

Abstract
An 18F-labeled caspase-3 sensitive nano-aggregation PET tracer ([18F]CSNAT) was prepared and evaluated for imaging caspase-3 activity in doxorubicin-treated tumor xenografts. This enzyme-activatable PET tracer is designed to function in a novel mechanism – enhanced retention of the 18F activity in apoptotic tumors is achieved through intramolecular macrocyclization and in situ aggregation upon caspase-3 activation.
Keywords: PET, apoptosis, cyclization, caspase-3
Positron emission tomography (PET) is a powerful non-invasive molecular imaging technique that uses molecules labeled with positron-emitting isotopes such as carbon-11, fluorine-18, and copper-64, to monitor biochemical processes in living subject in real time with excellent sensitivity.[1] A contrast-enhanced PET image is produced through selective retention of radioactivity at the location where the molecular target is present, and the common strategy is based on the binding of a radiolabeled ligand to the target receptor molecule. When the molecular target is an enzyme, the methods to generate PET imaging contrast are very few,[2] such as 2-[18F]fluoro-2-deoxy-glucose ([18F]FDG) for imaging hexokinase[3] and PET tracers for imaging herpes simplex virus-1 thymidine kinase.[4] This communication reports a new strategy for developing PET tracers for imaging caspase-3 activity in tumors treated with doxorubicin.
One of the most important pathways of chemotherapeutic-induced apoptosis leads to the activation of caspase-3, a central scavenging peptidase that cleaves a specific peptide sequence DEVD-X between X and D (where is X is any amino acid), committing the cell to programmed cell death—apoptosis.[5] While activatable fluorescent probes have been developed for imaging caspase-3 activity in cells and living mice,[6] there are only reports of radiolabeled caspase-3 inhibitors for PET imaging of caspase-3 in apoptotic cells.[7] There isn’t a general strategy to develop a PET tracer that is mechanistically similar to the activatable fluorescent probes in imaging caspase-3 activity with signal amplification.
We have previously described a biocompatible condensation reaction between 2-cyanobenzothiazole (CBT) and free cysteine for specific labeling of terminal cysteine residues in proteins[8] and controlled self-assembly of nano-aggregates in cells.[9] In this communication, we applied it to develop PET tracers for imaging caspase activity, as outlined in Scheme 1. The 18F-labeled caspase-sensitive nano-aggregation PET tracer ([18F]C-SNAT, or 1) has a 2-cyano-6-hydroxyquinoline (CHQ) and a cysteine residue whose amino group is coupled to the peptide substrate of caspase-3, DEVD,[10] and whose mercapto group of its side chain is converted to a disulfide bond. The 18F label is introduced to a propargylglycine residue between CHQ and the cysteine residue via the copper-catalyzed azide-alkyne 1,3-dipolar cyloaddition (CuAAC). Upon the activation of caspase-3 that cleaves the peptide DEVD, the amino group of the cysteine residue is released free, and the reductive intracellular environment reduces the disulfide to generate a free thiol. As shown before,[11] free cysteine and CHQ can undergo fast intramolecular cyclization through the condensation reaction (halflife at 119.8±10.2s). The cyclized products are more hydrophobic and self-assemble in situ into nanoparticles with a high density of 18F activity, resulting in prolonged retention in apoptotic cells and enhanced PET imaging contrast.
Scheme 1.
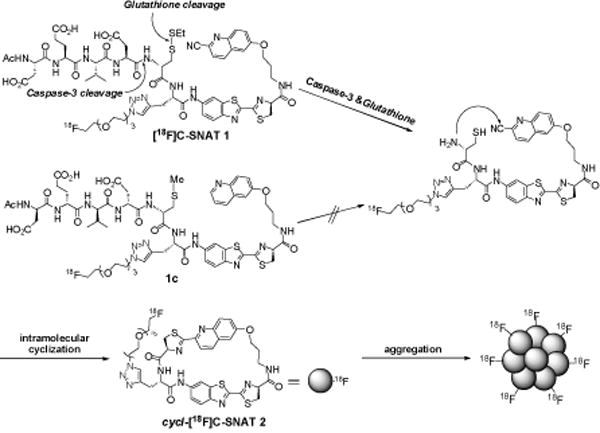
Activation, intramolecular cyclization of [18F] C-SNAT (1) and aggregation of cyclized product (2).
The control tracer 1-D is designed to have a similar structure and pharmacokinetics to 1 but cannot be cleaved by caspase-3 and undergo cyclization and aggregation. Therefore, it contains the same substrate sequence of caspase-3 but made in D-amino acids. In addition, 1-D does not have the cyano substitution on its quinoline and the mercapto group of the cysteine side chain is methylated.
