremia in the critically ill is an independent risk factor for mortality. Am J Kidney Dis. 2007;17:952–957. doi: 10.1053/j.ajkd.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Stelfox HT, Ahmed SB, Khandwala F, Zygun D, Shahpori R, Laupland K. The epidemiology of intensive care unit-acquired hyponatraemia and hypernatraemia in medical-surgical intensive care units. Crit Care. 2008;17:R162. doi: 10.1186/cc7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon M, Timsit JF, Francais A, Nguile-Makao M, Adrie C, Cohen Y, Garrouste-Orgeas M, Goldgran-Toledano D, Dumenil AS, Jamali S, Cheval C, Allaouchiche B, Souweine B, Azoulay E. Association between hypernatraemia acquired in the ICU and mortality: a cohort study. Nephrol Dial Transplant. 2010;17:2510–2515. doi: 10.1093/ndt/gfq067. [DOI] [PubMed] [Google Scholar]
- DeVita MV, Gardenswartz MH, Konecky A, Zabetakis PM. Incidence and etiology of hyponatremia in an intensive care unit. Clin Nephrol. 1990;17:163–166. [PubMed] [Google Scholar]
- Hoorn EJ, Betjes MG, Weigel J, Zietse R. Hypernatraemia in critically ill patients: too little water and too much salt. Nephrol Dial Transplant. 2008;17:1562–1568. doi: 10.1093/ndt/gfm831. [DOI] [PubMed] [Google Scholar]
- Aiyagari V, Deibert E, Diringer MN. Hypernatremia in the neurologic intensive care unit: how high is too high? J Crit Care. 2006;17:163–172. doi: 10.1016/j.jcrc.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Polderman KH, Schreuder WO, Strack van Schijndel RJ, Thijs LG. Hypernatremia in the intensive care unit: an indicator of quality of care? Crit Care Med. 1999;17:1105–1108. doi: 10.1097/00003246-199906000-00029. [DOI] [PubMed] [Google Scholar]
- Kovesdy CP, Lott EH, Lu JL, Malakauskas SM, Ma JZ, Molnar MZ, Kalantar-Zadeh K. Hyponatremia, hypernatremia, and mortality in patients with chronic kidney disease with and without congestive heart failure. Circulation. 2012;17:677–684. doi: 10.1161/CIRCULATIONAHA.111.065391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overgaard-Steensen C. Initial approach to the hyponatremic patient. Acta Anaesthesiol Scand. 2011;17:139–148. doi: 10.1111/j.1399-6576.2010.02311.x. [DOI] [PubMed] [Google Scholar]
- Clayton JA, Le Jeune IR, Hall IP. Severe hyponatraemia in medical inpatients: aetiology, assessment and outcome. QJM. 2006;17:505–511. doi: 10.1093/qjmed/hcl071. [DOI] [PubMed] [Google Scholar]
- Huda MS, Boyd A, Skagen K, Wile D, van HC, Watson I, Wong S, Gill G. Investigation and management of severe hyponatraemia in a hospital setting. Postgrad Med J. 2006;17:216–219. doi: 10.1136/pmj.2005.036947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman IS, Leibman J, O'Meara MP, Birkenfeld LW. Interrelations between serum sodium concentration, serum osmolarity and total exchangeable sodium, total exchangeable potassium and total body water. J Clin Invest. 1958;17:1236–1256. doi: 10.1172/JCI103712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overgaard-Steensen C, Larsson A, Bluhme H, Tonnesen E, Frokiaer J, Ring T. Edelman's equation is valid in acute hyponatremia in a porcine model: plasma sodium concentration is determined by external balances of water and cations. Am J Physiol Regul Integr Comp Physiol. 2010;17:R120–R129. doi: 10.1152/ajpregu.00412.2009. [DOI] [PubMed] [Google Scholar]
- Rose BD. New approach to disturbances in the plasma sodium concentration. Am J Med. 1986;17:1033–1040. doi: 10.1016/0002-9343(86)90401-8. [DOI] [PubMed] [Google Scholar]
- Mallie JP, Ait-Djaffer Z, Laroche F, Mario J, Perrier JC, Voltz C, Halperin ML. Variations in plasma sodium concentration in post-operative patients depend on an electrolyte-free water balance, part of a tonicity balance. Clin Nephrol. 1998;17:287–292. [PubMed] [Google Scholar]
