

Abstract
Objectives
To investigate the range of clinical presentations for Shwachman-Diamond syndrome (SDS) with the long-term goal of improving diagnosis.
Study design
We reviewed the North American Shwachman-Diamond Syndrome Registry. Genetic reports of biallelic SBDS mutations confirming the diagnosis of SDS were available for 37 patients.
Results
Neutropenia was the most common hematologic abnormality at presentation (30/37, 81%); however, only 51% (19/37) of patients presented with the classic combination of neutropenia and steatorrhea. Absence of pancreatic lipomatosis on ultrasound or CT scan, normal fecal elastase levels, and normal skeletal survey do not rule out the diagnosis of SDS. SDS was diagnosed in two asymptomatic siblings of SDS probands. Twenty-four of 37 patients (65%) had congenital anomalies.
Conclusion
Our cohort reveals a broad range of clinical presentation for SDS. Clues to the underlying diagnosis of SDS included cytopenias with a hypocellular marrow, congenital anomalies, family history, and myelodysplasia with clonal abnormalities frequently found in SDS. Reliance on classic clinical criteria for SDS would miss or delay diagnosis of a significant subset of patients with SDS.
Keywords: bone marrow failure, Shwachman Diamond Syndrome, ribosomes
Shwachman Diamond syndrome (SDS) is an autosomal recessive disorder characterized by congenital anomalies, exocrine pancreatic dysfunction, bone marrow failure and predisposition to myelodysplasia (MDS) and leukemia, particularly acute myeloid leukemia (AML)[1]. Previous studies have found mutations in the Shwachman-Bodian-Diamond Syndrome (SBDS) gene located on chromosome 7q11 can be found in approximately 90% of classically presenting patients with SDS. SBDS encodes an evolutionarily conserved protein which functions in ribosomal maturation as well as being implicated in additional cellular functions [2–4]. SDS is a multi-system disorder with potential manifestations in the skeletal, hepatic, cardiac, immune and central nervous systems [5].
Prior to the identification of the SBDS gene, SDS was diagnosed on the basis of clinical criteria consisting of the combination of marrow failure (typically manifested by neutropenia) and exocrine pancreatic dysfunction [5, 6]. With the advent of genetic testing for SBDS mutations, the range of SDS clinical phenotypes can now be explored. A recent case report of two patients with unusual presentations of SDS highlighted the need for a systematic evaluation of patients with SDS [7].
To investigate cryptic presentations of SDS, we conducted a retrospective review of medical records from the North American Shwachman-Diamond Syndrome Registry (SDSR). Data were extracted from clinic notes and from laboratory, pathology, and radiology reports. The SDSR was established in 2008 to elucidate the clinical spectrum, natural history, and molecular pathogenesis of SDS with the goal of improving diagnosis and therapy. The SDSR also provides an educational resource for patients, families, and healthcare providers. This registry is a collaborative effort between the Fred Hutchinson Cancer Research Center in Seattle, Washington, and Cincinnati Children’s Hospital Medical Center in Cincinnati, Ohio, and works in partnership with the Severe Chronic Neutropenia International Registry at University of Washington, in Seattle, Washington.
In this report, we investigate the initial clinical presentations of patients with genetically-confirmed SDS in the North American SDSR. We found a broad range of clinical presentations for SDS, and identified several features providing clues to the underlying diagnosis of SDS.
Methods
The study was a retrospective review of medical records including clinic notes, laboratory reports, pathology reports, and radiology reports collected through the SDSR. Informed consent was obtained in accordance with the study protocol approved by the Institutional Review Boards of the Fred Hutchinson Cancer Research Center and Cincinnati Children’s Hospital Medical Center. This report was limited to patients with SBDS gene mutations confirmed by review of the genetic testing reports. Neutropenia was defined as absolute neutrophil count (ANC) ≤ 1500/μL on at least 3 separate occasions, anemia as hemoglobin below the age-related normal range, and thrombocytopenia as platelet count ≤ 150,000 platelets//μL in the absence of other apparent etiologies for cytopenias [6]. Pancreatic insufficiency was defined as pancreatic isoamylase and/or trypsinogen below reference for age, or fecal elastase < 100 μg/g stool. Isoamylase was not utilized in patients < 3 years of age due to the age-dependent nature of isoamylase production, resulting in low levels in healthy children under 3 years of age [5, 6]. Similarly, trypsinogen was not utilized in patients >3 years of age due to the frequent normalization of trypsinogen with increasing age [5, 6]. Telomere length measurements were performed by Repeat Diagnostics Inc [8].