The precursor 3a for synthesizing 1 was prepared according to Scheme 2. N-Boc-propragylglycine was coupled with 6-amino-2-cyanobenzothiazole to afford 4. Deprotection of N-Boc group followed by coupling with protected D-cysteine provided the dipeptide 5. Condensation of 5 and 6a between the CBT group and cysteine under mild conditions, subsequent deprotection of N-Boc and S-Trt groups and protection of the thiol group gave intermediate 7a. The amino group of 7a was coupled with the protected caspase-3 substrate, Ac-Asp(tBu)-Glu(tBu)-Val-Asp(tBu)-OH 8a, and final deprotection of the t-Butyl group gave the desired precursor 3a. The precursor 3b for the control PET tracer 1-D consisted of D-peptide amino acids, and was prepared similarly.
Scheme 2.
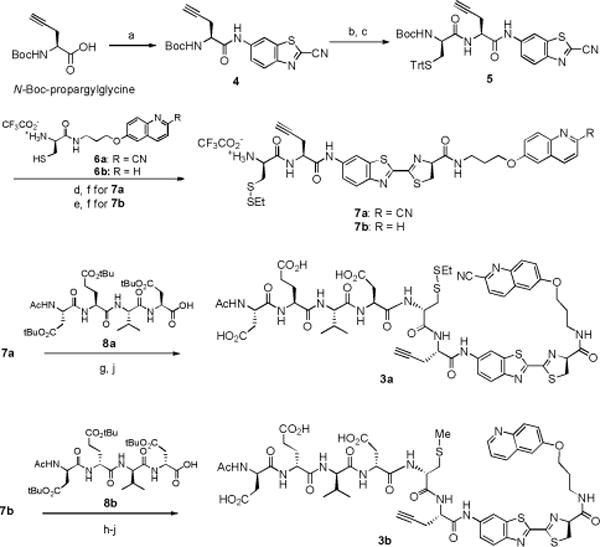
Synthesis of precursors 3a and 3b. a) i-butyl chloroformate, 4-methylmorpholine, THF, 0 °C, 2 h and then 6-amino-2-cyanobenzothiazole, THF, 0 °C to RT, 12 h; 85%. b) 60% TFA in DCM, RT, 1 h. c) N-Boc-S-Trt-D-Cysteine, HBTU, DIPEA, DMF, RT, 2 h; 80% from 4. d) 6a, DIPEA, DMF, RT, 30 min. e) 6b, DIPEA, DMF, 30 min. f) 60% TFA in DCM, RT, 1 h and then 2-(ethyldisulfanyl) pyridine, MeOH, RT, 2 h; 51% from 5 for (7a), 40% for 7b. g) Ac-DEVD-OH (8a), HBTU, DIPEA, THF, RT, 1 h. h) Ac-devd-OH (8b), HBTU, DIPEA, THF, RT, 1 h. i) CH3I, TCEP·HCl, DIPEA, DMF, 1 h. j) 50% TFA in DCM, RT, 2 h; 69% (3a) from 7a, 38% (3b) from 7b.
Both precursors were labeled with 18F through CuAAC, as reported before[12] (Scheme 3). The prosthetic group 10 was obtained in a one-step substitution of 9 by 18F in the presence of Kryptofix 222 (4,7,13,16,21,24-Hexaoxa-1,10-diazabicyclo[8.8.8] hexacosane) with a radiochemical yield (RCY) of 37±8% (n=16) (decay corrected to end of bombardment, EOB). The CuAAC labeling of 3a by 10 occurred in mild conditions (40–60 °C). When CuSO4 and sodium ascorbate were employed, the RCY (EOB) in this step was 15±6% (n=6) for 1, and 25±8% (n=4) for 1-D. When Cu(CH3)4PF6 and BPDS were applied, the RCY (EOB) increased to 30±5% (n=4) for 1 with less side products, probably due to improved stability of 1 in the absent of sodium ascorbate. The final product was purified on a semi-preparative HPLC and formulated in saline containing <10% EtOH. During the HPLC purification of 1-D, because both the precursor 3b and product 1-D had very close retention times (ΔtR<0.1 min), 3-azidopropan-1-amine hydrochloride was added to convert remaining 3b to a product with its retention time shifted to a much earlier time (ΔtR>10 min), which significantly improved the HPLC purification (Figure S1). In summary, 1 and 1-D were successfully obtained with an overall RCY (decay corrected to end of synthesis, EOS) and specific radioactivity (EOS) of 3.2±0.1% and 63±7.4 GBq/μmol (1.7±0.2 Ci/μmol) for 1, 1.8±0.8% and 99.9±33.3 GBq/μmol (2.7±0.9 Ci/μmol) for 1-D (radiochemical purity > 99%, chemical purity > 95%, Figure S2) after a total synthesis time of 3–3.5 h from EOB.