- Hillier TA, Abbott RD, Barrett EJ. Hyponatremia: evaluating the correction factor for hyperglycemia. Am J Med. 1999;17:399–403. doi: 10.1016/S0002-9343(99)00055-8. [DOI] [PubMed] [Google Scholar]
- Baylis PH. Osmoregulation and control of vasopressin secretion in healthy humans. Am J Physiol. 1987;17:R671–R678. doi: 10.1152/ajpregu.1987.253.5.R671. [DOI] [PubMed] [Google Scholar]
- Schrier RW. An odyssey into the milieu interieur: pondering the enigmas. J Am Soc Nephrol. 1992;17:1549–1559. doi: 10.1681/ASN.V2111549. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Kwon TH, Christensen BM, Promeneur D, Frokiaer J, Marples D. Physiology and pathophysiology of renal aquaporins. J Am Soc Nephrol. 1999;17:647–663. doi: 10.1681/ASN.V103647. [DOI] [PubMed] [Google Scholar]
- Lindner G, Kneidinger N, Holzinger U, Druml W, Schwarz C. Tonicity balance in patients with hypernatremia acquired in the intensive care unit. Am J Kidney Dis. 2009;17:674–679. doi: 10.1053/j.ajkd.2009.04.015. [DOI] [PubMed] [Google Scholar]
- Overgaard-Steensen C, Stodkilde-Jorgensen H, Larsson A, Broch-Lips M, Tonnesen E, Frokiaer J, Ring T. Regional differences in osmotic behavior in brain during acute hyponatremia: an in vivo MRI-study of brain and skeletal muscle in pigs. Am J Physiol Regul Integr Comp Physiol. 2010;17:R521–R532. doi: 10.1152/ajpregu.00139.2010. [DOI] [PubMed] [Google Scholar]
- Verbalis JG, Gullans SR. Hyponatremia causes large sustained reductions in brain content of multiple organic osmolytes in rats. Brain Res. 1991;17:274–282. doi: 10.1016/0006-8993(91)90806-7. [DOI] [PubMed] [Google Scholar]
- Pedersen TH, de PF, Nielsen OB. Increased excitability of acidified skeletal muscle: role of chloride conductance. J Gen Physiol. 2005;17:237–246. doi: 10.1085/jgp.200409173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom E, Hojer J, Ludwigs U, Pirskanen R. Fatal hyponatraemic brain oedema due to common gastroenteritis with accidental water intoxication. Intensive Care Med. 1997;17:348–350. doi: 10.1007/s001340050340. [DOI] [PubMed] [Google Scholar]
- Vexler ZS, Ayus JC, Roberts TP, Fraser CL, Kucharczyk J, Arieff AI. Hypoxic and ischemic hypoxia exacerbate brain injury associated with metabolic encephalopathy in laboratory animals. J Clin Invest. 1994;17:256–264. doi: 10.1172/JCI116953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battison C, Andrews PJ, Graham C, Petty T. Randomized, controlled trial on the effect of a 20% mannitol solution and a 7.5% saline/6% dextran solution on increased intracranial pressure after brain injury. Crit Care Med. 2005;17:196–202. doi: 10.1097/01.CCM.0000150269.65485.A6. [DOI] [PubMed] [Google Scholar]
- Sarnaik AP, Meert K, Hackbarth R, Fleischmann L. Management of hyponatremic seizures in children with hypertonic saline: a safe and effective strategy. Crit Care Med. 1991;17:758–762. doi: 10.1097/00003246-199106000-00005. [DOI] [PubMed] [Google Scholar]
- Worthley LI, Thomas PD. Treatment of hyponatraemic seizures with intravenous 29.2% saline. Br Med J (Clin Res Ed) 1986;17:168–170. doi: 10.1136/bmj.292.6514.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WL. Sodium and fluid management in acute brain injury. Curr Neurol Neurosci Rep. 2012;17:466–473. doi: 10.1007/s11910-012-0284-5. [DOI] [PubMed] [Google Scholar]
- Wijdicks EF, Sharbrough FW. New-onset seizures in critically ill patients. Neurology. 1993;17:1042–1044. doi: 10.1212/WNL.43.5.1042. [DOI] [PubMed] [Google Scholar]
