

Abstract
Importance of the field
Histone deacetylase (HDAC) inhibitors are being developed as a new, targeted class of anticancer drugs.
Area covered in this review
This review focuses on the mechanisms of action of the HDAC inhibitors, which selectively induce cancer cell death.
What the reader will gain
There are 11 zinc-dependent HDACs in humans and the biological roles of these lysine deacetylases are not completely understood. It is clear that these different HDACs are not redundant in their activity. This review focuses on the mechanisms by which HDAC inhibitors can induce transformed cell growth arrest and cell death, inhibit cell mobility and have antiangiogenesis activity. There are more than a dozen HDAC inhibitors, including hydroxamates, cyclic peptides, benzamides and fatty acids, in various stages of clinical trials and many more compounds in preclinical development. The chemically different HDAC inhibitors may target different HDACs.
Take home message
There are extensive preclinical studies with transformed cells in culture and tumor-bearing animal models, as well as limited clinical studies reported to date, which indicate that HDAC inhibitors will be most useful when used in combination with cytotoxic or other targeted anticancer agents.
Keywords: apoptosis, HDAC inhibitor, histone deacetylases, mechanism of action, reactive oxygen species, ROS, suberoylanilide hydroxamic acid
1. Histone deacetylases (HDACs)
Epigenetics refers to the regulation of gene expression though post-translational modification of protein complexes associated with DNA without changes in DNA sequence [1,2]. DNA is packaged in chromatin, which is structurally complex and dynamic, consisting of DNA, histones, and non-histone proteins. These proteins are subject to post-translational modification by acetylation of lysines, methylation of lysines and arginines, phosphorylation of serines, ubiquitination of lysines, sumoylation, proline, isomerization, and ADP-ribosylation. Acetylation and deacetylation, perhaps the best studied of these post-translational protein modifications, are regulated by the activity of two sets of enzymes, histone deacetylases (HDACs) and histone acetyltransferases (HATs) [3,4]. HATs catalyze the acetylation of lysines, which neutralizes the positive charge on histone and allow the negatively charged DNA to assume a transcription-competent conformation. HDACs remove acetyl groups from lysines, allowing interaction between negatively charged DNA and positively charged histone proteins, which can result in heterochromatin and transcriptional silencing of genes. Patterns of acetylation and methylation of lysines, as well as other post-translational modifications, appear to be a code for specific recruitment of protein complexes regulating gene expression and DNA replication and stability [5]. HDACs and HATs have many non-histone protein substrates including proteins of transcription complexes that have a role in regulating gene expression, and proteins in pathways that regulate cell proliferation, cell migration, cell death and angiogenesis [6,7].
Eighteen HDACs have been identified in humans that are classified based on their homology to yeast HDACs (Table 1). Eleven of these HDACs are zinc-dependent enzymes: class I, HDAC1, 2, 3, and 8 have homology to yeast RPD3; class IIa, HDACs 4, 5, 7, and 9 have homology to yeast HDA1; class IIb, HDACs 6 and 10 have two catalytic sites, and class IV, HDAC11, has conserved residues shared with both class I and class II deacetylases [6,8]. Class III HDACs include sirtuins 1 – 7, which have homology to yeast Sir2 and have an absolute requirement for NAD+.
Table 1.
Zinc-dependent histone deacetylases.
| HDAC | Yeast homology | Localization | Size (AA) | Catalytic sites | Chromosomal site |
|---|---|---|---|---|---|
| Class I | RPD3 | ||||
| HDAC1 | Nucleus | 483 | 1 | 1p34.1 | |
| HDAC2 | Nucleus | 488 | 1 | 6p21 | |
| HDAC3 | Nucleus | 423 | 1 | 5q31 | |
| HDAC8 | Nucleus | 377 | 1 | Xq13 | |
| Class IIa | HDA1 | ||||
| HDAC4 | Nuc/Cyt | 1084 | 1 | 2q372 | |
| HDAC5 | Nuc/Cyt | 1122 | 1 | 17q21 | |
| HDAC7 | Nuc/Cyt | 855 | 1 | 12q13 | |
| HDAC9 | Nuc/Cyt | 1011 | 1 | 7p21-p15 | |
| Class IIb | |||||
| HDAC6 | Mainly Cyt | 1215 | 2* | Xp11.22 – 33 | |
| HDAC10 | Mainly Cyt | 669 | 2 | 22q13.31 – 33 | |
| Class IV | |||||
| HDAC11 | Nuc/Cyt | 347 | 1 | 3p25.2 | |
Ubiquitin binding site toward C terminal end.
Analysis of lysine acetylation targets in transformed cells found 3600 acetylated lysines in 1750 proteins [7]. Inhibition of HDACs with vorinostat (suberoylanilide hydroxamic acid; SAHA, see Section 3) altered only about 10% of these acetylation sites. These sites were identified on proteins that regulate gene expression, RNA signaling, DNA damage repair, cell cycle progression, nuclear transport, cytoskeleton function, protein chaperones, and ribosome formation and function [7]. HDACs have many non-histone protein targets; phylogenic studies indicate that HDACs preceded histones in evolution of organisms [9].
Class I HDACs are primarily localized in the nucleus (Table 1). HDAC3 can shuttle between the nucleus and cytoplasm. Class II HDACs are primarily localized in the cytoplasm and shuttle between the nucleus and the cytoplasm. Class I HDACs are expressed in all tissue and have histone as substrates. Class I HDACs are found in protein complexes with transcription factors and co-repressors. HDACs do not bind directly to DNA and are recruited to target genes via association with transcriptional activators and repressors, incorporated into large multiprotein transcription complexes.
Class I HDACs have a simpler structure than class IIa or IIb HDACs, with relatively short amino acid and carboxy terminal extensions from the catalytic site. Class I HDACs have a primary role in cell survival and proliferation, based on, knockout studies and inhibition with class I-selective inhibitors [10,11]. Deletion of both HDAC1 and 2, but not either alone, causes cancer cell death and neural precursor maturation defects. HDAC1 and 2 redundantly regulate cardiac morphogenesis, growth and contractility [12]. HDAC2 is a regulator of chromatin compaction status and its downregulation or inhibition causes chromatin decondensation and sensitization to DNA-targeted anticancer drugs [13]. HDAC3 deletion causes early embryonic lethality. Inactivation of HDAC3 was associated with cessation in cell cycle progression, DNA damage, and impaired repair and apoptosis [14].
Class II HDACs have more tissue-specific regulatory function than class I HDACs [6,8,11]. Class IIa HDACs have conserved binding sites for transcription factors and the chaperone proteins 14-3-3, which are involved in regulation of the shuttling of these enzymes between the nucleus and cytoplasm. HDAC4 regulates neural survival in normal and diseased retinas [15]. The expression of HDAC4 is repressed by miRNA 206, facilitating reinnervation [16]. Mice lacking HDAC4 expression have premature ossification of developing bones, while overexpression of HDAC4 inhibits chondrocyte and osteocyte differentiation. HDAC5 knock-down mice have large hearts [8]. HDAC5 and HDAC9 are involved in the development of myocardium and skeletal muscle. HDAC7 is involved in the regulation of vascular endothelial development and vascular integrity [17].
HDAC6 is unique among the 11 zinc-dependent HDACs in having two catalytic sites and an ubiquitin binding site (see Section 4.7). Specific substrates of HDAC6 include α-tubulin, cortactin, transmembrane proteins such as IFN-αR, HSP90 and other chaperone proteins, and peroxiredoxins [18–21]. Paradoxically, HDAC6 inhibition can increase resistance of cancer cells to oxidative stress by causing the accumulation of acetylated peroxiredoxins. Acetylated peroxiredoxins have increased activity as H2O2 reductases [19]. The ubiquitin binding site toward the c-terminal end of HDAC6 plays a critical role in aggresome formation in the pathway of proteolysis of misfolded proteins [22,23].
Little is known about the function of HDAC10. HDAC11 negatively regulates expression of the gene encoding IL-10 in antigen-presenting cells which induce T-cell activation as well as T-cell tolerance [24]. These findings suggest that HDAC11 has molecular targets that influence immune activation.
Although HDACs are widely distributed in chromatin, inhibition of these enzymes alters the transcription of a relatively small proportion (2 – 10%) of expressed genes in transformed cells [25–28].
The regulation of HDAC activity can occur at multiple levels including protein–protein interaction, post-translational modification (sumoylation, phosphorylation, proteolysis, subcellular localization) and by metabolic co-factors [29,30]. For example, HDAC1 promoter is inducible by IL-2. HDAC4 promoter is regulated in part by P1/SP3 transcription factor. Phosphorylation and subsequent association with 14-3-3 regulates subcellular localization of HDAC4, HDAC5, HDAC7, and HDAC9.
The crystalline structure of the catalytic site of a histone deacetylase-like protein was solved with binding of inhibitors, trichostatin A (TSA) and SAHA [31]. More recently, the crystal structure of human HDAC4, 7 and 8 have been solved [32–35].
2. Aberrant HDACs and cancers
Structural mutations in HDACs associated with cancers appear to be rare. A mutation of HDAC2 has been reported in colon cancer and endometrial cancer cell lines [36]. HDAC4 mutations have been identified in breast cancer in a large scale sequencing study of breast and rectal colon cancers [37].