Scheme 3.
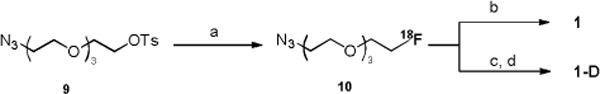
Radiosynthesis of 1 and 1-D, a) 18F/Kryptofix 222/K2CO3, DMSO, 110 °C, 20 min. b) 3a, Cu(CH3CN)4PF6, BPDS, DMSO/water, 60 °C, 30 min. c) 3b, CuSO4, sodium ascorbate, DMSO/water, 40 °C, 30 min. d) 3-azidopropan-1-amine hydrochloride, 40 °C, 30 min.
The serum stability of 1 and 1-D was tested in human and mouse serum. Both tracers showed > 90% intact in human serum after 2 h incubation at 37 °C. In mouse serum, 1-D displayed higher stability than 1: 90% of intact 1-D vs. 60% of intact 1 was observed after 1 h incubation—probably due to the replacement by the D-amino acids. The cyclization product 2 was also tested and showed >90% intact in mouse serum after 2 h incubation (Figure S3).
To evaluate the tracer 1 for caspase-3 detection, we first incubated it with recombinant caspase-3 in buffer. Within 1 h, 90 % of 1 was shown to cyclize and afford two cyclized isomers based on the Radio-HPLC chromatograph (Figure 1A)[13]. As expected, under the same condition, 1-D could not be cleaved by caspase-3 and no cyclization product but 1-D itself was observed.
Figure 1.
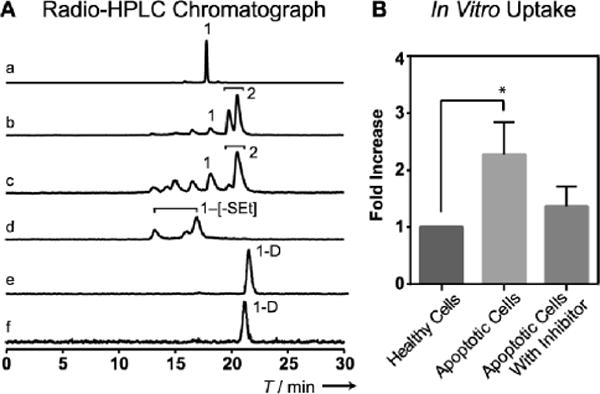
A) Analytical Radio-HPLC showing the radioactive signal composition: a) 1 in saline; b) 1 incubated with caspase-3 in solution for 1 h; c) extraction from 1 cellular uptake in apoptotic cell (4 h); d) extraction from 1 cellular uptake in healthy cell (4 h); No cyclization product 2 but disulfide bond cleaved intermediates (1-[-SEt]) were observed in healthy cells; e) 1-D in saline; f) 1-D incubated with caspase-3 for 1 h. B) In vitro uptake (fold over healthy cell, n=3) of 1 in healthy HeLa cells, apoptotic HeLa cells (treated with 2 μM Doxorubicin) and apoptotic HeLa cells with caspase-3 inhibitor (50 μM of Z-VAD-FMK) added. * indicates statistically significant P=0.045.
Next, we applied 1 to detect caspase-3 in apoptotic cells in vitro. HeLa cell was incubated with doxorubicin (Dox) (2 μM) for 24 h to induce apoptosis. Caspase-3 assays confirmed that the lysates of Dox-treated cells showed 9.5 fold higher caspase-3 activity than that of non-treated cells. (Figure S4A) Both Dox-treated and non-treated cells were then incubated with 1 for 4 h. The retained 18F activity in Dox-treated cells was 2.2-fold of that of non-treated cells. As control, a caspase-3 inhibitor (Z-VAD-fmk) was applied to a similar set of Dox-treated cells, which decreased the retained 18F activity to 1.3-fold (Figure 1B). Both Dox-treated and non-treated cells were subject to extraction and analyzed by Radio-HPLC. The major radioactive fractions from Dox-treated cells were cyclized products of 2 (Figure 1A), but it was not observed in the extraction from non-Dox-treated cells. These results demonstrate that caspase-3 activity can trigger the cyclization of 1 in living cells and enhance the retention of the radioactivity.