Results
The study subjects included 37 patients in the SDSR with genetic test reports confirming the presence of mutations in the SBDS gene (Table I). The SDS study cohort included 24 male and 13 female patients. Median patient age at clinical presentation was 3.5 years with a range of 0.02 – 18 years (Table II). The classic presentation of neutropenia associated with diarrhea was seen in only 19/37 (51%) of patients. One patient presented with isolated neutropenia without any history of diarrhea, failure to thrive, or congenital anomalies. Two patients presented with diarrhea without neutropenia. Three patients presented with failure to thrive without diarrhea or neutropenia. The absence of neutropenia or diarrhea at presentation does not rule out the diagnosis of SDS. It should also be noted that neutropenia may be intermittent and may develop over time.
Table 1.
SBDS Genetic Mutations
| Patient Number (n=37) | SBDS Mutations |
|---|---|
|
| |
| 25 | 258+2 T>C and 183_184 TA>CT |
| 5 | Homozygous 258+2 T>C |
| 2 | Homozygous 258+2 T>C and 183_184 TA>CT |
| 1 | 258+2 T>C and IVS2-2 A>G |
| 1 | 258+2 T>C and F57S (TTT>TCT) |
| 1 | 258+2 T>C and 18 delC |
| 1 | 258+2 T>C and 120 del G |
| 1 | 258+2 T>C and Q153R (458 A>G) |
Table 2.
Clinical Presentation of Shwachman Diamond Syndrome
| UPN | Sex | Neutropenia | Diarrhea | FTT | Congenital anomalies | Thrombocytopenia | Anemia |
|---|---|---|---|---|---|---|---|
| 1 | Male | + | + | + | + | ||
| 2 | Female | + | + | + | + | ||
| 3 | Female | + | + | + | |||
| 4 | Male | + | + | + | + | ||
| 5 | Male | + | + | + | |||
| 6 | Male | + | + | + (txn) | |||
| 7 | Female | + | + | + | + | + | |
| 8 | Male | + | + | ||||
| 9 | Male | + | + | + | |||
| 10 | Male | + | + | + | |||
| 11 | Female | + | |||||
| 12 | Male | + | |||||
| 13 | Male | + | + | + | + | ||
| 14 | Female | + | + | + | + | ||
| 15 | Male | + | + | + | + | ||
| 16 | Male | + | + | + | + | + | + (txn) |
| 17 | Female | + | + | + | + | ||
| 18 | Male | + | + | + | + | ||
| 19 | Female | + | + | + | + | + | |
| 20 | Female | + | + | + | + | ||
| 21 | Male | + | +( txn) | ||||
| 22 | Female | + | |||||
| 23 | Male | + | + | ||||
| 24 | Male | + | + | ||||
| 25 | Male | + | + | ||||
| 26 | Male | + | + | ||||
| 27 | Female | + | + | + | + | ||
| 28 | Female | + | + | + | + | + | |
| 29 | Male | + | + | + | + | ||
| 30 | Male | + | + | ||||
| 31 | Male | + | + | + | |||
| 32 | Male | + | + | + | |||
| 33 | Male | + | + | ||||
| 34 | Female | + | + | + | + | + | |
| 35 | Male | + | + | + | + | ||
| 36 | Female | + | + | + | |||
| 37 | Male | + |
FTT, failure to thrive; txn, transfusion dependent
Neutropenia (ANC <1500/μL), occurring in 81% (30 of 37 patients), was the most common hematologic abnormality at clinical presentation. Three patients presented with severe anemia requiring transfusion support together with neutropenia or pancytopenia. Strikingly, 5 patients (14%) came to medical attention with no history of cytopenias.
Non-classical presentations were also noted in several other patients. One patient presented with isolated mild thrombocytopenia at age 12 years with the initial diagnosis of hypocellular MDS with a del 20(q) cytogenetic clone without any history of steatorrhea or failure to thrive or cytopenias. Another proband presented at age 17 years without cytopenias or steathorrhea, but with a family history of a sibling who died from AML at age 20. Fatty pancreatic changes were noted in this sibling post-mortem and SBDS testing of the living proband was positive for SBDS mutations. Upon diagnosis, a bone marrow exam revealed clonal changes which subsequently progressed to MDS. Another SDS proband (had an asymptomatic sibling with normal stature and lacking a history of diarrhea; this sibling was subsequently found to carry the same SBDS mutations as the proband and the blood count showed red cell macrocytosis. One subject had very short telomeres (<1st percentile for age) across 3 lymphocyte subsets as well as in total lymphocytes (Figure), a pattern more typically associated with dyskeratosis congenita. This subject did have testing for mutations in DKC1 the most common gene affected in dyskeratosis congenita, and was negative.
