

Abstract
Haploinsufficiency for the HMG-box transcription factor SOX2 results in abnormalities of the human ventral forebrain and its derivative structures. These defects include anophthalmia (absence of eye), microphthalmia (small eye) and hypothalamic hamartoma (HH), an overgrowth of the ventral hypothalamus. To determine how Sox2 deficiency affects the morphogenesis of the ventral diencephalon and eye, we generated a Sox2 allelic series (Sox2IR, Sox2LP, and Sox2EGFP), allowing for the generation of mice that express germline hypomorphic levels (<40%) of SOX2 protein and that faithfully recapitulate SOX2 haploinsufficient human phenotypes. We find that Sox2 hypomorphism significantly disrupts the development of the posterior hypothalamus, resulting in an ectopic protuberance of the prechordal floor, an upregulation of Shh signaling, and abnormal hypothalamic patterning. In the anterior diencephalon, both the optic stalks and optic cups (OC) of Sox2 hypomorphic (Sox2HYP) embryos are malformed. Furthermore, Sox2HYP eyes exhibit a loss of neural potential and coloboma, a common phenotype in SOX2 haploinsufficient humans that has not been described in a mouse model of SOX2 deficiency. These results establish for the first time that germline Sox2 hypomorphism disrupts the morphogenesis and patterning of the hypothalamus, optic stalk, and the early OC, establishing a model of the development of the abnormalities that are observed in SOX2 haploinsufficient humans.
Keywords: Sox2, Prechordal floor, Optic cup, Optic stalk, Shh, Patterning
1. Introduction
SOX2 is a member of the SRY-related Box family of transcription factors. It plays a critical role in embryonic development, maintains pluripotency and self-renewal potential in embryonic stem cells, and was recently demonstrated to be able to cooperate with other factors to induce pluripotency in a variety of somatic cell types (Avilion et al., 2003; Takahashi et al., 2007; Lai et al., 2011). Consistent with the role of SOX2 in the maintenance of neural potential and self-renewal, conditional ablation of Sox2 in retinal progenitors shifts their fate from neural to ciliary epithelial, reducing their potential to differentiate into post-mitotic neurons (Taranova et al., 2006; Matsushima et al., 2011). As a key regulator of a wide array of morphogenetic processes, SOX2 is essential for the normal development of the CNS, the sensory cells of the inner ear and eye, taste bud cells, the division of early gut and respiratory structures, and branching and epithelial cell differentiation in the lung (Kelberman et al., 2006; Okubo et al., 2006; Taranova et al., 2006; Gontan et al., 2008; Domyan et al., 2011; Engelen et al., 2011). However, the precise role of SOX2 in the morphogenesis of many of these tissues has not been examined in detail. Specifically, there is a gap in our understanding of how reductions in SOX2 function translate into the morphological defects that are observed in humans with SOX2 haploinsufficiency. This genetic disorder accounts for a significant percentage of cases of anophthalmia, microphthalmia, and coloboma, the latter of which results from a failure of the embryonic optic fissure to fuse (Fantes et al., 2003; Kelberman et al., 2006; Bakrania et al., 2007; Schneider et al., 2009). Previous research using conditional ablation of Sox2 has demonstrated its importance for maintaining the neural potential of retinal progenitors, but the effects of germline Sox2 hypomorphism on the early stages of ocular development have not been defined (Taranova et al., 2006; Matsushima et al., 2011). SOX2 haploinsufficient humans also present with diencephalic abnormalities, such as hypothalamic hamartoma (HH), a benign overgrowth of the hypothalamus that is associated with a variety of developmental problems, cognitive deficiencies, and seizures (Freeman, 2003; Kelberman et al., 2006; Wallace et al., 2008). SOX2 and GLI3 mutations are the only reported genetic causes of human hamartomas; however, given that no animal model exists for HH, our current understanding of how these defects develop is extremely limited from both a morphological and molecular perspective.
Previous analyses of the role of SOX2 in the morphogenesis of the mammalian CNS have primarily relied on the examination of (i) Sox2 heterozygous mice, (ii) mouse lines in which Sox2 is ablated in a spatiotemporally-specific manner, or (iii) mouse lines with mutations affecting Sox2 enhancers (Dong et al., 2002; Ferri et al., 2004; Kiernan et al., 2005; Kelberman et al., 2006; Taranova et al., 2006; Miyagi et al., 2008; Matsushima et al., 2011). Each of these approaches is limited with respect to their ability to model human SOX2 haploinsufficiency. Sox2+/− mice do not typically exhibit ocular defects, which are almost ubiquitous in SOX2 haploinsufficient humans, precluding the use of Sox2+/− mice as a model of SOX2 haploinsufficiency (Avilion et al., 2003; Ellis et al., 2004; Kelberman et al., 2006; Taranova et al., 2006; Schneider et al., 2009). Moreover, the vast majority of reported human SOX2 haploinsufficiency cases result from mutations in the SOX2 open reading frame, most likely resulting in systemic attenuation of SOX2 function. Therefore, models that make use of restricted modification of Sox2 expression do not recapitulate the genetic environment associated with human SOX2 haploinsufficiency (Schneider et al., 2009).