The biodistribution of 1 was performed in nude mice that were euthanized at 120 minutes post injection. Organs were collected, weighed, and analyzed with a gamma counter for radioactivity uptake (Table S1). Briefly, kidney and bladder showed high levels of absolute uptake, suggesting primary renal clearance of the tracer. Low tracer activity in muscle promises minimal background signal in PET imaging. Uptake in brain was low, suggesting that 1 could not cross the blood-brain barrier.
Evaluation of 1 and 1-D for PET imaging of caspase-3 activity in vivo was performed in HeLa tumor xenograft-bearing nude mice. Tumors were implanted and grown for more than 10 days before intratumoral injection of 0.2 mg Dox (20 μL). 4 days post treatment, 1 or 1-D (5–15 MBq/135–405 μCi) was injected through the tail vein for PET imaging. Static PET scans (5 min) were performed 65, 125 and 182 min post tracer injection. Figure 2 shows representative PET images of the same mouse injected with 1 before and after Dox treatment and another mouse with 1-D after Dox treatment.
Figure 2.
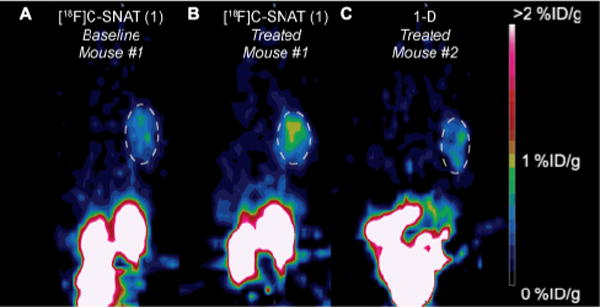
Representative PET images showing HeLa tumor xenografts (white dashed circles) on the right shoulder of mice 125 min after i.v. injection of tracer before (A) and after doxorubicin treatment (B & C). A) Mouse #1 before treatment imaged with 1 (7.8 MBq/211 μCi). B) Mouse #1 after treatment imaged with 1 (12 MBq/324 μCi). C) Mouse #2 after treatment imaged with 1-D (5.4 MBq/146 μCi). All images are normalized to the same scale.
Quantification of the PET images with the activatable PET tracer 1 revealed that the uptake (%ID/g) of the 18F activity in tumors significantly increased after Dox treatment: from 0.81 ± 0.28 (baseline) to 1.17 ± 0.17 (treated) at 65 min, from 0.67 ± 0.24 (baseline) to 1.29 ± 0.07 (treated) at 182 min (Figure 3A); this result correlates well with the caspase-3 level detected in tumors – a 1.9 fold increase in treated tumors (Figure S4B). The uptake difference between baseline and treated increased from 0.36 ± 0.15 at 65 min to 0.63 ± 0.11 at 182 min (Figure 3B), and the uptake ratio between tumor and muscle (T/M) increased from 3.30 fold at 65 min to 7.00 fold at 182 min in treated tumors (Figure 3C).
Figure 3.
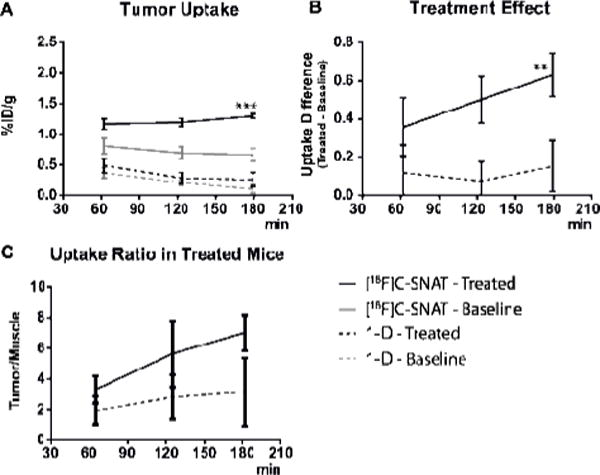
A) Uptake of 1 and 1-D (%ID/g ± sem) in xenograft HeLa tumor and muscle, before and after intratumor injection of Dox (0.2 mg) 4 days prior to the imaging. Uptake is calculated based on 5 min static PET scans at 65, 125 and 182 min. *** indicates p=0.002 at 182 min. B) The uptake of 1 and 1-D in treated tumors; ** shows a significant p=0.0037 at 182 min. C) The ratio between tumor and muscle uptake in treated tumors, calculated based on the uptake (average uptake in tumor/average uptake in muscle region).