Figure. Short telomeres in a patient with Shwachman Diamond syndrome.
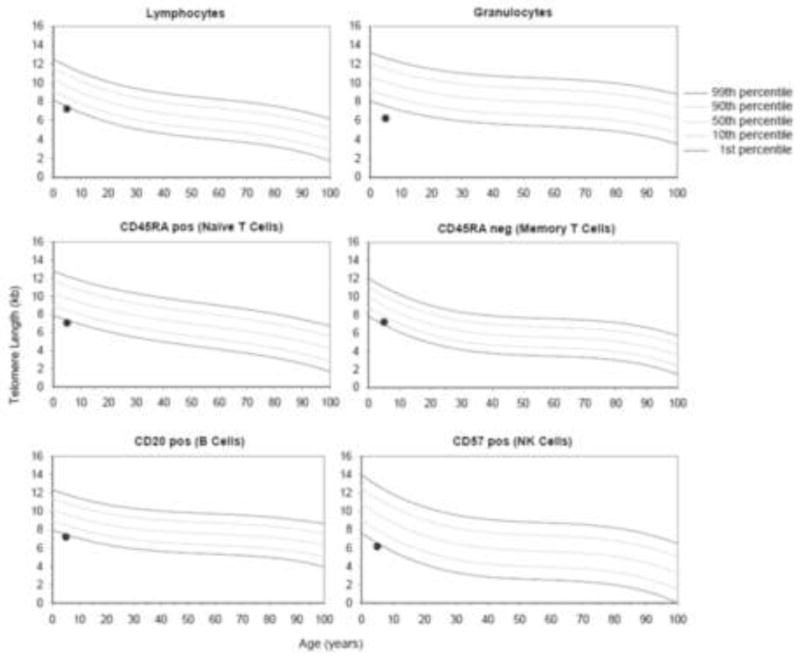
Extremely short telomeres in a patient with SDS. Telomeres were noted to be <1st percentile for age across 3 lymphocyte subsets as well as in total lymphocytes.
Bone marrow evaluations revealed hypocellularity for age in all 32 patients evaluated. Dysplasia was noted in 27 of 31 evaluable records. Marrow dysplasia was most significant in the myeloid lineage in 22 of 31 patients, although erythroid and megakaryocytic involvement were noted in 11 and 8 cases respectively. Seven of 31 patients demonstrated cytogenetic clones with del 20q noted in 5/37 subjects. Three of these patients had complex clonal abnormalities of whom two are alive and well after hematopoietic stem cell transplant and one remains stable with observation.
Twenty-seven of 37 patients (73%) presented with failure to thrive. Only 21/37 patients (58%) had a history of diarrhea. Radiologic reports of pancreatic imaging studies were available for 24 patients. 17 patients had pancreatic ultrasound reports available. Of these, 14 had pancreatic lipomatosis noted on ultrasound. One patient initially had a normal pancreatic ultrasound at age 2.6 years, but pancreatic lipomatosis was noted on a subsequent ultrasound 3 years later. One patient had an initial pancreatic ultrasound that reported lipomatosis but a subsequent ultrasound report did not comment on pancreatic lipomatosis. Two patients had no pancreatic lipomatosis noted on ultrasound; one of these patients had a small pancreatic size. Two patients had ultrasound reports that lacked any comments regarding the pancreas. Ten patients had CT reports available. Pancreatic lipomatosis was noted in nine patients. For one patient, an initial CT scan showed a small pancreas without lipomatosis at age 1.3 years but a subsequent CT scan showed pancreatic lipomatosis 4 years later. The pancreas was small or atretic for 5 patients and enlarged for one. One patient had pancreatic lipomatosis noted by MRI.
Fourteen of seventeen patients tested (82%) had low fecal elastase levels (<100 ug/g wet stool), and 3 had normal fecal elastase levels. Two of the three patients with normal levels were on pancreatic enzyme supplements at the time the fecal elastase levels were evaluated but one patient had normal fecal elastase levels without pancreatic enzyme supplements. Either serum trypsinogen (age <3 years) or pancreatic isoamylase (age > 3years) were low in all 17 patients tested.
Twenty-four of 37 patients (65%) had congenital or endocrine anomalies (Table III). Skeletal abnormalities were the most common abnormality reported in 38% of patients, either noted on physical exam or by skeletal survey. A wide variety of congenital anomalies involving the cardiac, gastrointestinal, renal, neurologic, urologic, and other systems were reported.