To determine how reduced SOX2 dosage impacts the development of the neural ectoderm derivatives that are affected in SOX2 haploinsufficient humans, we generated a series of knockin Sox2 hypomorphic (Sox2LP and Sox2IR) and null (Sox2EGFP) alleles. Sox2EGFP/LP and Sox2EGFP/IR (collectively referred to as Sox2HYP) mice express less than 40% of wild type SOX2 protein levels and exhibit variable anophthalmia and microphthalmia, similar to SOX2 haploinsufficient humans (Ellis et al., 2004; Taranova et al., 2006). Sox2HYP mice exhibit a global reduction in SOX2 levels, a genetic environment that mimics that of SOX2 haploinsufficient humans. Here, we present a detailed analysis of Sox2HYP mice and ascertain the temporal onset of the ocular and diencephalic morphological defects that result from reduced SOX2 dosages. We find that Sox2HYP embryos exhibit morphological and molecular abnormalities of the posterior prechordal floor, including an upregulation of Shh and a downregulation of Gli3. E10.5 Sox2HYP embryos exhibit morphological defects of the optic stalk and optic cup (OC), and molecular patterning abnormalities in the retinal primordium. Sox2HYP eyes exhibit decreased levels of Pax2 and are colobomic, a phenotype that has not been described in a mouse model of Sox2 deficiency. Taken together, these results (i) represent the first characterization of the molecular and morphological phenotype of the SOX2 mutant hypothalamus, a region known to be affected in SOX2 haploinsufficient humans; (ii) establish the morphological and molecular onset of the ocular defects observed in Sox2HYP mice; and (iii) establish an animal model for SOX2 deficiency-related coloboma.
2. Materials and methods
2.1. Animals
The generation of the Sox2IR, Sox2LP, Sox2EGFP alleles has been previously described (Ellis et al., 2004; Taranova et al., 2006). Sox2IR/+ and Sox2LP/+ females were bred to Sox2EGFP/+ stud males to generate Sox2EGFP/IR, Sox2EGFP/LP, Sox2+/LP, Sox2+/IR and Sox2+/+ embryos (Taranova et al., 2006). No phenotypic differences were observed between Sox2+/+, Sox2IR/+, or Sox2LP/+ embryos, and these embryos were considered controls (Sox2CONT). Sox2EGFP/LP embryos were reported to express approximately 10% lower levels of SOX2 protein than Sox2-EGFP/IR embryos; however, no clear phenotypic differences were observed between these genotypes with respect to any of the described phenotypes. Sox2EGFP/LP and Sox2EGFP/IR embryos are therefore are collectively referred to as Sox2HYP. All of the molecular and scanning electron microscopy (SEM) analyses were performed on a mixed C57BL/6-CD1 (Jackson Laboratories) background. Sox2HYP mice on this background mice display less severe but similar phenotypes to Sox2HYP C57BL/6 mice. Whole mount morphological analyses of E13-E14 embryos were performed on a C57BL/6 background. All of the animal work was performed in accordance with University of North Carolina at Chapel Hill DLAM and IACUC approval. The date of vaginal plug was considered E0.5, and precise embryo stages were determined using established somite reference points (Chan et al., 2004).
2.2. Genotyping protocols
Sox2 alleles were genotyped using the following primer sequences and protocols: for the Sox2 wildtype allele, 5′-GCTCTGTTATTGGAATCAGGCTGC-3′ and 5′-CTGCTCAGGGAAGGAGGGG-3′, (35 cycles of 15 s at 94 °C, 30 s at 55 °C, 30 s at 72 °C); for the Sox2IR allele, 5′-GGCTCTCCTCAAGCGTATTCAA-3′ and 5’-TTGTAGTCGGGGATGCGGC-3’, (35 cycles of 15 s at 94 °C, 30 s at 58 °C, 45 at 72 °C); for the Sox2LP allele, 5′-CAGCAGCCTCTGTTCCACATACAC-3′ and 5′-CAACGCATTTCAGTTCCCCG-3′, (35 cycles of 15 s at 94 °C, 30 s at 55 °C, 30 at 72 °C). The presence of the Sox2EGFP allele was determined visually using a UV fluorescent microscope.