In contrast, the uptake of 1-D in both treated and non-treated tumors was much lower than that of 1 (Figure 3A), and the uptake difference between before and after treatment (<0.2%ID/g) was also much smaller (Figure 3B). The ratio of T/M did not show significant increases either (Figure 3C). Our PET imaging results demonstrate that 1 can image caspase-3 activity in drug-treated tumors in vivo and that both caspase-3 activation and cyclization are required for the enhanced imaging contrast in apoptotic tumors.
[18F]C-SNAT (1) compares favorably to known apoptosis PET tracers (Table S2) with both high tumor/muscle ratio in apoptotic tumors and high uptake value (%ID/g) in apoptotic tumors. Consistent with the mechanism, [18F]C-SNAT showed a trend of increasing uptake over the time (Figure 3A) in apoptotic tumors and thus increased differences between treated apoptotic and non-treated tumors at later time points. This trend has not been observed with other apoptosis PET tracers; for example, with [18F]ICMT-11, a PET tracer that binds active caspase-3, the uptake at the apoptotic tumors decreased over the time after injection.[7a] Furthermore, our probe designing principle is not limited to caspase-3 but may serve as a general strategy for developing PET tracers for imaging the activity of other enzymes (i.e. furin, MMPs).
In conclusion, we have successfully designed and synthesized an 18F-labeled caspase-3 triggered nano-aggregation PET tracer ([18F]C-SNAT), and demonstrated its application for imaging caspase-3 activity in doxorubicin-treated tumor xenografts. This activatable PET tracer undergoes intramolecular cyclization and subsequent aggregation upon caspase-3 activation to achieve enhanced retention in apoptotic tumors. Applications of this strategy for other enzyme targets as well as translation of [18F]C-SNAT into clinical studies are currently under investigation.
Supplementary Material
Footnotes
This work has been supported by the Stanford University National Cancer Institute (NCI) Centers of Cancer Nanotechnology Excellence (1U54CA151459-01), the NCI ICMIC@Stanford (1P50CA114747-06), and an IDEA award from Department of Defense Breast Cancer Research Program (W81XWH-09-1-0057). MP is grateful to the postdoctoral fellowship support from the Danish Cancer Foundation, and AS is supported by a postdoctoral fellowship from the Susan Komen Breast Cancer Foundation. The use of Stanford Small Animal Imaging Facility is acknowledged.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Hoffman EJ, Phelps ME. Med Instrum. 1979;13:147–151. [PubMed] [Google Scholar]; b) Gambhir SS. Nat Rev Cancer. 2002;2:683–693. doi: 10.1038/nrc882. [DOI] [PubMed] [Google Scholar]
- 2.a) Ren G, Blum G, Verdoes M, Liu HG, Syed S, Edgington LE, Gheysens O, Miao Z, Jiang H, Gambhir SS, Bogyo M, Cheng Z. PLoS One. 2011;6:e28029. doi: 10.1371/journal.pone.0028029. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Razgulin A, Ma N, Rao J. Chem Soc Rev. 2011;40:4186–4216. doi: 10.1039/c1cs15035a. [DOI] [PubMed] [Google Scholar]
- 3.a) Ido T, Wan CN, Casella V, Fowler JS, Wolf AP, Reivich M, Kuhl DE. J Labelled Compd Radiopharm. 1978;14:175–183. [Google Scholar]; b) Pacak J, Tocik Z, Cerny M. J Chem S D. 1969:77. [Google Scholar]
- 4.a) Tjuvajev JG, Doubrovin M, Akhurst T, Cai S, Balatoni J, Alauddin M, Finn R, Conti P, Tjuvajev JG, Blasberg R. J Nucl Med. 2002;43:1072–1083. [PubMed] [Google Scholar]; b) Iyer M, Barrio JR, Namavari M, Bauer E, Satyamurthy N, Nguyen K, Toyokuni T, Phelps ME, Herschman HR, Gambhir SS. J Nucl Med. 2001;42:96–105. [PubMed] [Google Scholar]; c) Tjuvajev J, Avril N, Safer M, Joshi R, Oku T, Sasajima T, Miyagawa T, Beattie B, Daghighian F, Augenson F, DiResta G, Koutcher J, Zweit J, Finn R, Larson S, Blasberg R. J Nucl Med. 1997;38:239p. [Google Scholar]