- Murphy N, Auzinger G, Bernel W, Wendon J. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology. 2004;17:464–470. doi: 10.1002/hep.20056. [DOI] [PubMed] [Google Scholar]
- Tuttolomondo A, Pinto A, Parrinello G, Licata G. Intravenous high-dose furosemide and hypertonic saline solutions for refractory heart failure and ascites. Semin Nephrol. 2011;17:513–522. doi: 10.1016/j.semnephrol.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Cansick J, Rees L, Koffman G, Van't Hoff W, Bockenhauer D. A fatal case of cerebral oedema with hyponatraemia and massive polyuria after renal transplantation. Pediatr Nephrol. 2009;17:1231–1234. doi: 10.1007/s00467-008-1100-y. [DOI] [PubMed] [Google Scholar]
- Baran D, Hutchinson TA. The outcome of hyponatremia in a general hospital population. Clin Nephrol. 1984;17:72–76. [PubMed] [Google Scholar]
- Hew-Butler T, Ayus JC, Kipps C, Maughan RJ, Mettler S, Meeuwisse WH, Page AJ, Reid SA, Rehrer NJ, Roberts WO, Rogers IR, Rosner MH, Siegel AJ, Speedy DB, Stuempfle KJ, Verbalis JG, Weschler LB, Wharam P. Statement of the Second International Exercise-Associated Hyponatremia Consensus Development Conference, New Zealand, 2007. Clin J Sport Med. 2008;17:111–121. doi: 10.1097/JSM.0b013e318168ff31. [DOI] [PubMed] [Google Scholar]
- Arieff AI, Llach F, Massry SG. Neurological manifestations and morbidity of hyponatremia: correlation with brain water and electrolytes. Medicine (Baltimore) 1976;17:121–129. doi: 10.1097/00005792-197603000-00002. [DOI] [PubMed] [Google Scholar]
- Restuccia T, Gómez-Ansón B, Guevara M, Alessandria C, Torre A, Alayrach ME, Terra C, Martín M, Castellví M, Rami L, Sainz A, Ginès P, Arroyo V. Effects of dilutional hyponatremia on brain organic osmolytes and water content in patients with cirrhosis. Hepatology. 2004;17:1613–1622. doi: 10.1002/hep.20237. [DOI] [PubMed] [Google Scholar]
- Videen JS, Michaelis T, Pinto P, Ross BD. Human cerebral osmolytes during chronic hyponatremia. A proton magnetic resonance spectroscopy study. J Clin Invest. 1995;17:788–793. doi: 10.1172/JCI117728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson BE, Pierides AM, Bradley WG. Central pontine myelinolysis. Two cases with associated electrolyte disturbance. Q J Med. 1976;17:373–386. [PubMed] [Google Scholar]
- Sterns RH, Riggs JE, Schochet SS Jr. Osmotic demyelination syndrome following correction of hyponatremia. N Engl J Med. 1986;17:1535–1542. doi: 10.1056/NEJM198606123142402. [DOI] [PubMed] [Google Scholar]
- Brunner JE, Redmond JM, Haggar AM, Kruger DF, Elias SB. Central pontine myelinolysis and pontine lesions after rapid correction of hyponatremia: a prospective magnetic resonance imaging study. Ann Neurol. 1990;17:61–66. doi: 10.1002/ana.410270110. [DOI] [PubMed] [Google Scholar]
- Sterns RH, Cappuccio JD, Silver SM, Cohen EP. Neurologic sequelae after treatment of severe hyponatremia: a multicenter perspective. J Am Soc Nephrol. 1994;17:1522–1530. doi: 10.1681/ASN.V481522. [DOI] [PubMed] [Google Scholar]
- Mount DB. The brain in hyponatremia: both culprit and victim. Semin Nephrol. 2009;17:196–215. doi: 10.1016/j.semnephrol.2009.03.021. [DOI] [PubMed] [Google Scholar]
- Lohr JW. Osmotic demyelination syndrome following correction of hyponatremia: association with hypokalemia. Am J Med. 1994;17:408–413. doi: 10.1016/0002-9343(94)90166-X. [DOI] [PubMed] [Google Scholar]
- Adrogue HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;17:1581–1589. doi: 10.1056/NEJM200005253422107. [DOI] [PubMed] [Google Scholar]