2.3. Tissue preparation for SEM analyses
The embryos were processed in a manner similar to that described in Sulik et al., 1994. Briefly, the embryos were fixed in 2.5% glutaraldehyde, cut manually through the forebrain at or near the level of the optic primordium, and post-fixed for 1–2 h in 2% osmium tetroxide. The samples were then dehydrated in a graded ethanol series and critical point dried using liquid CO2. The samples were then mounted on aluminum stubs, manually cleaned of debris, coated with a gold–palladium sputter-coater, and imaged on a JEOL 6300 scanning electron microscope.
2.4. Tissue preparation for immunohistochemical and in situ hybridization analyses
The embryos were fixed at 4 °C in a solution of phosphate buffered saline (PBS) and 4% paraformaldehyde for periods of 2 h to overnight, depending on the stage of the embryo. Following three washes in PBS, the embryos were cryoprotected in a sucrose gradient (10%, 20%, and 30% sucrose in PBS) and mounted in Optimum Cutting Temperature mounting medium (OCT, Tissue-Tek). For immunohistochemical (IHC) analyses, frontal 10 μm sections were blocked in a solution of 1–4% heat-inactivated goat serum and 0.1% Triton X-100 (Sigma). The sections were incubated with primary antibodies overnight at 4 °C and with secondary antibodies for 1 h at room temperature. For in situ analyses, 20 μm frontal or horizontal sections were incubated with digoxigenin (DIG)-labeled probes and visualized using enzymatic detection, following the manufacturer’s protocol (Roche). The following antibodies were used for this study: rabbit anti-PH3 (1:1000, Abcam), rabbit anti-cleaved capase-3 (1:250, Cell Signaling), and Alexa Fluor 546-conjugated anti-rabbit (1:1000, Molecular Probes). The following in situ hybridization probes were used for this study: Nkx2.1 [Dr. A. Lamantia, (Shimamura et al., 1995)], Otx1 [Dr. J. P. Martinez-Barbera, (Simeone et al., 1992)], Pax2 [Dr. J.P. Martinez-Barbera, (Sajedi et al., 2008)], Pax6 [Dr. A. LaMantia, (Anchan et al., 1997)], Rax [Dr. C. Cepko, (Furukawa et al., 1997)], Shh (Dr. E. Tucker), Tbx3, [Dr. M. Pontecorvi, (Pontecorvi et al., 2008)], and Vax2 [Dr. G. Lemke, (Mui et al., 2005)]. At least three Sox2CONT and Sox2HYP embryos were analyzed for each probe.
2.5. Measurements and statistics
All of the optic stalk length measurements were made relative to the head width of the embryo at the optic level. E10.5 head-width measurements were made between the points on the surface ectoderm where the maxillary process emerges, immediately ventral to the eye. The optic stalk lengths were measured linearly. All of the distance measurements were made in triplicate using ImageJ software and averaged. The Mendelian ratios were examined with the two-tailed squared test using three degrees of freedom. The cell death and proliferation analyses were performed by computing the ratio of positively stained cells to the total number of cells in a predefined region. Cells were only considered PH3-positive if they were located along the apical edge of the retina. Cells were considered positive for cleaved caspase-3 if staining could be directly associated with a condensed or fragmented nucleus. The results are reported as the mean ± the standard error of the mean. At least three Sox2CONT and Sox2HYP embryos were analyzed for cell death and proliferation and for all in situ gene expression analyses. Once the penetrance of the hypothalamic defect was determined, the gene expression analyses were performed on severely affected embryos only (see Fig. 2 legend).
Fig. 2.