Table 3.
Congenital Anomalies and Medical Co-morbidities Identified in Patients with Shwachman-Diamond Syndrome
| Congenital anomalies | Number of patients n= 37 |
|---|---|
| Cardiac | 7 (19%) |
| VSD | 3 |
| PFO/ASD | 5 |
| PDA | 5 |
| Gastrointestinal | 3 (8%) |
| Malrotation | 1 |
| Bilateral inguinal hernia | 1 |
| Imperforate anus | 1 |
| Musculoskeletal | 14 (38%) |
| Thoracic dystrophy (rib abnormalities) | 9 |
| Short arms/legs | 2 |
| Metaphyseal dysplasia | 4 |
| Other legs (knock knees, bowing legs) | 2 |
| Pelvic dysostosis - absent pubic ramus | 1 |
| Scoliosis | 3 |
| Neurologic | 2 (5%) |
| Chiari Malformation, type I | 1 |
| Cerebellar tonsillar ectopia | 1 |
| Myopathy/hypotonia | 1 |
| Urologic | 2 (5%) |
| Testicular atrophy | 2 |
| Hypospadias | 1 |
| Other | 5 (14%) |
| Subglottic stenosis | 1 |
| Eye anomaly | 1 |
| Ear anomalies/hearing loss | 4 |
| Medical comorbidities | Number of patients n= 37 |
| Eczema | 11 |
| Elevated Liver Function Tests | 15 |
| Adrenal insufficiency | 1 |
| Hypopituitarism | 1 |
| Type I diabetes | 1 |
| Pulmonary HTN | 1 |
| Hypothyroid | 1 |
Discussion
Prior to the advent of genetic testing, the diagnosis of SDS was based largely on the clinical criteria of neutropenia and exocrine pancreatic insufficiency. We studied the presenting clinical phenotypes of 37 patients with genetically confirmed SDS and discovered an unexpectedly broad range in clinical phenotype at presentation. The frequency of cryptic presentations of SDS raises the likelihood that SDS is an underdiagnosed disorder. Serum trypsinogen and pancreatic isoamylase were the most sensitive measures of clinically asymptomatic pancreatic dysfunction in SDS. Normal pancreatic imaging studies and normal fecal elastase do not rule out the diagnosis of SDS.
The timely diagnosis of SDS carries implications for medical management and treatment. Routine monitoring of blood counts and marrow cytogenetics will allow early detection of marrow failure and transformation to MDS, preventing progression to AML. The incidence of AML in SDS ranges from 18–36% over 20–30 years) [1], and often those who do progress to AML are older. Severe marrow failure, MDS or leukemia are treated with a hematopoietic stem cell transplant. Patients with SDS require reduced intensity conditioning regimens to avoid undue regimen-related toxicities, thus the prior recognition of the underlying diagnosis of SDS is critical [9, 10]. Transplant outcomes are superior if initiated prior to the development of leukemia, so regular monitoring of the blood and bone marrow are of great value. The diagnosis of SDS also permits genetic testing of potential sibling donors prior to transplant, particularly key if the sibling is the stem cell donor.
It is important to note that hematologic abnormalities such as cytopenias, marrow hypocellularity and marrow dysplasia are not static but evolve over time even if absent at initial presentation. Similarly, skeletal abnormalities may evolve over time. Our study demonstrates that the absence of these findings at presentation does not rule out the diagnosis of SDS. In our cohort of patients with genetically-confirmed SDS, 31/31 marrows examined were hypocellular for age. Normocellular or hypercellular marrows have been reported in the literature for patients diagnosed with SDS on clinical grounds prior to the availability of genetic testing. Given the clinical overlap between SDS and other marrow failure syndromes, further study of the range of marrow abnormalities in patients with SDS with or without SBDS mutations is of high interest.
The differential diagnosis of neutropenia presenting without diarrhea is broad, and SDS is a rare disorder. Clues to cryptic presentations of SDS in our cohort included bone marrow hypocellularity, mild marrow dysmorphologies, congenital anomalies, family history of SDS, and the del20q clonal marrow abnormality. SBDS genetic testing should be considered for patients with idiopathic hypoproductive cytopenias together with any of these additional features.
This study raises the importance of genetic testing of clinically asymptomatic siblings of SDS probands. Furthermore, the diagnosis of SDS should also be considered in adults because cryptic presentations may be missed during childhood. Thus far, the Registry has not found any reports of aplastic anemia arising in obligate SBDS heterozygotes such as parents or grandparents.