Altered expression and aberrant recruitment of HDACs have been reported in many human cancers [6,8,38,39]. Systematic analysis of cultured cancer cell lines (primarily cultures of human cancer cells and various human tumor biopsy samples) found that many had higher levels of expression of the zinc-dependent HDACs than in corresponding normal tissues. Overexpression of class I HDACs has been reported in esophageal, prostate, non-small cell lung, gastrointestinal, and oral cancers [38–42]. Analysis of the expression of histone deacetylases in lymphomas found that the most frequently altered HDAC was HDAC6, which was weakly expressed or undetected in 9 out of 14 lymphoid cell lines and in 83 of 89 primary lymphoma tissue specimens [43]. HDAC5 and HDAC10 have been reported as decreased in expression in lung cancer associated with poor prognosis. HDAC6 is overexpressed in many cutaneous T-cell lymphoma (CTCL) and peripheral T-cell lymphoma (PTCL) cells. HDAC8 expression correlates with poor outcome in neuroblastoma and inhibition of HDAC8 induces differentiation of transformed cells [44].
Fusion proteins involving chromosomal translocation have been identified in acute myelocytic leukemia (AML) and acute lymphoblastic leukemia (ALL) [45,46]. The chromosomal translocation results in a fusion protein (MLL-CBP) consisting of the CBP and mixed linage protein, MLL. Aberrant recruitment of HDACs to the transcription factors that involve oncogenetic DNA binding fusion proteins resulting from chromosomal translocations or overexpression of repressive transcription factors also include oncogenetic PMA–RARα or PLZF–RARα and AML1–ETO fusion proteins in acute pro-myelocytic and acute myelogenous leukemia.
3. Histone deacetylase inhibitors (HDACi)
HDACi have been discovered that have different chemical structures including hydroxamic acids, cyclic peptides, electrophilic ketones, short-chain fatty acids, benzamides, boronic acid-based compounds, benzofuranone- and sulfonamide-containing molecules, and α/β peptide structures (Table 2) [47–53]. Various chemical modifications have incorporated solubilizing functional groups, which may improve pharmacokinetic properties. Considerable efforts are being made to develop isoform-selective deacetylase inhibitors such as tubacin, which selectively inhibits HDAC6, and PC-34051, a selective HDAC8 inhibitor [18,50,53].
Table 2.
Histone deacetylase inhibitors in clinical trials as monotherapy and combination (partial list)*.
| Drug name | Structure | Dose range | Clinical trials | Ref. |
|---|---|---|---|---|
| Hydroxamates | ||||
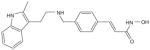
| Vorinostat (SAHA, zolinza) |
|
μmol/l | FDA approved CTCL Phll-Ill (p.o.) |
[6,45,105,126,127,134–146] |
| Panobinostat (LBH589) |
|
nmol/l | Phl-ll (p.o.), Phll (i.v.) | [107,128] |
| Belinostat (PXD101) |
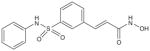
|
μmol/l | Phll (i.v.) Phl (p.o.) | [109,147–154] |
| Givinostat (ITF-2357) |
|
nmol/l | Phl-ll (p.o.) | [48,150] |
| PCI-24781 (CRA-024781) |
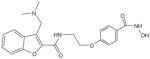
|
nmol/l | Phl (p.o.) | [48,151] |
| JNJ-26481585 |
|
μmol/l | Phl-ll (p.o.) | [48,152] |
| SB-639 |
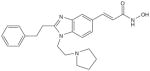
|
μmol/l | Phl (p.o.) | [48,153] |
| Cyclic peptide | ||||
| Romidepsin (depsipeptide, FK228) |
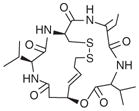
|
nmol/l | FDA approved CTCL Phl-ll (i.v.) |
[109,110,154–158] |
| Aliphatic acids | ||||
| Valproic acid (baceca) (savicol) |
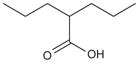
|
mmol/l | Ph ll-lll (p.o) Ph II (top) |
[159] |
| Phenylbutyrate (VP-101, EI-532) |
|
mmol/l | Ph II (p.o.) | [160–163] |
| Pivanex (AN-9) |
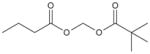
|
mmol/l | Phl-ll (i.v.) | [164] |
| Benzamides | ||||
| Entinostat (MS 275, SNDX 275) |
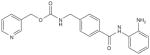
|
μmol/l | Phl-ll (p.o.) | [165,166] |
| MGCD0103 |
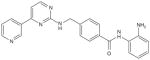
|
μmol/l | Phl-ll (p.o.) | [166,167] |
For reviews of clinical trials with HDACi see references in text.
The hydroxamic class of HDACi, of which vorinostat (SAHA) is the first HDACi developed and approved by the US FDA for clinical use [47], generally have common structural characteristics: zinc-binding moiety (ZBM) in the catalytic pocket, opposite capping group, and straight-chain alkyl, vinyl, or aryl linker connecting the two. These functional groups interact with three relatively conserved regions of the catalytic pocket of HDACs. The zinc facilitates hydrolysis of the acetyl–lysine bond and is at the bottom of the narrow catalytic pocket. A hydrophobic chain with six carbons is generally the optimal link of the ZBM and the opposite capping group. The capping group about the catalytic site opening is generally a hydrophobic structure that interacts with the rim amino acids. The amino acid sequence of the rim surrounding the catalytic site of the different HDACs has greater sequence diversity compared with the other domains, and thus may have the most potential to be manipulated to develop selective HDACi.
A new concept in HDACi structures is combining inhibition of protein kinases and HDACs in one molecule [54]. That structurally diverse compounds are effective inhibitors of HDACs suggests that the mechanism of action of these compounds may involve not only blocking the catalytic site but also binding of the rim of the enzyme with other proteins independent of the deacetylase activity.
4. Mechanism of action of histone deacetylase inhibitors
HDACi can induce transformed cell death by one or more pathways (Figure 1). HDACi can cause transformed cell growth arrest, cell death and inhibition of angiogenesis. The pathway leading to cell death of transformed cells induced by HDACi depends, in part, on the HDACi, concentration and time of exposure to the agent and the molecular characteristics of a particular transformed cell. Normal cells are relatively resistant to HDACi-induced cell death [55,56].
Figure 1.

HDACi can affect one or more pathways, leading to transformed cell death.
4.1 DNA damage and repair
There is no evidence that HDACi are directly mutagenic. Histone acetylation, induced by HDACi, does cause structural alterations in chromatin, which can expose DNA to damaging agents, such as UV, radiation, cytotoxic drugs, and reactive oxygen, resulting in DNA double-strand breaks (DSB) [6,8,57].
HDACi can induce the accumulation of reactive oxygen species (ROS), resulting in DNA damage [55,56,58]. HDACi induce the accumulation of the phosphorylated form of H2AX, a marker of DSB [59]. HDACi can downregulate the expression of genes for DNA repair proteins involved in homologous recombination, including, RAD51, BRACA1, and BRAC2, and in non-homologous DSB repair including Ku70, Ku86 and DNA-PKcs [60–65]. The accumulation of DSB causes altered gene expression and leads to apoptotic cell death. Transformed cells may have many defects in pathways of DSB repair and, unlike normal cells, lack the capacity to repair DNA damage.
The synergy of HDACi and DNA-damaging agents, such as cytotoxic drugs or radiation, may result from the combined effects of HDACi in inhibiting DNA repair processes as well as activating intrinsic and extrinsic cell apoptotic death pathways (see Section 4.4).
4.2 Altered gene expression
HDACi altered gene transcription by inducing acetylation of histones, transcription factors and other proteins regulating gene expression [6–8]. Early experiments with lymphoid cell lines cultured with TSA showed that only about 2% of expressed genes were altered, either increased or decreased, compared with untreated cells [25]. More recent studies, using cDNA arrays, found as many as 10 – 20% of expressed genes altered in their transcription in cell lines of leukemia, multiple myeloma, and colon, kidney, prostate, and breast cancer cells cultured with the HDACi [26–28]. The number of genes with altered expression increased with time of culture and concentration of HDACi. Some changes in gene expression are probably direct effects of the HDACi, while many may be downstream effects. The pattern of alterations of gene expression are similar for different HDAC inhibitors, but there are differences induced by different agents in various transformed cells [28].
The cyclin-dependent kinase (CDK) inhibitor, p21 (WAF1/CIP1), is one of the most commonly induced genes by HDACi [66]. HDACi-induced expression of p21 is independent of p53. In ARP-1 cells, vorinostat caused specific modifications in the pattern of acetylation and methylation of lysines in histones H3 and H4 associated with the proximal promoter region of the p21 gene [67]. Histone acetylation or methylation in the promoter region of the expressed p27 (KIP1) or the silent epsilon globin gene in HDACi-cultured ARP-1 cells were not altered, nor was the expression of these genes. Vorinostat caused a marked decrease in HDAC1 and Myc and recruitment of RNA polymerase II in the protein complex associated with the proximal promoter region of the p21 gene, with little detectable change in HDAC2, Brg1, GCN5, P300 pr Sp1 proteins in the complex. These findings suggest that the selective alteration of transcription of a gene by HDACi may be determined by the composition of proteins in the transcription factor complex including the HDACs.