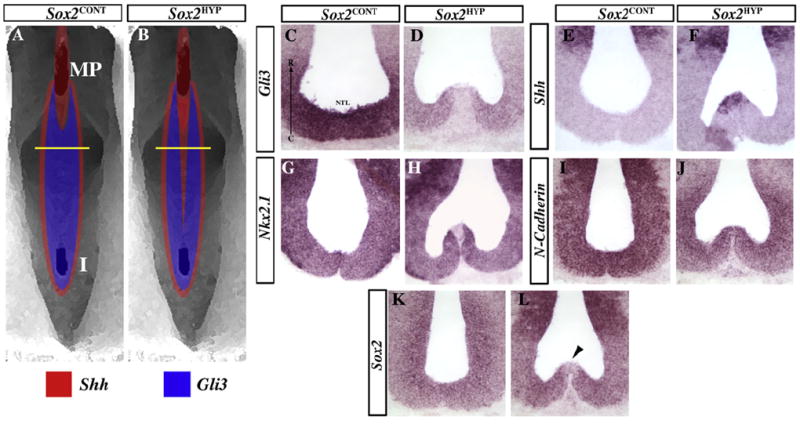
Disrupted Shh signaling and hypothalamic patterning in E12.5 Sox2HYP embryos. (A and B) Diagram illustrating Shh and Gli3 expression in Sox2CONT (A) and Sox2HYP (B) embryos. The yellow lines indicate the approximate levels of the cuts in C– L. (C and D) The Shh signaling antagonist Gli3 is expressed in the prechordal floor of Sox2CONT (C) embryos between the mammillary region and the infundibulum, but is downregulated in this region in Sox2HYP (D) embryos. (E and F) Shh expression in the prechordal plate of Sox2CONT (E) and Sox2HYP (F) embryos. Shh is normally excluded fromthe prechordal floor between the mammillary region and the infundibulum (E), but remains expressed in this region in Sox2HYP embryos (F). (G and H) Nkx2.1 is normally expressed throughout the ventral hypothalamus (G) but is excluded from the prechordal floor in Sox2HYP (H) embryos. (I and J) N-cadherin is normally expressed in all neural progenitors (I), and this expression pattern is largely maintained in Sox2HYP (J) embryos. (K and L) Sox2 is also expressed in all neural progenitors in Sox2CONT (K) embryos, but is strongly downregulated in the prechordal floor of Sox2HYP (L, arrowhead) embryos. A clear protuberance is present in most Sox2HYP embryos (13/15 examined).
2.6. Anatomic nomenclature
The anatomical features referred to below are defined as follows and are diagrammed in Fig. S1. According to the revised prosomeric model of Puelles and Rubenstein, 2003 the prechordal floor is defined as the ventral-most aspect of the diencephalon and telencephalon, with a caudal limit at the posterior edge of the retromammillary area. The mammillary area is defined as the region surrounding the mammillary body anlage, which is located near the posterior limit of the prechordal floor. The mammillary pouch is defined as the invagination of the prechordal floor within the mammillary area and was used as amorphological landmark for the expression analyses (García-Calero et al., 2006). The tuberal hypothalamus is defined as the region of the prechordal floor anterior to the mammillary area and posterior to the infundibulum (Shimamura et al., 1995). The ciliary margin of both E10.5 and E16.5 retinas is defined as the distal aspect of the retina.
3. Results
3.1. Sox2HYP embryos are recovered in Mendelian ratios and do not exhibit global developmental delays
Sox2HYP embryos carry a Sox2EGFP null reporter allele and either the Sox2IR or Sox2LP hypomorphic allele. When expressed in mice, these alleles have been shown to result in less than 40% of wild-type SOX2 protein levels (Taranova et al., 2006). Given that SOX2 is required for the survival of the peri-implantation embryo, we analyzed genotypic ratios at different embryonic stages (E9.5–E18) to determine whether Sox2HYP embryos display increased embryonic lethality. Sox2-HYP embryos are recovered at the expected Mendelian ratio (Table S1). To determine whether overall development is delayed in Sox2HYP embryos, somite counts for E9.5 and E10.5 litters were tabulated and found to be similar between Sox2CONT and Sox2HYP embryos (Table S1). These results indicate that (i) Sox2 deficiency does not lead to embryonic lethality, and (ii) the morphological defects observed in Sox2HYP mice are not the result of a developmental delay (See Table S1).
3.2 The diencephalic prechordal floor is laterally expanded in E10.5 Sox2HYP embryos
SOX2 haploinsufficient humans exhibit HH, an overgrowth of the posteroventral diencephalon. Given the complex and dynamic morphology of the developing diencephalon, we used SEM to analyze this region in E9.5 and E10.5 Sox2HYP embryos. The SEM data revealed that the posterior prechordal floor is broader in Sox2HYP embryos as early as E9.5 (Fig. S1D vs. E and F). By E10.5, two evaginations are visible in the Sox2-CONT prechordal floor; the infundibulum, which is the precursor of the posterior and infundibular lobes of the pituitary, and the more posterior mammillary pouch (Fig. 1 and Fig. S2). SEM images of E10.5 Sox2HYP embryos revealed that the infundibulum is broadened (Fig. 1D vs. E and F). The infundibular widths were determined from measurements of histological sections, which confirmed a statistically significant expansion of the infundibulum in Sox2HYP embryos (Sox2CONT: 147.0 ± 3.7 μm, Sox2HYP: 183.8 ± 10.3 μm, p < 0.05, n = a minimum of 5 for each group, Fig. S3). With respect to the mammillary pouch, Sox2CONT embryos exhibit a singular, deep invagination of the posterior prechordal floor at E10.5 (Fig. 1A and D). The mammillary pouch of Sox2HYP embryos, however, is often bifurcated by a region of ectopic tissue (Fig. 1A, D vs. B and E, arrow). Unlike E10.5 Sox2CONT embryos, a subset of age-matched Sox2HYP embryos exhibit protuberances in the posterior tuberal hypothalamus (Fig. 1D vs. F, arrowhead). These SEM analyses provide the first insight into the effects SOX2 deficiency on the morphological development of the posterior prechordal floor, a tissue that is affected in SOX2 haploinsufficient humans (Kelberman et al., 2006).