- Verbalis JG, Goldsmith SR, Greenberg A, Schrier RW, Sterns RH. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;17:S1–21. doi: 10.1016/j.amjmed.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Bissram M, Scott FD, Liu L, Rosner MH. Risk factors for symptomatic hyponatraemia: the role of pre-existing asymptomatic hyponatraemia. Intern Med J. 2007;17:149–155. doi: 10.1111/j.1445-5994.2006.01294.x. [DOI] [PubMed] [Google Scholar]
- Lin CM, Po HL. Extrapontine myelinolysis after correction of hyponatremia presenting as generalized tonic seizures. Am J Emerg Med. 2008;17:632–636. doi: 10.1016/j.ajem.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Vu T, Wong RF, Hamblin PS, Zajac JF, Grossmann MF. Patients presenting with severe hypotonic hyponatremia: etiological factors, assessment, and outcomes. Hosp Pract (Minneap) 2009;17:128–136. doi: 10.3810/hp.2009.12.266. [DOI] [PubMed] [Google Scholar]
- Sterns RH, Nigwekar SU, Hix JK. The treatment of hyponatremia. Semin Nephrol. 2009;17:282–299. doi: 10.1016/j.semnephrol.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Moritz ML, Ayus JC. 100 cc 3% sodium chloride bolus: a novel treatment for hyponatremic encephalopathy. Metab Brain Dis. 2010;17:91–96. doi: 10.1007/s11011-010-9173-2. [DOI] [PubMed] [Google Scholar]
- Liamis G, Kalogirou M, Saugos V, Elisaf M. Therapeutic approach in patients with dysnatraemias. Nephrol Dial Transplant. 2006;17:1564–1569. doi: 10.1093/ndt/gfk090. [DOI] [PubMed] [Google Scholar]
- Perianayagam A, Sterns RH, Silver SM, Grieff M, Mayo R, Hix J, Kouides R. DDAVP is effective in preventing and reversing inadvertent overcorrection of hyponatremia. Clin J Am Soc Nephrol. 2008;17:331–336. doi: 10.2215/CJN.03190807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann M, Dickie H, Tovey L, Treacher D. Management of sodium disorders during continuous haemofiltration. Crit Care. 2010;17:418. doi: 10.1186/cc9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendland EM, Kaplan AA. A proposed approach to the dialysis prescription in severely hyponatremic patients with end-stage renal disease. Semin Dial. 2012;17:82–85. doi: 10.1111/j.1525-139X.2011.00981.x. [DOI] [PubMed] [Google Scholar]
- Chung HM, Kluge R, Schrier RW, Anderson RJ. Clinical assessment of extracellular fluid volume in hyponatremia. Am J Med. 1987;17:905–908. doi: 10.1016/0002-9343(87)90649-8. [DOI] [PubMed] [Google Scholar]
- Hoyle GE, Chua M, Soiza RL. Volaemic assessment of the elderly hyponatraemic patient: reliability of clinical assessment and validation of bioelectrical impedance analysis. QJM. 2011;17:35–39. doi: 10.1093/qjmed/hcq157. [DOI] [PubMed] [Google Scholar]
- Shapiro DS, Sonnenblick M, Galperin I, Melkonyan L, Munter G. Severe hyponatraemia in elderly hospitalized patients: prevalence, aetiology and outcome. Intern Med J. 2010;17:574–580. doi: 10.1111/j.1445-5994.2010.02217.x. [DOI] [PubMed] [Google Scholar]
- Fenske W, Maier SK, Blechschmidt A, Allolio B, Stork S. Utility and limitations of the traditional diagnostic approach to hyponatremia: a diagnostic study. Am J Med. 2010;17:652–657. doi: 10.1016/j.amjmed.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;17:673–680. doi: 10.1053/j.ajkd.2007.07.015. [DOI] [PubMed] [Google Scholar]
- Kamel KS, Halperin ML. Managing overly rapid correction of chronic hyponatremia: an ounce of prevention or a pound of cure. J Am Soc Nephrol. 2010;17:2015–2016. doi: 10.1681/ASN.2010101062. [DOI] [PubMed] [Google Scholar]
- Glover M, Clayton J. Thiazide-induced hyponatraemia: epidemiology and clues to pathogenesis. Cardiovasc Ther. 2012;17:e219–226. doi: 10.1111/j.1755-5922.2011.00286.x. [DOI] [PubMed] [Google Scholar]
- Steele A, Gowrishankar M, Abrahamson S, Mazer CD, Feldman RD, Halperin ML. Postoperative hyponatremia despite near-isotonic saline infusion: a phenomenon of desalination. Ann Intern Med. 1997;17:20–25. doi: 10.7326/0003-4819-126-1-199701010-00003. [DOI] [PubMed] [Google Scholar]