HDACi can inhibit gene expression mediated by STAT5 [68]. HDACi can repress the transcription of the androgen receptor (AR) gene [69] and block AR-mediated transcriptional activation of genes. HDACi, such as SAHA, can alter the miRNA expression profile in transformed cells [70]. These miRNAs have target genes related to angio-genesis, apoptosis, chromatin modification cell proliferation and differentiation. HDACi can activate a Sp1/Sp3- mediated induction of multiple immediate early (fos, Juh, egr1, egr3, atj3, arc, mr4a1) and stress response genes (mdrg4, Mt1B, MtiE, Mt1f, ME1H) associated with apoptosis of colon cancer cells [71].
4.3 Cell growth arrest
HDACi can induce cell growth arrest in both normal and transformed cells in culture. Vorinostat causes predominantly G1 arrest at low concentration and both G1 and G2/M arrests at higher concentration [66].
In cells cultured with HDACi, increased levels of the CDK inhibitors and decreased levels of cyclins may account for reduced CDK activity, causing dephosphorylation of Rb, blocking E2F activities in the transcription of genes for G1 progression and G1/S transition [72,73]. HDACi can kill both proliferation and non-proliferating transformed cells [56,74]. This is in contrast to the action of many chemotherapy drugs, which are effective only on proliferating transformed cells.
4.4 Induced apoptosis
HDACi can induce death of transformed cells by activating the extrinsic and/or intrinsic apoptotic pathways [6,8,11,75–80]. A number of downstream components, such as activation of caspase-3, are shared in the extrinsic and intrinsic pathways [76]. The extrinsic apoptotic pathway is initiated by binding of death receptor, including Fas, TNF receptor-1 (TNFR-1), TNF-related apoptosis-inducing ligand (TRAIL) receptor (DR-4 and −5), DR-3 (Apo3) and DR-6, to their ligands leading to activation of caspase-8 and −10 [80]. HDACi can upregulate expression of both the death receptors and their ligands in vitro and in vivo in transformed cells, but not in normal cells. For example, M-carboxycinnamic acid, bishydroxamide (CBHA) induced Fas and Fas ligand in neuroblastoma cells [79]. TNF was upregulated by depsipeptide in HL-60 and K562 cells, and C-FLIP, an inhibitor of the death receptor pathway, was downregulated. Sequential treatment with vorinostat followed by TRAIL was shown to target multiple pathways in tumor progression, angiogenesis and metastasis. HDACi can cause TNF-related apoptosis inducing ligand protein degradation by inhibiting the ubiquitin-dependent pathway, an effect that may be the basis of the effectiveness of the combination of HDACi with a proteasome inhibitor in inducing transformed cell death [80]. Taken together, these studies indicate that the extrinsic apoptotic pathway can account for HDACi-induced cell death in many transformed cells. Combination therapy of HDACi with factors inducing the extrinsic apoptotic pathway have a potential for effective therapeutic strategies.
Intrinsic apoptotic pathway is mediated by disruption of mitochondria with the release of mitochondrial intermembrane proteins, including cytochrome c, AIF and Smac, leading to activation of caspases [6,8,11,81,82]. HDACi induce intrinsic apoptotic pathway by inactivation or suppression of antiapoptotic proteins and activation of pro-apoptotic proteins. HDACi can induce the pro-apoptotic cleavage of Bid, which initiates the intrinsic pathway leading to mitochondrial disruption in transformed cells. High levels of expression of Bcl-2 or Bcl-XL, which protect mitochondria, have been found in some transformed cells resistant to HDACi-induced cell death [77]. Inhibition of Bcl-2 by a chemical inhibitor, such as HA14 – 1, can increase sensitivity to HDACi-induced cell death. HDACi upregulate pro-apoptotic proteins of the Bcl-2 family, such as Bim, Bmf, Bax and Bik, and decrease antiapoptotic proteins of the Bcl-2 family, such as Bcl-2, Bcl-XL, Bcl-w and Mcl-1, as well as the pro-survival gene inhibitor of apoptosis, XIAP; it also induces degradation of survivin.
The HDACi, vorinostat or butyrate, induced autophagic cell death with vacuoles in the cytoplasm of transformed cells with Apaf-1 knockout or overexpression of Bcl-XL [83]. A senescence phenotype with polyploidy was induced by vorinostat in HCT1165 colon cancer cells [84].
4.5 Mitotic disruption
HDACi can cause aberrant accumulation of acetylated histones in heterochromatin and centromere domains with consequent death of transformed cells [85–88]. In transformed cell cultures with TSA, histones in newly synthesized chromatin remain acetylated, and disrupt the structure and function of the centromere and the pericentric heterochromatin with loss of binding to heterochromatin binding proteins. Histone acetylation also blocks histone phosphorylation, disrupting the function of mitotic spindle checkpoint proteins, such as BubR1, hBUB1, CENP-F and CENP-E, causing transient arrest at pro-metaphase and aberrant mitosis with chromosomal disruption resulting in cell death.
4.6 ROS and altered redox pathways
HDACi cause an accumulation of ROS in transformed cells but not in normal cells [55,56,89–91]. Increased cellular ROS can occur within 2 h of culture with HDACi, before disruption of mitochondria. Free radical scavengers such as N-acetylcysteine decrease HDACi-induced apoptosis, indicating that the generation of ROS is a factor in facilitating transformed-cell death. Thioredoxin (Trx) is a hydrogen donor that is required for activation of several proteins, including, ribonucleotide reductases, essential for DNA synthesis, and transcription factors, such as NF-κB. Reduced Trx is an antioxidant scavenger of ROS [92]. Vorinostat upregulates the expression of TBP-2 [56,58], which binds and inhibits reduced Trx activity, causing a downregulation of Trx in transformed but not in normal cells. Trx is an inhibitor of apoptosis signal-regulating kinase 1 (ASK1). Inhibition of Trx by binding to TBP2 activates ASK1 which, in turn, promotes apoptosis by inducing SET1-JNK and MKK3/MKK6-p38 signaling cascades, and enhancing the expression of pro-apoptotic protein Bim [93].
4.7 Inhibition of HDAC6 and target proteins
HDAC6 is unique among zinc-containing HDACs in having two catalytic sites: a ubiquitin binding site and cytoplasmic localization where it associates with non-histone substrates, such as HSP90 and α-tubulin [18–22,94–96]. Overexpression of HDAC6 leads to deacetylation of α-tubulin and an increase of cell motility. HDAC6 can bind both mono- and poly-ubiquitinated proteins and promotes its own ubiquitination. Specific inhibition of HDAC6 activity with tubacin or downregulation by SiRNAs cause accumulation of acetylated α-tubulin, HSP90, peroxiredoxins and other client proteins. HSP90 acetylation causes loss of chaperone function and exposes its client proteins, such as pro-survival and pro-proliferation proteins, Akt, Bcr-Abl, c-Raf and ErB2 to poly-ubiquitination and degradation via proteasome pathway [22,23,97]. HSP90 chaperone function is essential for the stability and function of several proteins, such as steroid hormone receptors and protein kinases involved in cell signaling pathways and cellular homeostasis. Recent studies have demonstrated both a direct physical interaction between HDAC6 and HSP90 and HDAC6 as a regulator of HSP90 activity through its deacetylation [22,95–98]. HDAC6 can bind directly to protein phosphatase (PP1) and cause simultaneous changes in cellular protein phosphorylation and acetylation; given the number of client proteins of HSP90, many molecular alterations can be a result of HSP90 inactivation through HDAC6 inhibition by HDACi.
HDAC6 is a component of the aggresome, a cellular structure that constitutes the major site of degradation for misfolded protein aggregates, both non-ubiquitinated and ubiquitinated misfolded proteins [22,23]. Misfolded proteins are susceptible to forming cytotoxic aggregates that can interfere with normal cell function. HDAC6 acts as a bridge between the dynein motors and the ubiquitination process, directing the poly-ubiquitinated proteins to the aggresome. The BUZ domain of HDAC6 has high affinity for ubiquitin molecule and is involved in the transport of poly-ubiquitinated proteins. Loss of HDAC6 function increases the sensitivity of transformed cells to misfolded protein stress induced by proteasome inhibitor.
Taken together, these findings have implications for developing therapeutic strategies combining HDACi and proteasome inhibitors and/or HSP90 inhibitors in the treatment of certain malignancies. Indeed, there is an ongoing substantial effort to develop HDAC6 isoform selective inhibitors that may be clinically useful in treating cancers and possibly other diseases [50].
4.8 Antiangiogenesis
HDACi can exert anticancer activity by inhibiting tumor angiogenesis [99]. Solid tumors are frequently angiogenesis-dependent. Tumor angiogenesis can be mediated by hypoxia secondary to tumor growth or by increased oncogenic signaling and, as a consequence, induce hypoxia-inducible factor-1α (HIF-1α) and its transcriptional target, VEGF. HDACi inhibit angiogenesis via the suppression of HIF-1α and its target, VEGF, in animal models [100–102]. Under normoxic conditions, HIF-1α binds to von Hippel–Lindau protein (pVHL) and is inactivated by ubiquitination-proteasome degradation. Hypoxic conditions can increase transcription of HDACs 1, 2, and 3 in transformed cells, causing decreased expression of pVHL and, as a consequence, increased expression of HIF-1α, which promotes angiogenesis – a sequence of events that may be clocked by HDACi. HDACi can also induce HIF-1α degradation in a VHL-independent mechanism. Class II HDACs, HDAC4 or HDAC6, physically associate with HIF-1α and their selective inhibition by siRNA induces HIF-1α degradation. HIF-1α binds to HSP90 and disrupts the HSP90 chaperone function by acetylation, exposing HIF-1α to proteasomal degradation. These observations support the development of combination therapies of HDACi with antiangiogenesis drugs.