Fig. 1.
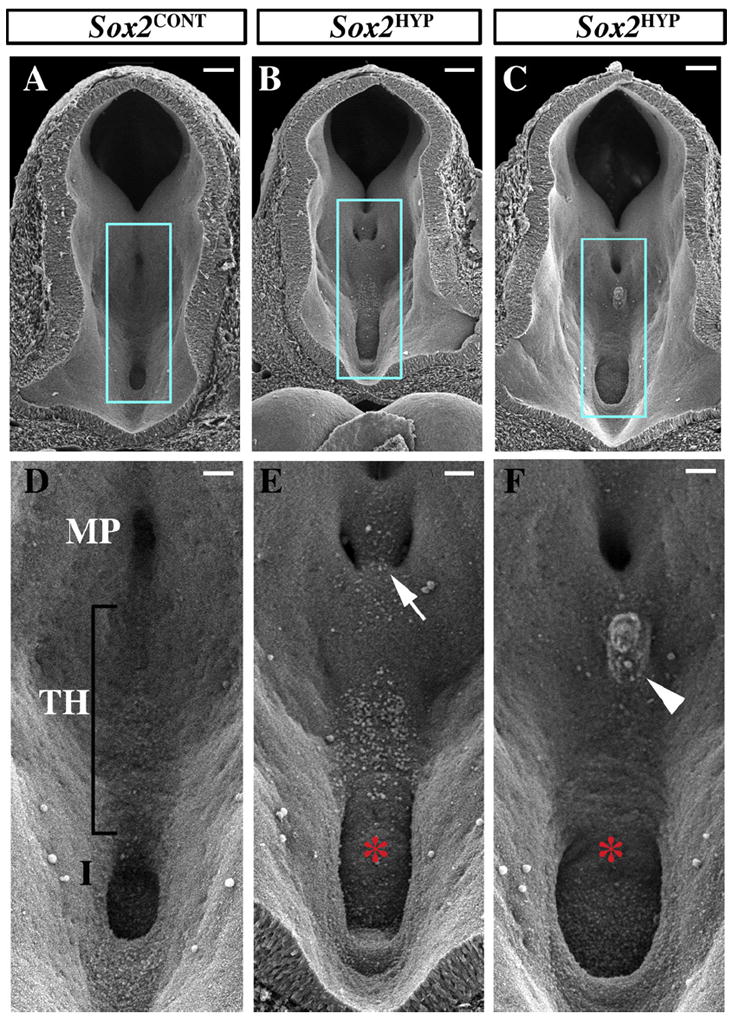
Scanning electron analyses of the posterior ventral diencephalon in E10.5 Sox2CONT and Sox2HYP embryos. (A–C) Posterior-facing views of Sox2CONT (A) and Sox2HYP (B and C) E10.5 diencephala. Scale bars for A–C: 100 μm. (D–F) Magnified images of the regions outlined in blue in (A-C) showing the mammillary region, tuberal hypothalamus, and infundibulum of Sox2CONT (D) and Sox2HYP (E and F) embryos. Sox2CONT mammillary pouches appear as singular midline invaginations. (D) In contrast, Sox2HYP mammillary pouches are often divided by ectopic, undescended tissue (arrow in E). The infundibula of Sox2HYP embryos are expanded relative to those of Sox2CONT embryos (D vs. E, F, red asterisks). A subset of Sox2HYP embryos exhibit a protuberance in the tuberal hypothalamus by E10.5 (arrowhead). Scale bars for D–F: 50 μm. MP: mammillary pouch; TH: tuberal hypothalamus; I: infundibulum.