- Clark BA, Shannon RP, Rosa RM, Epstein FH. Increased susceptibility to thiazide-induced hyponatremia in the elderly. J Am Soc Nephrol. 1994;17:1106–1111. doi: 10.1681/ASN.V541106. [DOI] [PubMed] [Google Scholar]
- Friedman E, Shadel M, Halkin H, Farfel Z. Thiazide-induced hyponatremia. Reproducibility by single dose rechallenge and an analysis of pathogenesis. Ann Intern Med. 1989;17:24–30. doi: 10.7326/0003-4819-110-1-24. [DOI] [PubMed] [Google Scholar]
- Lee WH, Packer M. Prognostic importance of serum sodium concentration and its modification by converting-enzyme inhibition in patients with severe chronic heart failure. Circulation. 1986;17:257–267. doi: 10.1161/01.CIR.73.2.257. [DOI] [PubMed] [Google Scholar]
- Schrier RW. Use of diuretics in heart failure and cirrhosis. Semin Nephrol. 2011;17:503–512. doi: 10.1016/j.semnephrol.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Konstam MA, Gheorghiade M, Burnett JC Jr, Grinfeld L, Maggioni AP, Swedberg K, Udelson JE, Zannad F, Cook T, Ouyang J, Zimmer C, Orlandi C. Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;17:1319–1331. doi: 10.1001/jama.297.12.1319. [DOI] [PubMed] [Google Scholar]
- Cho C, Logan JL, Lien YH. Massive aquaresis after tolvaptan administration and albumin infusion in a patient with alcoholic cirrhosis. Am J Med. 2012;17:e5–e6. doi: 10.1016/j.amjmed.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Hew-Butler T. Arginine vasopressin, fluid balance and exercise: is exercise-associated hyponatraemia a disorder of arginine vasopressin secretion. Sports Med. 2010;17:459–479. doi: 10.2165/11532070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML. Cerebral salt wasting: truths, fallacies, theories, and challenges. Crit Care Med. 2002;17:2575–2579. doi: 10.1097/00003246-200211000-00028. [DOI] [PubMed] [Google Scholar]
- Moritz ML. Syndrome of inappropriate antidiuresis and cerebral salt wasting syndrome: are they different and does it matter. Pediatr Nephrol. 2012;17:689–693. doi: 10.1007/s00467-012-2112-1. [DOI] [PubMed] [Google Scholar]
- Connolly ES Jr, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, Patel AB, Thompson BG, Vespa P. American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the Management of Aneurysmal Subarachnoid Hemorrhage: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2012;17:1711–1737. doi: 10.1161/STR.0b013e3182587839. [DOI] [PubMed] [Google Scholar]
- Luft FC, Rankin LI, Bloch R, Weyman AE, Willis LR, Murray RH, Grim CE, Weinberger MH. Cardiovascular and humoral responses to extremes of sodium intake in normal black and white men. Circulation. 1979;17:697–706. doi: 10.1161/01.CIR.60.3.697. [DOI] [PubMed] [Google Scholar]
- Andersen LJ, Andersen JL, Pump B, Bie P. Natriuresis induced by mild hypernatremia in humans. Am J Physiol Regul Integr Comp Physiol. 2002;17:R1754–R1761. doi: 10.1152/ajpregu.00732.2001. [DOI] [PubMed] [Google Scholar]
- Yee AH, Burns JD, Wijdicks EF. Cerebral salt wasting: pathophysiology, diagnosis, and treatment. Neurosurg Clin N Am. 2010;17:339–352. doi: 10.1016/j.nec.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Ellison DH, Berl T. Clinical practice. The syndrome of inappropriate antidiuresis. N Engl J Med. 2007;17:2064–2072. doi: 10.1056/NEJMcp066837. [DOI] [PubMed] [Google Scholar]
- Fenske W, Stork S, Koschker AC, Blechschmidt A, Lorenz D, Wortmann S, Allolio B. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;17:2991–2997. doi: 10.1210/jc.2008-0330. [DOI] [PubMed] [Google Scholar]