4.9 Antimetastatic
HDACi upregulate metastasis suppressor genes, such as Kangai (KAII), Ras homologue genes, RhoB, reversion-inducing cysteine-rich protein with KAZAL motifs (RECK) and tissue inhibitor of metalloproteinases (TIMP-1) [103]. Metastasis-promoting genes can be downregulated by HDACi, including genes for MMPs, integrin-α5 and collagen proteins. These findings suggest that HDACi may be effective in reducing the metastatic potential of primary tumors.
4.10 Glucose metabolism
HDACi target glucose transporter 1 (GLUT1-mediated glucose transport) and hexokinase I (HXK1) enzymatic activity, inhibiting glucose utilization in transformed cells [104]. This HDACi effect may have importance in selectively ‘starving’ transformed cells, contributing to cell growth arrest and death.
The present evidence, based primarily on studies with transformed cells in culture, indicates that HDACs have multiple targets that are involved in almost every cellular pathway critical to cell survival, differentiation, proliferation, migration, and death (Figure 1). In cultured cell studies, HDACi induce cell death of transformed but not normal cells, which may reflect the capacity of normal but not transformed cells to ‘recover’ from exposure to reversible HDACi.
5. HDACi in combination with other anticancer agents
In preclinical studies with cultured cells and tumor-bearing animal models, and in clinical trials, HDACi have been used in combination with radiation; antimetabolites, such as 5-flurodeoxyuridine and gemcitabine; antitubulin agents, such as docetaxel, paclitaxel and epothilone B; topoisomerase (Topo) II inhibitors, doxorubicin, epirubicin, VP-16 (etoposide) and ellipticine; DNA cross-linking agent, cisplatin; HSP90 antagonist, 17-ally-amino-demethoxy gelda-namycin; and targeted agents, such as rituximab, trastuzumab, and EFGR inhibitor, erlotinib. Synergy or additive effects have been found with many of these combinations in inducing cancer cell death [6,8,105–112]. Romidepsin is synergistic with several conventional antileukemia/lymphoma drugs in leukemia and lymphoma cell line studies [103,109]. In preclinical studies, vorinostat was found to be synergistic with cytosine arabinoside and etoposide in treatment of acute leukemias [106]. HDACi have also been reported to have synergy with the transcription modulator, all-trans retinoic acid (ATRAL): DNA demethylating agent, 5-aza-2′ deoxycytidine, and the Bcr-Abl kinase inhibitor, imatinib. Upregulation of death receptors, and/or reducing the inhibitory regulators of death receptor pathway by HDACi, sensitizes tumor cells to TRAIL. HDACi can achieve synergy with TRAIL by simultaneous activation of the intrinsic and the extrinsic apoptotic pathways without changing the expression of TRAIL receptors or the inhibitory protein c-FLIP. Kinase inhibitors, including CDK inhibitor flavopiridol, phosphatidylinositol 3 kinase inhibitor, LY294002, FLT3 inhibitor, PKC412 and MEK1/2 inhibitor PD184352, potentiate the cell-killing effect of HDACi. Blocking NF-κB activation by the IκBα phosphorylation inhibitor, BAY 11-7082, markedly increased HDACi-induced apoptosis.
The synergistic effects of HDACi in combination with other drugs may depend on the sequence of drug administration. For example, prior treatment with HDACi, which induced chromatin decondensation, followed by Topo II inhibitors resulted in synergistic antitumor cell activity. The reverse order of administration of the drugs resulted in antagonistic effects, or had no more effect than each drug alone. Pretreatment with HDACi had greater antitumor effect than the reverse, in combination with cisplatin. These studies have guided, in part, clinical development of HDACi in combination therapeutic clinical trials.
A better understanding of the biological roles of the HDACs, the mechanisms of action of HDACi, and the development of selectively targeted HDACi should lead to increasingly effective combination HDACi therapeutic strategies.
6. Development of HDACi for therapy of non-oncologic diseases
Several studies have shown that HDACi have therapeutic potential in several non-oncologic diseases including inflammatory lesions, autoimmune disorders, neurological disorders, sickle cell anemia and malaria [6,8,113–120]. Vorinostat slowed the progression of Huntington-like syndrome in trans-genic mice [119] and increased expression of SNM protein in spinal muscular atrophy fibroblasts [117]. HDAC2 modulates synaptic plasticity and long-lasting changes of neural circuits and, in turn, plays a role in regulating learning and memory. HDAC2-selective inhibition may be useful in memory impairment [118]. HDACi have been reported to be potent inducers of γ-globin gene expression with therapeutic value in the treatment of sickle cell anemia [120]. HDACi have anti-rheumatic activity in rodent models [121]. HDACi modulate stem cell survival and mobilization in in vitro studies [122]. HDACi have been shown to decrease multilineage differentiation potential of human mesenchymal stem cells [123]. HDACi have been found to improve animal survival after hemorrhagic shock [124].
7. Clinical development of HDACi as anticancer drugs
Over a dozen structurally different HDACi are in clinical trials either as monotherapy or in combination therapy for various hematologic and solid tumors (Table 2). Four major chemical classes of HDACi are currently in clinical trials, including short-chain fatty acid (butyrates and valproic acid), hydroxamates (vorinostat, panobinostat, belinostat, givinostat, PCI24781 and JNJ26481585), benzamides (entinostat and MGCD-103), and cyclic tetrapeptide (romidepsin). There are ongoing clinical trials with HDACi in combination therapy with radiation, cytotoxic agents, and different targeted anticancer agents (ClinicalTrials.gov [6,8,11,105–112,125]). These clinical trials include patients with cancer of lung, breast, pancreas, renal and bladder, melanoma, glioblastoma, leukemias, lymphomas, and multiple myeloma.
Vorinostat was the first of the HDACi to be approved for clinical use in the therapy of CTCL by the US FDA. In a Phase II study, orally administered vorinostat in 33 previously treated patients with refractory CTCL achieved partial response in eight patients (24.2%); 14 of 31 evaluable patients (45.2%) had pruritus relief. More recently, romidepsin received FDA approval for the therapy of CTCL [109,110].
Vorinostat is being evaluated in Phase II and III clinical trials as monotherapy and in combination with various anticancer agents for both hematologic and solid tumors [47,105,126,127]. Ongoing clinical trials in combination therapy for vorinostat include azacitidine, decitabine, the proteasome inhibitor, bortezomib, and taxanes.
Panobinostat (LBH589) is more potent than vorinostat in preclinical models [107,128]. It is in clinical trials for hematologic and solid tumors as monotherapy and various combination therapy protocols, including with proteasome inhibitors as well as with the DNA methylase inhibitor, azacitidine.
Other hydroxamic acid-based HDACi in clinical trials include belinostat (PDX101), givinostat (ITF2357) and JNJ26481585 (Table 2). Belinostat is in Phase I and II clinical trials for hematological and solid malignancies, including metastatic and refractory ovarian cancer. Givinostat is an orally administrated hydroxamate that is being investigated in a clinical trial in patients with pretreated refractory Hodgkin’s disease. Each of the hydroxamic acid-based HDACi in clinical trials has shown antitumor activity, including stable disease, partial response and in a few cases, complete responses of transient duration at doses generally well tolerated by the patients.
Adverse effects observed with the hydroxamic class of HDACi include fatigue, nausea, dehydration, diarrhea, and thrombocytopenia. With certain hydroxamic acid-based HDACi, electrocardiogram changes have occurred. These side effects have been reversible upon cessation of the administration of the drug.
Two benzamide HDACi are in clinical trials, entinostat (MS275, Sndx-275) and MGCD103 (Table 2). These agents are being evaluated as monotherapy and in combination with other anticancer drugs. Recently, clinical trials with MGCD103 were suspended owing to the development of pericarditis as a possible adverse effect. Entinostat is in clinical trials in patients with advanced acute leukemia and in patients with solid tumors, including Phase II clinical trials in patients with refractory metastatic melanoma.
Romidepsin, a cyclic peptide HDACi, is in clinical trials as monotherapy as well as in combination with gemcitabine. Romidepsin, FDA-approved for CTCL, is being evaluated in a Phase II study with patients with high-risk myelodysplastic syndrome and acute myelogenous leukemia [109,110]. Another Phase II clinical trial with depsipeptide is ongoing in patients with refractory lung cancer.
The fatty acids, including valproic acid, are relatively weaker HDACi than hydroxamic acids, benzamides or cyclic peptides, and are in clinical trials as monotherapy and combination therapy with various cancer agents (Table 2).
8. Biomarkers predicting response to HDACi
In essentially all the clinical trials with HDACi in which anti-cancer activities is observed, only a portion of patients respond. The identification and development of assays for biomarkers that can predict resistance or sensitivity to HDACi is a major challenge in development of these agents [129]. High levels of expression of class I HDACs (HDACs 1, 2, and/or 3) have been found in colon and rectal cancers and prostate cancer in patients with a poor prognosis. Evaluation of HDAC expression in tumors in relation to the therapeutic response to HDACi is an ongoing area of investigation.