3.3. Shh signaling and hypothalamic patterning are disrupted in the E12.5 Sox2HYP prechordal floor
E12.5 Sox2HYP embryos were analyzed histologically at the level of the tuberal hypothalamus, revealing a protuberance along the prechordal floor. Given that GLI3 has been implicated in human HHs, we examined Gli3 expression in the E12.5 Sox2HYP hypothalamic anlage (Wallace et al., 2008). In Sox2CONT embryos, Gli3 is not expressed along the prechordal floor from its caudal limit to approximately 60 lm anterior to the mammillary pouch; however, Gli3 is expressed rostral to this point (Fig. 2A and C). In Sox2HYP embryos, however, Gli3 is absent from the prechordal floor along nearly the entire length of the tuberal hypothalamus (Fig. 2B and D). Gli3 and Shh have been demonstrated to be mutually antagonistic in several CNS regions, including the developing spinal cord and telencephalon (Litingtung and Chiang, 2000; Rallu et al., 2002). Consistent with these previous findings, Shh exhibits a nearly mutually exclusive expression pattern to Gli3 along the prechordal floor of E12.5 Sox2CONT embryos, i.e., it is expressed only at low levels in the mammillary region but is downregulated approximately 60 μm anterior to the mammillary pouch (Fig. 2A and E). In contrast, Shh expression is observed as far as 300 μm anterior to the mammillary pouch in Sox2HYP embryos (Fig. 2B and F).
To determine the cell identity of the hypothalamic protuberance in Sox2HYP embryos, we examined the expression of hypothalamic and neural progenitor markers. Examination of the expression of the hypothalamic progenitor marker Nkx2.1 revealed that this gene is expressed anterior to the mammillary region along the prechordal floor in Sox2CONT embryos but is absent in the protuberance of the prechordal floor observed in Sox2HYP embryos (Fig. 2G vs. H). We next examined Sox2CONT and Sox2HYP embryos for the expression of markers that define and maintain neural progenitor identity: N-cadherin and Sox2 itself (Graham et al., 2003; Zhang et al., 2010). N-cadherin is expressed normally along the prechordal floor of Sox2HYP embryos, indicating that these cells have maintained a neural progenitor identity (Fig. 2I vs. J). Uniform expression of Sox2 is observed along the prechordal floor of Sox2CONT embryos (Fig. 2K). Sox2 expression is also present throughout most of the neuroepithelium of Sox2HYP embryos but is nearly undetectable along the prechordal floor of the tuberal hypothalamus (Fig. 2K vs. L). At E14.5, the protuberance in the Sox2HYP tuberal hypothalamus remains the same size as at E12.5; however, the Nkx2.1 and Shh expression patterns are unaffected (Fig. S4B and D). These analyses indicate that morphological defects observed in the prechordal floor of Sox2HYP embryos are associated with disrupted Shh signaling and abnormal hypothalamic patterning.
3.4. The retinal primordium of E10.5 Sox2HYP embryos is morphologically abnormal
To determine the onset of the optic malformations observed in Sox2HYP mice, we performed whole-mount and/or SEM analyses of embryos between E9.5 and E14.5. At E9.5, no clear differences can be discerned between the optic vesicles of whole mount Sox2CONT and Sox2HYP embryos (Fig. S2A–C). Sox2HYP embryos first exhibit readily discernable morphological defects at E10.5, during formation of the OC. In Sox2-EGFP/+ embryos, the EGFP reporter permits visualization of the neural retina as a ring of tissue surrounding the circular, SOX2-positive invaginating lens (Fig. 3A). In Sox2HYP embryos, however, the ventral aspect of the retina cannot be discerned through the surface ectoderm (Fig. 3D). By E13.5 Sox2HYP embryos exhibit coloboma, a persistence of the ventral optic fissure (Fig. 3B vs. E). Severely affected Sox2HYP embryos develop anophthalmia by E14.5, with only pigmented epithelial cells remaining (Fig. 3C vs. F).
Fig. 3.