- Maesaka JK, Imbriano LJ, Ali NM, Ilamathi E. Is it cerebral or renal salt wasting. Kidney Int. 2009;17:934–938. doi: 10.1038/ki.2009.263. [DOI] [PubMed] [Google Scholar]
- Sterns RH, Silver SM. Cerebral salt wasting versus SIADH: what difference. J Am Soc Nephrol. 2008;17:194–196. doi: 10.1681/ASN.2007101118. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Rosenthal SM, Vargas GA, Fenwick RG, Huang EA, Matsuda-Abedini M, Lustig RH, Mathias RS, Portale AA, Miller WL, Gitelman SE. Nephrogenic syndrome of inappropriate antidiuresis. N Engl J Med. 2005;17:1884–1890. doi: 10.1056/NEJMoa042743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Moore K, Tormey W, Baylis PH, Thompson CJ. Downward resetting of the osmotic threshold for thirst in patients with SIADH. Am J Physiol Endocrinol Metab. 2004;17:E1019–E1023. doi: 10.1152/ajpendo.00033.2004. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Johnson AK. The physiological regulation of thirst and fluid intake. News Physiol Sci. 2004;17:1–6. doi: 10.1152/nips.01470.2003. [DOI] [PubMed] [Google Scholar]
- Anderson RJ, Chung HM, Kluge R, Schrier RW. Hyponatremia: a prospective analysis of its epidemiology and the pathogenetic role of vasopressin. Ann Intern Med. 1985;17:164–168. doi: 10.7326/0003-4819-102-2-164. [DOI] [PubMed] [Google Scholar]
- Decaux G. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) Semin Nephrol. 2009;17:239–256. doi: 10.1016/j.semnephrol.2009.03.005. [DOI] [PubMed] [Google Scholar]
- Schwartz WB, Bennett W, Curelop S, Bartter FC. A syndrome of renal sodium loss and hyponatremia probably resulting from inappropriate secretion of antidiuretic hormone. Am J Med. 1957;17:529–542. doi: 10.1016/0002-9343(57)90224-3. [DOI] [PubMed] [Google Scholar]
- Fenske W, Allolio B. The syndrome of inappropriate secretion of antidiuretic hormone: diagnostic and therapeutic advances. Horm Metab Res. 2010;17:691–702. doi: 10.1055/s-0030-1255117. [DOI] [PubMed] [Google Scholar]
- Decaux G, Andres C, Gankam KF, Soupart A. Treatment of euvolemic hyponatremia in the intensive care unit by urea. Crit Care. 2010;17:R184. doi: 10.1186/cc9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soupart A, Penninckx R, Stenuit A, Decaux G. Azotemia (48 h) decreases the risk of brain damage in rats after correction of chronic hyponatremia. Brain Res. 2000;17:167–172. doi: 10.1016/S0006-8993(99)02259-3. [DOI] [PubMed] [Google Scholar]
- Gross PA, Wagner A, Decaux G. Vaptans are not the mainstay of treatment in hyponatremia: perhaps not yet. Kidney Int. 2011;17:594–600. doi: 10.1038/ki.2011.78. [DOI] [PubMed] [Google Scholar]
- Kamoi K, Tamura T, Tanaka K, Ishibashi M, Yamaji T. Hyponatremia and osmoregulation of thirst and vasopressin secretion in patients with adrenal insufficiency. J Clin Endocrinol Metab. 1993;17:1584–1588. doi: 10.1210/jc.77.6.1584. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Fukuyama J, Kabayama Y, Harada H. Dual facets of hyponatraemia and arginine vasopressin in patients with ACTH deficiency. Clin Endocrinol (Oxf) 1998;17:785–792. doi: 10.1046/j.1365-2265.1998.00621.x. [DOI] [PubMed] [Google Scholar]
- Arlt W, Allolio B. Adrenal insufficiency. Lancet. 2003;17:1881–1893. doi: 10.1016/S0140-6736(03)13492-7. [DOI] [PubMed] [Google Scholar]
- Liamis G, Milionis HJ, Elisaf M. Endocrine disorders: causes of hyponatremia not to neglect. Ann Med. 2011;17:179–187. doi: 10.3109/07853890.2010.530680. [DOI] [PubMed] [Google Scholar]
- Park CW, Shin YS, Ahn SJ, Kim SY, Choi EJ, Chang YS, Bang BK. Thyroxine treatment induces upregulation of renin-angiotensin-aldosterone system due to decreasing effective plasma volume in patients with primary myxoedema. Nephrol Dial Transplant. 2001;17:1799–1806. doi: 10.1093/ndt/16.9.1799. [DOI] [PubMed] [Google Scholar]