The accumulation of acetylated histones in peripheral mononuclear cells is a marker for the biological activity of HDACi but does not correlate with therapeutic response. The level of acetylated histones in peripheral mononuclear cells returns to base levels within 8 – 10 h following a single oral dose of the HDACi, vorinostat [126].
A biomarker that may inform on the therapeutic response to the HDACi is HR23B, a protein that shuffles ubiquitinated cargo proteins to the protostome and has been validated as sensitivity determinate of HDACi induced apoptosis [129]. HR23B is found expressed at high levels in CTCL that respond favorably to the HDACi, vorinostat.
9. Factors associated with resistance to HDACi
Development of resistance to HDACi is a major concern, as with any new antitumor therapy [130]. The mechanism of resistance to HDACi are not well understood. Based on pre-clinical studies, resistance to HDACi has been found to be associated with epigenetic alterations, altered stress response mechanisms and antiapoptotic survival mechanisms. High levels of Bcl-2 [77], Trx [131] and peroxiredoxins [132] have been associated with resistance of transformed cells to chemotherapy, and may play a role in resistance to HDACi-induced cell death. Overexpression of Bcl-2 blocks HDACi-induced cell death [71]. Peroxiredoxins reduced H2O2 and may protect transformed cells from HDACi-induced cell death, which is associated with ROS production.
Pharmacokinetic properties of the HDACi, such as poor solubility and relatively short half-life, are factors limiting therapeutic efficacy.
HDACi treatment of cancer cells can induce P-glycoprotein (P-gp), resulting in multidrug resistance of cancer cells to other chemotherapy agents [133]. Romidepsin is the only HDACi in clinical trials where P-gp may be a factor in resistance to this agent. It has been reported that the specific downregulation of HDAC1 but not HDAC2 by RNA silencing caused an induction of P-gp expression in HeLa cells, suggesting that HDAC1 inhibition by HDACi can induce P-gp.
10. Expert opinion
HDACi are a promising group of targeted anticancer agents with potential applications in the therapy of hematologic and solid neoplasms, as well as in several non-oncologic diseases. There is a great deal of competition in the HDACi field; several new and, hopefully, more effective compounds are being developed and entering clinical trials.
Histones and many non-histone proteins are targets of HDACs. This group of enzymes are more properly designated lysine deacetylases. The target proteins include factors regulating gene expression, cell proliferation, cell migration and cell death, and have a role in angiogenesis and immune response. While not completely understood, it is clear that the mechanisms of HDACi-induced transformed cell death may involve more than one pathway.
Normal cells are relatively resistant to HDACi-induced cell death. A possible explanation for this therapeutic window is that the majority of cancer cells have multiple genetic and molecular defects. Normal cells, compared with transformed cells, have the capacity to reverse the adverse effects of HDACi. This suggests that an intermittent regimen of exposure to the drugs may minimize occurrence of toxic effects on normal cells.
Understanding the biological activities of the 11 zinc-dependent HDACs is evolving. An important question is whether pan HDACi – such as vorinostat, which inhibits class I HDACs and class IIb HDAC6 – are potentially more effective therapeutic agents than an HDAC-selective inhibitor. The development of HDAC isoform-selective inhibitors will certainly be useful in further dissecting their biological functions.
HDACs inhibitors developed for clinical use to date are structurally diverse and inhibit more than a single zinc-dependent HDAC. HDACi can have antitumor activity across a broad variety of hematologic and solid tumors, but only a proportion of patients with a given diagnosis show a therapeutic response. This highlights the need to identify markers of potential response or resistance to one or another HDACi.
Optimization of the pharmacokinetic properties, including water solubility and oral availability, is a challenging area in the future development of HDACi.
Preclinical and clinical studies to date indicate that HDACi may be most therapeutically effective in combination with other anticancer agents including radiation and cytotoxic and targeted drugs. As we gain more understanding of the mechanisms of action of the zinc-dependent HDACs, and the different HDACi, we should be able to develop more effective therapeutic strategies. Current evidence indicates that HDACi can not only block the catalytic activity of the enzymes but also affect the protein–protein interactions of specific HDACs with various critical protein partners. The large number of defects present in most cancer cells suggests that therapeutic strategies targeting multiple biological pathways are likely to be more effective than a drug targeted at a single pathway. This concept supports the need for continued clinical development of HDACi in combination with other anticancer agents.
Article highlights.
There are 11 zinc-dependent histone deacetylases in humans.
HDACs have histone and many non-histone substrates.
HDAC inhibitors (HDACi) can selectively induce cancer cell death.
HDACi can induce cancer cell death by one or more pathways.
HDACi are in clinical trials as monotherapy and combination therapy for hematologic and solid tumors.
HDACi, vorinostat and romidepsin are approved for therapy of cutaneous T-cell lymphoma.
This box summarizes key points contained in the article.
Acknowledgments
The author is grateful to Joann Perrone, Mable Miranda, and Megan Choy for their excellent assistance in the preparation of this manuscript.
Footnotes
Declaration of interest
Studies referred to in this review from the author’s laboratory were supported, in part, by the National Institute of Cancer Grant P30CA08748-44, The David Koch Foundation, The Jack and Susan Rudin Foundation, The CapCure Foundation, and Experimental Therapeutics at Memorial Sloan-Kettering Cancer Center. Memorial Sloan-Kettering Cancer Center and Columbia University hold patents on suberoylani-lide hydroxamic acid (SAHA, vorinostat) and related compounds that were exclusively licensed to ATON Pharma, a biotechnology start-up that was wholly acquired by Merck, Inc. in April 2004. The author was a founder of ATON and has a potential royalty interest in the further development of vorinostat by Merck.
Bibliography
- 1.Handel AE, Ebers GC, Ramagopalan SV. Epigenetics: molecular mechanisms and implications for disease. Trends Mol Med. 2009;16(1):7–16. doi: 10.1016/j.molmed.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 2.Ho DH, Burggren WW. Epigenetics and transgenerational transfer: a physiological perspective. J Exp Biol. 2010;213(Pt 1):3–16. doi: 10.1242/jeb.019752. [DOI] [PubMed] [Google Scholar]
- 3.Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13(2):143–53. doi: 10.1016/s0959-437x(03)00015-7. [DOI] [PubMed] [Google Scholar]
- 4.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 5.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 6.Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J cell biochem. 2009;107(4):600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 8.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10(1):32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338(1):17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Lagger G, O’Carroll D, Rembold M, et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002;21(11):2672–81. doi: 10.1093/emboj/21.11.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5(10):981–9. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 12.Montgomery RL, Davis CA, Potthoff MJ, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21(14):1790–802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marchion DC, Bicaku E, Turner JG, et al. HDAC2 regulates chromatin plasticity and enhances DNA vulnerability. Mol Cancer Ther. 2009;8(4):794–801. doi: 10.1158/1535-7163.MCT-08-0985. [DOI] [PubMed] [Google Scholar]
- 14.Bhaskara S, Chyla BJ, Amann JM, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell. 2008;30(1):61–72. doi: 10.1016/j.molcel.2008.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323(5911):256–9. doi: 10.1126/science.1166226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams AH, Valdez G, Moresi V, et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. 2009;326(5959):1549–54. doi: 10.1126/science.1181046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang S, Young BD, Li S, et al. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126(2):321–34. doi: 10.1016/j.cell.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 18.Haggarty SJ, Koeller KM, Wong JC, et al. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA. 2003;100(8):4389–94. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parmigiani RB, Xu WS, Venta-Perez G, et al. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci USA. 2008;105(28):9633–8. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X, Yuan Z, Zhang Y, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27(2):197–213. doi: 10.1016/j.molcel.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovacs JJ, Murphy PJ, Gaillard S, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18(5):601–7. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 22.Kawaguchi Y, Kovacs JJ, McLaurin A, et al. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115(6):727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 23.Boyault C, Gilquin B, Zhang Y, et al. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25(14):3357–66. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villagra A, Cheng F, Wang HW, et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009;10(1):92–100. doi: 10.1038/ni.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5(4–5):245–53. [PMC free article] [PubMed] [Google Scholar]