It is important to emphasize that the absence of a gene mutation does not rule out the diagnosis of SDS. Although 90% of patients with classically presenting SDS have mutations in the SBDS gene, there remain a proportion of patients with clinically diagnosed SDS without known mutations. However, it is currently unclear whether these SBDS mutation-negative patients represent a different genetic variant of SDS or instead might constitute a mixture of different undefined disorders with overlapping clinical features. Therefore, this analysis was restricted to patients with SBDS mutation-positive SDS. Additional studies are necessary to further characterize these SBDS mutation-negative patients both clinically and molecularly.
Current diagnosis and management of SDS is largely based on case series and consensus reports, as its natural history remains poorly characterized due to the rarity of this disease. Controlled clinical studies to inform diagnosis and therapy of SDS are lacking. Although this study represents a large cohort of patients with SDS evaluated for initial presentation, the small size of the current registry cohort remains a limitation that should be considered in interpretation. Longitudinal cohort studies stand to better delineate diagnostic criteria, phenotypic range, complications, and treatment outcomes to improve the medical care of patients with SDS. This study highlights the continued importance of systematic study through registries for rare disorders.
Acknowledgments
Supported by the National Institute of Allergy and Infectious Diseases (NIAID; 1 R03 AI079734-02 to A.S.), National Heart, Lung, and Blood Institute ( 5 R01 HL079582-11 to A.S.), NIAID (5 R24 AI049363-09 to D.D.), Schwachman-Diamond Project Ltd, Schwachman-Diamond Syndrome Foundation, Schwachman-Diamond Syndrome America, The Butterfly Guild at Seattle Children’s Hospital, and family donors.
We thank Nicholas Dobbins for setting up the database programs and data query pulls for the SDSR and Joan Moore for her help with patient enrollment, data collection, data extraction, and analysis. We thank the FHCRC Collaborative Data Services for initial work with the SDSR database.
Abbreviations
- SDS
Shwachman-Diamond syndrome
- SDSR
Shwachman-Diamond Syndrome Registry
- MDS
myelodysplasia
- SBDS
Shwachman-Bodian-Diamond syndrome
- ANC
absolute neutrophil count
- AML
acute myeloid leukemia
Footnotes
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Myers KC, Davies SM, Shimamura A. Clinical and Molecular Pathophysiology of Shwachman–Diamond Syndrome: An Update. Hematology/oncology clinics of North America. 2013;27:117–28. doi: 10.1016/j.hoc.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Menne TF, Goyenechea B, Sanchez-Puig N, Wong CC, Tonkin LM, Ancliff PJ, et al. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39:486–95. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- 3.Finch AJ, Hilcenko C, Basse N, Drynan LF, Goyenechea B, Menne TF, et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011;25:917–29. doi: 10.1101/gad.623011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganapathi KA, Austin KM, Lee CS, Dias A, Malsch MM, Reed R, et al. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007;110:1458–65. doi: 10.1182/blood-2007-02-075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dror Y, Donadieu J, Koglmeier J, Dodge J, Toiviainen-Salo S, Makitie O, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci. 2011;1242:40–55. doi: 10.1111/j.1749-6632.2011.06349.x. [DOI] [PubMed] [Google Scholar]
- 6.Rothbaum R, Perrault J, Vlachos A, Cipolli M, Alter BP, Burroughs S, et al. Shwachman-Diamond syndrome: report from an international conference. J Pediatr. 2002;141:266–70. doi: 10.1067/mpd.2002.125850. [DOI] [PubMed] [Google Scholar]
- 7.Andolina J, Morrison C, Thompson A, Chaudhury S, Mack A, Proytcheva M, et al. Shwachman-Diamond Syndrome: Diarrhea, no Longer Required? J Pediatr Hematol Oncol. 2012 doi: 10.1097/MPH.0b013e3182667c13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat Protocols. 2006;1:2365–76. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- 9.Myers KC, Davies SM. Hematopoietic stem cell transplantation for bone marrow failure syndromes in children. Biol Blood Marrow Transplant. 2009;15:279–92. doi: 10.1016/j.bbmt.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 10.Bhatla D, Davies SM, Shenoy S, Harris RE, Crockett M, Shoultz L, et al. Reduced-intensity conditioning is effective and safe for transplantation of patients with Shwachman-Diamond syndrome. Bone Marrow Transplant. 2008;42:159–65. doi: 10.1038/bmt.2008.151. [DOI] [PubMed] [Google Scholar]