- Liamis G, Tsimihodimos V, Doumas M, Spyrou A, Bairaktari E, Elisaf M. Clinical and laboratory characteristics of hypernatraemia in an internal medicine clinic. Nephrol Dial Transplant. 2008;17:136–143. doi: 10.1093/ndt/gfm376. [DOI] [PubMed] [Google Scholar]
- Moritz ML, Ayus JC. Preventing neurological complications from dysnatremias in children. Pediatr Nephrol. 2005;17:1687–1700. doi: 10.1007/s00467-005-1933-6. [DOI] [PubMed] [Google Scholar]
- Chassagne P, Druesne L, Capet C, Menard JF, Bercoff E. Clinical presentation of hypernatremia in elderly patients: a case control study. J Am Geriatr Soc. 2006;17:1225–1230. doi: 10.1111/j.1532-5415.2006.00807.x. [DOI] [PubMed] [Google Scholar]
- Palevsky PM, Bhagrath R, Greenberg A. Hypernatremia in hospitalized patients. Ann Intern Med. 1996;17:197–203. doi: 10.7326/0003-4819-124-2-199601150-00002. [DOI] [PubMed] [Google Scholar]
- Lindner G, Schwarz C, Funk GC. Osmotic diuresis due to urea as the cause of hypernatraemia in critically ill patients. Nephrol Dial Transplant. 2012;17:962–967. doi: 10.1093/ndt/gfr428. [DOI] [PubMed] [Google Scholar]
- Raman A, Schoeller DA, Subar AF, Troiano RP, Schatzkin A, Harris T, Bauer D, Bingham SA, Everhart JE, Newman AB, Tylavsky FA. Water turnover in 458 American adults 40-79 yr of age. Am J Physiol Renal Physiol. 2004;17:F394–F401. doi: 10.1152/ajprenal.00295.2003. [DOI] [PubMed] [Google Scholar]
- Marko NF. Hypertonic saline, not mannitol, should be considered gold-standard medical therapy for intracranial hypertension. Crit Care. 2012;17:113. doi: 10.1186/cc11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn T. Hypernatremia without water depletion. Clin Nephrol. 2011;17:130–135. doi: 10.5414/cn106866. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Takaya A, Nakagawa T, Sakaguchi I, Nishi K. Fatal hypernatremia due to drinking a large quantity of shoyu (Japanese soy sauce) J Forensic Leg Med. 2011;17:91–92. doi: 10.1016/j.jflm.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Bhave G, Neilson EG. Volume depletion versus dehydration: how understanding the difference can guide therapy. Am J Kidney Dis. 2011;17:302–309. doi: 10.1053/j.ajkd.2011.02.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn A, Brachet E, Blum D. Controlled fall in natremia and risk of seizures in hypertonic dehydration. Intensive Care Med. 1979;17:27–31. doi: 10.1007/BF01738999. [DOI] [PubMed] [Google Scholar]
- Lee JH, Arcinue E, Ross BD. Brief report: organic osmolytes in the brain of an infant with hypernatremia. N Engl J Med. 1994;17:439–442. doi: 10.1056/NEJM199408183310704. [DOI] [PubMed] [Google Scholar]
- Fang C, Mao J, Dai Y, Xia Y, Fu H, Chen Y, Wang Y, Liu A. Fluid management of hypernatraemic dehydration to prevent cerebral oedema: a retrospective case control study of 97 children in China. J Paediatr Child Health. 2010;17:301–303. doi: 10.1111/j.1440-1754.2010.01712.x. [DOI] [PubMed] [Google Scholar]
- Righini A, Ramenghi L, Zirpoli S, Mosca F, Triulzi F. Brain apparent diffusion coefficient decrease during correction of severe hypernatremic dehydration. AJNR Am J Neuroradiol. 2005;17:1690–1694. [PMC free article] [PubMed] [Google Scholar]
- Alshayeb HM, Showkat A, Babar F, Mangold T, Wall BM. Severe hypernatremia correction rate and mortality in hospitalized patients. Am J Med Sci. 2011;17:356–360. doi: 10.1097/MAJ.0b013e31820a3a90. [DOI] [PubMed] [Google Scholar]
- Moritz ML, Ayus JC. The changing pattern of hypernatremia in hospitalized children. Pediatrics. 1999;17:435–439. doi: 10.1542/peds.104.3.435. [DOI] [PubMed] [Google Scholar]