- 26.Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci USA. 2004;101(2):540–5. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gray SG, Qian CN, Furge K, et al. Microarray profiling of the effects of histone deacetylase inhibitors on gene expression in cancer cell lines. Int J Oncol. 2004;24(4):773–95. doi: 10.3892/ijo.24.4.773. [DOI] [PubMed] [Google Scholar]
- 28.Peart MJ, Smyth GK, van Laar RK, et al. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102(10):3697–702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dokmanovic M, Perez G, Xu W, et al. Histone deacetylase inhibitors selectively suppress expression of HDAC7. Mol Cancer Ther. 2007;6(9):2525–34. doi: 10.1158/1535-7163.MCT-07-0251. [DOI] [PubMed] [Google Scholar]
- 30.Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93(1):57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- 31.Finnin MS, Donigian JR, Cohen A, et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401(6749):188–93. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 32.Somoza JR, Skene RJ, Katz BA, et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12(7):1325–34. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 33.Vannini A, Volpari C, Filocamo G, et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci USA. 2004;101(42):15064–9. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bottomley MJ, Lo Surdo P, Di Giovine P, et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem. 2008:M803514200. doi: 10.1074/jbc.M803514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ficner R. Novel structural insights into class I and II histone deacetylases. Curr Top Med Chem. 2009;9(3):235–40. doi: 10.2174/156802609788085304. [DOI] [PubMed] [Google Scholar]
- 36.Ropero S, Fraga MF, Ballestar E, et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet. 2006;38(5):566–9. doi: 10.1038/ng1773. [DOI] [PubMed] [Google Scholar]
- 37.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 38.Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8(6):1409–20. doi: 10.1158/1535-7163.MCT-08-0860. [DOI] [PubMed] [Google Scholar]
- 39.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakagawa M, Oda Y, Eguchi T, et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18(4):769–74. [PubMed] [Google Scholar]
- 41.Zhang Z, Yamashita H, Toyama T, et al. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast. Breast Cancer Res Treat. 2005;94(1):11–6. doi: 10.1007/s10549-005-6001-1. [DOI] [PubMed] [Google Scholar]
- 42.Chang H-H, Chiang C-P, Hung H-C, et al. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009;45(7):610–14. doi: 10.1016/j.oraloncology.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 43.Gloghini A, Buglio D, Khaskhely NM, et al. Expression of histone deacetylases in lymphoma: implication for the development of selective inhibitors. Br J Haematol. 2009;147(4):515–25. doi: 10.1111/j.1365-2141.2009.07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oehme I, Deubzer HE, Lodrini M, et al. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin Investig Drugs. 2009;18(11):1605–17. doi: 10.1517/14728220903241658. [DOI] [PubMed] [Google Scholar]
- 45.Lin RJ, Sternsdorf T, Tini M, Evans RM. Transcriptional regulation in acute promyelocytic leukemia. Oncogene. 2001;20(49):7204–15. doi: 10.1038/sj.onc.1204853. [DOI] [PubMed] [Google Scholar]
- 46.Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20(40):5695–707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- 47.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25(1):84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 48.Wang H, Dymock BW. New patented histone deacetylase inhibitors. Expert Opin Ther Pat. 2009;19(12):1727–57. doi: 10.1517/13543770903393789. [DOI] [PubMed] [Google Scholar]
- 49.Schemies J, Sippl W, Jung M. Histone deacetylase inhibitors that target tubulin. Cancer Lett. 2009;280(2):222–32. doi: 10.1016/j.canlet.2009.01.040. [DOI] [PubMed] [Google Scholar]
- 50.Varghese S, Senanayake T, Murray-Stewart T, et al. Polyaminohydroxamic acids and polyaminobenzamides as isoform selective histone deacetylase inhibitors. J Med Chem. 2008;51(8):2447–56. doi: 10.1021/jm701384x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marson C. Histone deacetylase inhibitors: design, structure-activity relationships and therapeutic implications for cancer. Anti-cancer agents in medicinal chemistry. 2009;9(6):661–92. doi: 10.2174/187152009788679976. [DOI] [PubMed] [Google Scholar]
- 52.Butler KV, Kozikowski AP. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr Pharm Des. 2008;14(6):505–28. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]
- 53.Balasubramanian S, Verner E, Buggy JJ. Isoform-specific histone deacetylase inhibitors: the next step? Cancer Lett. 2009;280(2):211–21. doi: 10.1016/j.canlet.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 54.Mahboobi S, Dove S, Sellmer A, et al. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: a series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rbeta, and histone deacetylases. J Med Chem. 2009;52(8):2265–79. doi: 10.1021/jm800988r. [DOI] [PubMed] [Google Scholar]
- 55.Ruefli AA, Ausserlechner MJ, Bernhard D, et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci USA. 2001;98(19):10833–8. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ungerstedt JS, Sowa Y, Xu WS, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102(3):673–8. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer Lett. 2009;274(2):169–76. doi: 10.1016/j.canlet.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 58.Butler LM, Zhou X, Xu WS, et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc Natl Acad Sci USA. 2002;99(18):11700–5. doi: 10.1073/pnas.182372299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaymes TJ, Padua RA, Pla M, et al. Histone deacetylase inhibitors (HDI) cause DNA damage in leukemia cells: a mechanism for leukemia-specific HDI-dependent apoptosis? Mol Cancer Res. 2006;4(8):563–73. doi: 10.1158/1541-7786.MCR-06-0111. [DOI] [PubMed] [Google Scholar]
- 60.Adimoolam S, Sirisawad M, Chen J, et al. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci USA. 2007;104(49):19482–7. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Munshi A, Kurland JF, Nishikawa T, et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res. 2005;11(13):4912–22. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- 62.Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009;280(2):125–33. doi: 10.1016/j.canlet.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 63.Chen CS, Wang YC, Yang HC, et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res. 2007;67(11):5318–27. doi: 10.1158/0008-5472.CAN-06-3996. [DOI] [PubMed] [Google Scholar]
- 64.Fernandez-Capetillo O, Nussenzweig A. Linking histone deacetylation with the repair of DNA breaks. Proc Natl Acad Sci USA. 2004;101(6):1427–8. doi: 10.1073/pnas.0307342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Munshi A, Tanaka T, Hobbs ML, et al. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther. 2006;5(8):1967–74. doi: 10.1158/1535-7163.MCT-06-0022. [DOI] [PubMed] [Google Scholar]
- 66.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Nat Acad Sci USA. 2000;97(18):10014–19. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gui CY, Ngo L, Xu WS, et al. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. ProcNatl Acad Sci USA. 2004;101(5):1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rascle A, Johnston JA, Amati B. Deacetylase activity is required for recruitment of the basal transcription machinery and transactivation by STAT5. Mol Cell Biol. 2003;23(12):4162–73. doi: 10.1128/MCB.23.12.4162-4173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang LG, Ossowski L, Ferrari AC. Androgen receptor level controlled by a suppressor complex lost in an androgen-independent prostate cancer cell line. Oncogene. 2004;23(30):5175–84. doi: 10.1038/sj.onc.1207654. [DOI] [PubMed] [Google Scholar]
- 70.Lee E-M, Shin S, Cha H, et al. Suberoylanilide hydroxamic acid (SAHA) changes microRNA expression profiles in A549 human non-small cell lung cancer cells. Intl J Mol Med. 2009;24(1):45–50. doi: 10.3892/ijmm_00000204. [DOI] [PubMed] [Google Scholar]
- 71.Wilson AJ, Chueh AC, Togel L, et al. Apoptotic sensitivity of colon cancer cells to histone deacetylase inhibitors is mediated by an Sp1/Sp3-activated transcriptional program involving immediate-early gene induction. Cancer Res. 2010;70(2):609–20. doi: 10.1158/0008-5472.CAN-09-2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rosato RR, Grant S. Histone deacetylase inhibitors: insights into mechanisms of lethality. Expert Opin Ther Targets. 2005;9(4):809–24. doi: 10.1517/14728222.9.4.809. [DOI] [PubMed] [Google Scholar]
- 73.Zhao Y, Tan J, Zhuang L, et al. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci USA. 2005;102(44):16090–5. doi: 10.1073/pnas.0505585102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burgess A, Ruefli A, Beamish H, et al. Histone deacetylase inhibitors specifically kill nonproliferating tumour cells. Oncogene. 2004;23(40):6693–701. doi: 10.1038/sj.onc.1207893. [DOI] [PubMed] [Google Scholar]