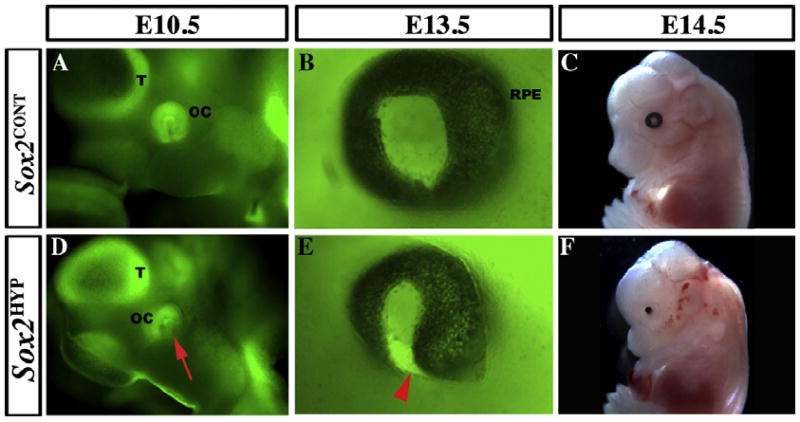
Temporal sequence of the ocular phenotype in whole-mount Sox2HYP embryos. (A and D) Lateral views of E10.5 whole mount Sox2EGFP/+ (A) and Sox2HYP (D) heads. In Sox2EGFP/+ embryos, the ring of the distal optic cup (OC) can be seen ventral and posterior to the telencephalic vesicle (T), contacting the surface ectoderm. The SOX2-positive lens is visible in the center of the ring. In contrast, the ventral aspect of the distal OC is not visible in the Sox2HYP embryo (red arrow, D). (B and E) Lateral views of E13.5 whole mount Sox2EGFP/+ (B) and Sox2HYP (E) eyes. In the Sox2EGFP/+ embryo, the black retinal pigment epithelium surrounds the entire outer surface of the eye. In contrast, the ventral optic fissure of the Sox2HYP eye has failed to close, a defect referred to as coloboma (red arrowhead, E). (C and F) E14.5 whole mount Sox2CONT (C) and Sox2HYP (F) embryos. A round, symmetric eye with a visible lens is present in the Sox2CONT embryo, whereas only a remnant of the retinal pigmented epithelium can be seen in the Sox2HYP embryo.
To determine the nature of the ventral OC defects observed in whole mount E10.5 Sox2HYP embryos, we performed SEM analysis of the optic anlage. In Sox2CONT embryos, the ventral aspect of the OC is in close opposition to the surface ectoderm (Fig. 4A and D). Sox2HYP OCs, however, often exhibit an abnormal morphology, such that there is an increased separation between the ventral OC and the surface ectoderm (Fig. 4A, D vs. B, C, E and F), explaining why the ventral OC cannot be discerned through the surface ectoderm in Sox2HYP embryos (Fig. 3B).
Fig. 4.
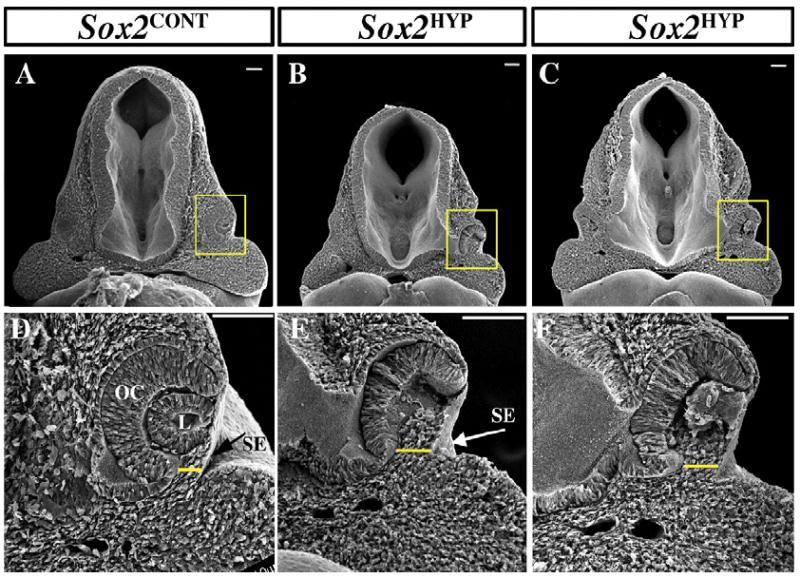
Scanning electron analyses of the optic anlage of E10.5 Sox2CONTand Sox2HYP embryos. (A and D) Low- (A) and high- (D) magnification views of an E10.5 Sox2CONT OC. The distal OC is in close contact with the surface ectoderm, with a slight ventral separation (yellow line). B, C, E and F) Low- (B and C) and high- (E and F) magnification views of E10.5 Sox2HYP OCs. The distance between the ventral OC and the surface ectoderm is increased in Sox2HYP embryos (yellow lines in E–F). Scale bars: 100 μm. L: lens, OC: optic cup, SE: surface ectoderm.
Given that E14.5 and later-stage Sox2HYP embryos exhibit microphthalmia or anophthalmia, we analyzed cell number, proliferation and apoptosis in E9.5 and E10.5 retinal progenitors using Hoechst, phospho-histone H3 (PH3), and cleaved caspase-3, respectively. We find that cell number, proliferation, and apoptosis are not significantly affected in the presumptive retina of E9.5 or E10.5 Sox2HYP embryos (Fig. S5A–M).