- 5.a) Rosado JA, Lopez JJ, Gomez-Arteta E, Redondo PC, Salido GM, Pariente JA. J Cell Physiol. 2006;209:142–152. doi: 10.1002/jcp.20715. [DOI] [PubMed] [Google Scholar]; b) Schwarz K, Simonis G, Yu XJ, Wiedemann S, Strasser RH. Mol Cell Biochem. 2006;281:45–54. doi: 10.1007/s11010-006-0233-1. [DOI] [PubMed] [Google Scholar]; c) Lei W, Yu R, Mandlekar S, Kong ANT. Cancer Res. 1998;58:2102–2106. [PubMed] [Google Scholar]
- 6.a) Barnett EM, Zhang X, Maxwell D, Chang Q, Piwnica-Worms D. Proc Natl Acad Sci USA. 2009;106:9391–9396. doi: 10.1073/pnas.0812884106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Edgington LE, Berger AB, Blum G, Albrow VE, Paulick MG, Lineberry N, Bogyo M. Nat Med. 2009;15:967–974. doi: 10.1038/nm.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang Z, Fan J, Cheney PP, Berezin MY, Edwrds WB, Akers WJ, Shen D, Liang K, Culver JP, Achilefu S. Mol Pharmaceutics. 2009;6:416–427. doi: 10.1021/mp800264k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Nguyen QD, Smith G, Glaser M, Perumal M, Arstad E, Aboagye EO. Proc Natl Acad Sci USA. 2009;106:16375–16380. doi: 10.1073/pnas.0901310106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou D, Chu WH, Chen DL, Wang Q, Reichert DE, Rothfuss J, D’Avignon A, Welch MJ, Mach RH. Org Biomol Chem. 2009;7:1337–1348. doi: 10.1039/b819024k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Faust A, Wagner S, Law MP, Hermann S, Schnockel U, Keul P, Schober O, Schafers M, Levkau B, Kopka K. Q J Nucl Med Mol Imaging. 2007;51:67–73. [PubMed] [Google Scholar]; d) Reshef A, Shirvan A, Akselrod-Ballin A, Wall A, Ziv I. J Nucl Med. 2010;51:837–840. doi: 10.2967/jnumed.109.063917. [DOI] [PubMed] [Google Scholar]; e) Su H, Chen G, Gangadharmath U, Gomez LF, Liang Q, Mu F, Mocharla VP, Szardenings AK, Walsh JC, Xia CF, Yu C, Kolb HC. Mol Imaging Biol. 2013 doi: 10.1007/s11307-013-0644-9. [DOI] [PubMed] [Google Scholar]
- 8.a) Ren HJ, Xiao F, Zhan K, Kim YP, Xie HX, Xia ZY, Rao J. Angew Chem. 2009;121:9838–9842. doi: 10.1002/anie.200903627. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:9658–9662. doi: 10.1002/anie.200903627. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jeon J, Shen B, Xiong LQ, Miao Z, Lee KH, Rao J, Chin FT. Bioconjugate Chem. 2012;23:1902–1908. doi: 10.1021/bc300273m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Liang GL, Ren HJ, Rao J. Nat Chem. 2009;2:54–60. doi: 10.1038/nchem.480. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liang GL, Ronald J, Chen YX, Ye DJ, Pandit P, Ma ML, Rutt B, Rao J. Angew Chem. 2011;123:6407–6410. doi: 10.1002/anie.201007018. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:6283–6286. doi: 10.1002/anie.201007018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caspase-7, another effector caspase can aslo recognize this peptide sequence.
- 11.Ye DJ, Liang GL, Ma ML, Rao J. Angew Chem. 2011;123:2323–2327. [Google Scholar]; Angew Chem Int Ed. 2011;50:2275–2279. doi: 10.1002/anie.201006140. [DOI] [PubMed] [Google Scholar]
- 12.a) Mamat C, Ramenda T, Wuest FR. Mini-Rev Org Chem. 2009;6:21–34. [Google Scholar]; b) Gill HS, Marik J. Nat Protoc. 2011;6:1718–1725. doi: 10.1038/nprot.2011.390. [DOI] [PubMed] [Google Scholar]; c) Nwe K, Brechbiel MW. Cancer Biother Radiopharm. 2009;24:289–302. doi: 10.1089/cbr.2008.0626. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Glaser M, Robins EG. J Labelled Compd Radiopharm. 2009;52:407–414. [Google Scholar]
- 13.Two cyclized isomers are likely diastereomers due to two different ring-closing orientations, as observed in our previous study (ref 11).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.