- 75.Nakata S, Yoshida T, Horinaka M, et al. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23(37):6261–71. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- 76.Insinga A, Monestiroli S, Ronzoni S, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11(1):71–6. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 77.Xu W, Ngo L, Perez G, et al. Intrinsic apoptotic and thioredoxin pathways in human prostate cancer cell response to histone deacetylase inhibitor. Proc Natl Acad Sci USA. 2006;103(42):15540–5. doi: 10.1073/pnas.0607518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 79.Coffey DC, Kutko MC, Glick RD, et al. The histone deacetylase inhibitor, CBHA, inhibits growth of human neuroblastoma xenografts in vivo, alone and synergistically with all-trans retinoic acid. Cancer Res. 2001;61(9):3591–4. [PubMed] [Google Scholar]
- 80.Borbone E, Berlingieri MT, De Bellis F, et al. Histone deacetylase inhibitors induce thyroid cancer-specific apoptosis through proteasome-dependent inhibition of TRAIL degradation. Oncogene. 2010;29(1):105–16. doi: 10.1038/onc.2009.306. [DOI] [PubMed] [Google Scholar]
- 81.Rosato RR, Maggio SC, Almenara JA, et al. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Mol Pharmacol. 2006;69(1):216–25. doi: 10.1124/mol.105.017145. [DOI] [PubMed] [Google Scholar]
- 82.Zhang XD, Gillespie SK, Borrow JM, Hersey P. The histone deacetylase inhibitor suberic bishydroxamate regulates the expression of multiple apoptotic mediators and induces mitochondria-dependent apoptosis of melanoma cells. Mol Cancer Ther. 2004;3(4):425–35. [PubMed] [Google Scholar]
- 83.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2004;101(52):18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xu WS, Perez G, Ngo L, et al. Induction of polyploidy by histone deacetylase inhibitor: a pathway for antitumor effects. Cancer Res. 2005;65(17):7832–9. doi: 10.1158/0008-5472.CAN-04-4608. [DOI] [PubMed] [Google Scholar]
- 85.Cimini D, Mattiuzzo M, Torosantucci L, Degrassi F. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol Biol Cell. 2003;14(9):3821–33. doi: 10.1091/mbc.E03-01-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taddei A, Maison C, Roche D, Almouzni G. Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat Cell Biol. 2001;3(2):114–20. doi: 10.1038/35055010. [DOI] [PubMed] [Google Scholar]
- 87.Dowling M, Voong KR, Kim M, et al. Mitotic spindle checkpoint inactivation by trichostatin a defines a mechanism for increasing cancer cell killing by microtubule-disrupting agents. Cancer Biol Ther. 2005;4(2):197–206. [PubMed] [Google Scholar]
- 88.Robbins AR, Jablonski SA, Yen TJ, et al. Inhibitors of histone deacetylases alter kinetochore assembly by disrupting pericentromeric heterochromatin. Cell Cycle. 2005;4(5):717–26. doi: 10.4161/cc.4.5.1690. [DOI] [PubMed] [Google Scholar]
- 89.Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res. 2003;63(13):3637–45. [PubMed] [Google Scholar]
- 90.Lillig CH, Holmgren A. Thioredoxin and related molecules – from biology to health and disease. Antioxid Redox Signal. 2007;9(1):25–47. doi: 10.1089/ars.2007.9.25. [DOI] [PubMed] [Google Scholar]
- 91.Marks PA. Thioredoxin in cancer: role of histone deacetylase inhibitors. Semin Cancer Biol. 2006;16(6):436–43. doi: 10.1016/j.semcancer.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nishiyama A, Matsui M, Iwata S, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999;274(31):21645–50. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- 93.Tan J, Zhuang L, Jiang X, et al. Apoptosis signal-regulating kinase 1 is a direct target of E2F1 and contributes to histone deacetylase inhibitor-induced apoptosis through positive feedback regulation of E2F1 apoptotic activity. J Biol Chem. 2006;281(15):10508–15. doi: 10.1074/jbc.M512719200. [DOI] [PubMed] [Google Scholar]
- 94.Zhang YY, Gilquin BB, Khochbin SS, Matthias PP. Two catalytic domains are required for protein deacetylation. J Biol Chem. 2006;281(5):2401–4. doi: 10.1074/jbc.C500241200. [DOI] [PubMed] [Google Scholar]
- 95.Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. JBiol Chem. 2005;280(29):26729–34. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 96.Westendorf JJ, Zaidi SK, Cascino JE, et al. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol Cell Biol. 2002;22(22):7982–92. doi: 10.1128/MCB.22.22.7982-7992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen CS, Weng SC, Tseng PH, Lin HP. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J Biol Chem. 2005;280(46):38879–87. doi: 10.1074/jbc.M505733200. [DOI] [PubMed] [Google Scholar]
- 98.Solit DB, Rosen N. Hsp90: a novel target for cancer therapy. Curr Top Med Chem. 2006;6(11):1205–14. doi: 10.2174/156802606777812068. [DOI] [PubMed] [Google Scholar]
- 99.Ellis L, Hammers H, Pili R. Targeting tumor angiogenesis with histone deacetylase inhibitors. Cancer Lett. 2009;280(2):145–53. doi: 10.1016/j.canlet.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liang D, Kong X, Sang N. Effects of histone deacetylase inhibitors on HIF-1. Cell Cycle. 2006;5(21):2430–5. doi: 10.4161/cc.5.21.3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kong X, Lin Z, Liang D, et al. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1 alpha. Mol Cell Biol. 2006;26(6):2019–28. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qian DZ, Kachhap SK, Collis SJ, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66(17):8814–21. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]
- 103.Kano Y, Akutsu M, Tsunoda S, et al. Cytotoxic effects of histone deacetylase inhibitor FK228 (depsipeptide, formally named FR901228) in combination with conventional anti-leukemia/lymphoma agents against human leukemia/lymphoma cell lines. Invest New Drugs. 2007;25(1):31–40. doi: 10.1007/s10637-006-9000-0. [DOI] [PubMed] [Google Scholar]
- 104.Wardell SE, Ilkayeva OR, Wieman HL, et al. Glucose metabolism as a target of histone deacetylase inhibitors. Mol Endocrinol. 2009;23(3):388–401. doi: 10.1210/me.2008-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Richon VM, Garcia-Vargas J, Hardwick JS. Development of vorinostat: current applications and future perspectives for cancer therapy. Cancer Lett. 2009;280(2):201–10. doi: 10.1016/j.canlet.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 106.Shiozawa K, Nakanishi T, Tan M, et al. Preclinical studies of vorinostat (suberoylanilide hydroxamic acid) combined with cytosine arabinoside and etoposide for treatment of acute leukemias. Clin Cancer Res. 2009;15(5):1698–707. doi: 10.1158/1078-0432.CCR-08-1587. [DOI] [PubMed] [Google Scholar]
- 107.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 2009;280(2):233–41. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 108.Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res. 2009;15(12):3970–7. doi: 10.1158/1078-0432.CCR-08-2786. [DOI] [PubMed] [Google Scholar]
- 109.Piekarz RL, Bates SE. Epigenetic modifiers: basic understanding and clinical development. Clin Cancer Res. 2009;15(12):3918–26. doi: 10.1158/1078-0432.CCR-08-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27(32):5410–17. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ma X, Ezzeldin HH, Diasio RB. Histone deacetylase inhibitors: current status and overview of recent clinical trials. Drugs. 2009;69(14):1911–34. doi: 10.2165/11315680-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 112.Nolan L, Johnson PW, Ganesan A, et al. Will histone deacetylase inhibitors require combination with other agents to fulfil their therapeutic potential? Br J Cancer. 2008;99(5):689–94. doi: 10.1038/sj.bjc.6604557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jennifer LB, Yongyao X, Susanne JS, et al. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 2007;109(3):1123–30. doi: 10.1182/blood-2006-04-019711. [DOI] [PubMed] [Google Scholar]
- 114.Adcock IM. HDAC inhibitors as anti-inflammatory agents. Br J Pharmacol. 2007;150(7):829–31. doi: 10.1038/sj.bjp.0707166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Villagra A, Sotomayor EM, Seto E. Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene. 2009;29(2):157–73. doi: 10.1038/onc.2009.334. [DOI] [PubMed] [Google Scholar]
- 116.Andrews KT, Tran TN, Wheatley NC, Fairlie DP. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr Top Med Chem. 2009;9(3):292–308. doi: 10.2174/156802609788085313. [DOI] [PubMed] [Google Scholar]
- 117.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev. 2008;7(10):854–68. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 118.Antonello M, Rotili D, Valente S, Kazantsev AG. Histone deacetylase inhibitors and neurodegenerative disorders: holding the promise. Curr Pharm Des. 2009;15(34):3940–57. doi: 10.2174/138161209789649349. [DOI] [PubMed] [Google Scholar]
- 119.Hockly E, Richon VM, Woodman B, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci USA. 2003;100(4):2041–6. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cao H, Jung M, Stamatoyannopoulos G. Hydroxamide derivatives of short-chain fatty acid have erythropoietic activity and induce [gamma] gene expression in vivo. Exp Hematol. 2005;33(12):1443–9. doi: 10.1016/j.exphem.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 121.Lin HS, Hu CY, Chan HY, et al. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br J Pharmacol. 2007;150(7):862–72. doi: 10.1038/sj.bjp.0707165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Romagnani P, Lasagni L, Mazzinghi B, et al. Pharmacological modulation of stem cell function. Curr Med Chem. 2007;14(10):1129–39. doi: 10.2174/092986707780362880. [DOI] [PubMed] [Google Scholar]
- 123.Lee S, Park JR, Seo MS, et al. Histone deacetylase inhibitors decrease proliferation potential and multilineage differentiation capability of human mesenchymal stem cells. Cell Prolif. 2009;42(6):711–20. doi: 10.1111/j.1365-2184.2009.00633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Butt MU, Sailhamer EA, Li Y, et al. Pharmacologic resuscitation: cell protective mechanisms of histone deacetylase inhibition in lethal hemorrhagic shock. J Surg Res. 2009;156(2):290–6. doi: 10.1016/j.jss.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 125.Botrugno OA, Santoro F, Minucci S. Histone deacetylase inhibitors as a new weapon in the arsenal of differentiation therapies of cancer. Cancer Lett. 2009;280(2):134–44. doi: 10.1016/j.canlet.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 126.Kelly WK, Marks PA. Drug insight: histone deacetylase inhibitors – development of the new targeted anticancer agent suberoylanilide hydroxamic acid. Nat Clin Pract Oncol. 2005;2(3):150–7. doi: 10.1038/ncponc0106. [DOI] [PubMed] [Google Scholar]
- 127.Duvic M, Vu J. Vorinostat: a new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin Invest Drugs. 2007;16(7):1111–20. doi: 10.1517/13543784.16.7.1111. [DOI] [PubMed] [Google Scholar]
- 128.Giles F, Fischer T, Cortes J, et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res. 2006;12(15):4628–35. doi: 10.1158/1078-0432.CCR-06-0511. [DOI] [PubMed] [Google Scholar]
- 129.Stimson L, La Thangue NB. Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett. 2009;280(2):177–83. doi: 10.1016/j.canlet.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 130.Fantin VR, Richon VM. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res. 2007;13(24):7237–42. doi: 10.1158/1078-0432.CCR-07-2114. [DOI] [PubMed] [Google Scholar]
- 131.Powis G, Mustacich D, Coon A. The role of the redox protein thioredoxin in cell growth and cancer. Free Radic Biol Med. 2000;29(3–4):312–22. doi: 10.1016/s0891-5849(00)00313-0. [DOI] [PubMed] [Google Scholar]
- 132.Chung YM, Yoo YD, Park JK, et al. Increased expression of peroxiredoxin II confers resistance to cisplatin. Anticancer Res. 2001;21(2A):1129–33. [PubMed] [Google Scholar]
- 133.Kim SN, Kim NH, Lee W, et al. Histone deacetylase inhibitor induction of P-glycoprotein transcription requires both histone deacetylase 1 dissociation and recruitment of CAAT/enhancer binding protein beta and pCAF to the promoter region. Mol Cancer Res. 2009;7(5):735–44. doi: 10.1158/1541-7786.MCR-08-0296. [DOI] [PubMed] [Google Scholar]
- 134.Kelly WK, O’Connor OA, Krug LM, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23(17):3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fujiwara Y, Yamamoto N, Yamada Y, et al. Phase I and pharmacokinetic study of vorinostat (suberoylanilide hydroxamic acid) in Japanese patients with solid tumors. Cancer Sci. 2009;100(9):1728–34. doi: 10.1111/j.1349-7006.2009.01237.x. [DOI] [PubMed] [Google Scholar]
- 136.Modesitt SC, Sill M, Hoffman JS, Bender DP. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;109(2):182–6. doi: 10.1016/j.ygyno.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 137.Vansteenkiste J, Van Cutsem E, Dumez H, et al. Early phase II trial of oral vorinostat in relapsed or refractory breast, colorectal, or non-small cell lung cancer. Invest New Drugs. 2008;26(5):483–8. doi: 10.1007/s10637-008-9131-6. [DOI] [PubMed] [Google Scholar]
- 138.Blumenschein GR, Jr, Kies MS, Papadimitrakopoulou VA, et al. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs. 2008;26(1):81–7. doi: 10.1007/s10637-007-9075-2. [DOI] [PubMed] [Google Scholar]
- 139.Ramalingam SS, Parise RA, Ramanathan RK, et al. Phase I and pharmacokinetic study of vorinostat, a histone deacetylase inhibitor, in combination with carboplatin and paclitaxel for advanced solid malignancies. Clin Cancer Res. 2007;13(12):3605–10. doi: 10.1158/1078-0432.CCR-07-0162. [DOI] [PubMed] [Google Scholar]
- 140.Luu TH, Morgan RJ, Leong L, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res. 2008;14(21):7138–42. doi: 10.1158/1078-0432.CCR-08-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Woyach JA, Kloos RT, Ringel MD, et al. Lack of therapeutic effect of the histone deacetylase inhibitor vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J Clin Endocrinol Metab. 2009;94(1):164–70. doi: 10.1210/jc.2008-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27(12):2052–8. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Watanabe T, Kato H, Kobayashi Y, et al. Potential efficacy of the oral histone deacetylase inhibitor vorinostat in a phase I trial in follicular and mantle cell lymphoma. Cancer Sci. 2010;101(1):196–200. doi: 10.1111/j.1349-7006.2009.01360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.O’Connor OA, Heaney ML, Schwartz L, et al. Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol. 2006;24(1):166–73. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- 145.Crump M, Coiffier B, Jacobsen ED, et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncol. 2008;19(5):964–9. doi: 10.1093/annonc/mdn031. [DOI] [PubMed] [Google Scholar]
- 146.Garcia-Manero G, Yang H, Bueso-Ramos C, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111(3):1060–6. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 147.Ramalingam SS, Belani CP, Ruel C, et al. Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma. J Thorac Oncol. 2009;4(1):97–101. doi: 10.1097/JTO.0b013e318191520c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Steele NL, Plumb JA, Vidal L, et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res. 2008;14(3):804–10. doi: 10.1158/1078-0432.CCR-07-1786. [DOI] [PubMed] [Google Scholar]
- 149.Zain JM, Foss F, Kelly WK, et al. Final results of a phase I study of oral belinostat (PXD101) in patients with lymphoma. (Meeting Abstracts) J Clin Oncol. 2009;27(15S):8580. [Google Scholar]
- 150.Golay J, Cuppini L, Leoni F, et al. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia. 2007;21(9):1892–900. doi: 10.1038/sj.leu.2404860. [DOI] [PubMed] [Google Scholar]
- 151.Evens AM, Weiyun A, Balasubramanian S, et al. Phase I analysis of the safety and pharmacodynamics of the broad spectrum histone deacetylase inhibitor (HDACi) PCI-24781 in relapsed and refractory lymphoma. Blood (ASH Annual Meeting) Abstract, Nov 2009, Am Society Hematol. 2009;114:2726. [Google Scholar]
- 152.Arts J, King P, Marien A, et al. JNJ-26481585, a novel ‘second-generation’ oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin Cancer Res. 2009;15(22):6841–51. doi: 10.1158/1078-0432.CCR-09-0547. [DOI] [PubMed] [Google Scholar]
- 153.Novotny-Diermayr V, Sangthongpitag K, Hu CY, et al. SB939, a novel potent and orally active histone deacetylase inhibitor with high tumor exposure and efficacy in mouse models of colorectal cancer. Mol Cancer Ther. 2010;9(3):642–52. doi: 10.1158/1535-7163.MCT-09-0689. [DOI] [PubMed] [Google Scholar]
- 154.Schrump DS, Fischette MR, Nguyen DM, et al. Clinical and molecular responses in lung cancer patients receiving Romidepsin. Clin Cancer Res. 2008;14(1):188–98. doi: 10.1158/1078-0432.CCR-07-0135. [DOI] [PubMed] [Google Scholar]
- 155.Shah MH, Binkley P, Chan K, et al. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12(13):3997–4003. doi: 10.1158/1078-0432.CCR-05-2689. [DOI] [PubMed] [Google Scholar]
- 156.Fouladi M, Furman WL, Chin T, et al. Phase I study of depsipeptide in pediatric patients with refractory solid tumors: a Children’s Oncology Group report. J Clin Oncol. 2006;24(22):3678–85. doi: 10.1200/JCO.2006.06.4964. [DOI] [PubMed] [Google Scholar]
- 157.Piekarz RL, Frye AR, Wright JJ, et al. Cardiac studies in patients treated with depsipeptide, FK228, in a phase II trial for T-cell lymphoma. Clin Cancer Res. 2006;12(12):3762–73. doi: 10.1158/1078-0432.CCR-05-2095. [DOI] [PubMed] [Google Scholar]
- 158.Reid T, Weeks A, Vakil M, et al. Dose escalation study of pivanex (a histone deacetylase inhibitor) in combination with docetaxel for advanced non-small cell lung cancer. (Meeting Abstracts) J Clin Oncol. 2004;22(14 Suppl):7279. [Google Scholar]
- 159.Carducci MA, Gilbert J, Bowling MK, et al. A Phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-h infusion schedule. Clin Cancer Res. 2001;7(10):3047–55. [PubMed] [Google Scholar]
- 160.Gilbert J, Baker SD, Bowling MK, et al. A phase I dose escalation and bioavailability study of oral sodium phenylbutyrate in patients with refractory solid tumor malignancies. Clin Cancer Res. 2001;7(8):2292–300. [PubMed] [Google Scholar]
- 161.Phuphanich S, Baker SD, Grossman SA, et al. Oral sodium phenylbutyrate in patients with recurrent malignant gliomas: a dose escalation and pharmacologic study. Neuro Oncol. 2005;7(2):177–82. doi: 10.1215/S1152851704000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Camacho LH, Olson J, Tong WP, et al. Phase I dose escalation clinical trial of phenylbutyrate sodium administered twice daily to patients with advanced solid tumors. Invest New Drugs. 2007;25(2):131–8. doi: 10.1007/s10637-006-9017-4. [DOI] [PubMed] [Google Scholar]
- 163.Kummar S, Gutierrez M, Gardner ER, et al. Phase I trial of MS-275, a histone deacetylase inhibitor, administered weekly in refractory solid tumors and lymphoid malignancies. Clin Cancer Res. 2007;13(18 Pt 1):5411–17. doi: 10.1158/1078-0432.CCR-07-0791. [DOI] [PubMed] [Google Scholar]
- 164.Atmaca A, Al-Batran SE, Maurer A, et al. Valproic acid (VPA) in patients with refractory advanced cancer: a dose escalating phase I clinical trial. Br J Cancer. 2007;97(2):177–82. doi: 10.1038/sj.bjc.6603851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gore L, Rothenberg ML, O’Bryant CL, et al. A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS-275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res. 2008;14(14):4517–25. doi: 10.1158/1078-0432.CCR-07-1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Siu LL, Pili R, Duran I, et al. Phase I study of MGCD0103 given as a three-times-per-week oral dose in patients with advanced solid tumors. J Clin Oncol. 2008;26(12):1940–7. doi: 10.1200/JCO.2007.14.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]