3.5. Sox2HYP ventral optic stalks are shorter and exhibit increased cell death
We hypothesized that increased separation between the ventral aspect of the Sox2HYP OC and the surface ectoderm results from a hypoplastic ventral optic stalk; i.e., a decrease in the length of the ventral optic stalk could prevent the ventral aspect of the OC from reaching the surface ectoderm. To test this hypothesis, we measured the length of the ventral optic stalk, identifying a clear trend that, relative to the width of the head, the E10.5 optic stalk is shorter in Sox2HYP than in age-matched Sox2CONT embryos at the level of the mid OC (Fig. 5E, p = 0.06).
Fig. 5.
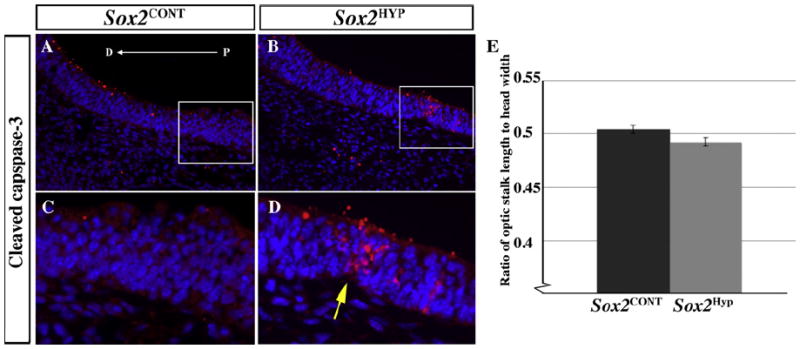
Morphological and cell death analyses in the ventral optic stalk of E10.5 Sox2CONT and Sox2HYP embryos. (A and B) Sox2CONT (A) and Sox2HYP (B) optic stalks stained for cleaved caspase-3. Positively stained cells are more common in Sox2HYP optic stalks (arrow in D). Red staining on the dorsal surface of the optic stalk does was not associated with nuclei and was not included in the analysis. (C and D) Magnified images of the regions outlined in (A) and (B), respectively. (E) Quantification of optic stalk length (optic stalk length/head width = 0.504 ± 0.004 and 0.492 ± 0.004 for Sox2CONT and Sox2HYP optic stalks, respectively, p = 0.06, n = 11 optic stalks for both groups). D: distal, P: proximal.
To determine whether the observed shortening of the ventral optic stalk is the result of increased cell death, cleaved caspase-3 staining was analyzed in E9.5 and E10.5 optic stalks. At E9.5, both Sox2CONT and Sox2HYP embryos exhibit cell death in the central portion of the ventral optic stalk, but no statistically significant difference was observed between Sox2CONT and Sox2HYP embryos (Fig. S5 G–M). At E10.5, however, cell death is rarely observed in the ventral optic stalk of Sox2CONT embryos, whereas cleaved caspase-3-positive cells are common in this tissue in Sox2HYP embryos (Fig. 5A, C vs. B and D). Regions of cell death in E10.5 Sox2HYP ventral optic stalks occasionally result in a “pinched” appearance, which is never observed in the ventral optic stalk of Sox2CONT embryos (Fig. 5C vs. D [arrow]). These findings are the first to characterize the early optic phenotype in mice with reduced SOX2 signaling and support a differential role for SOX2 in the development of the retinal primordium and the ventral optic stalk. Moreover, these results are the first to define a requirement for SOX2 in the development of the mammalian optic stalk.
3.6. Characterization of the patterning of Sox2HYP neural retinas at two developmental stages
Given that many animal models of ocular defects exhibit abnormal molecular patterning of the retina and/or optic stalk, we next determined whether molecular patterning was disrupted in the E10.5 Sox2HYP optic anlage. We examined the expression of proximal/ventral markers (Pax2, Vax2), which label the ventral optic stalk and ventral retina, the dorsal retina marker Tbx3, and a marker of the ciliary margin and retinal pigmented epithelium (RPE) (Otx1) (Nornes et al., 1990; Barbieri et al., 1999; Martinez-Morales et al., 2001; Sowden et al., 2001). The expression of Pax6, which is present in the retina in a high-distal/low-proximal gradient, and Rax, which marks all retinal progenitors at this stage, were also examined (Mathers et al., 1997; Bäumer et al., 2002: Matsushima et al., 2011). In the dorsal and ventral OC of Sox2HYP embryos, Tbx3 (Fig. 6A and B) and Vax2 (Fig. 6C and D), respectively, mimic the expression patterns observed in Sox2CONT embryos. Within the Sox2HYP OC, Pax6 expression is increased (Fig. 6E and F) and Rax expression is largely unaffected (Fig. 6G and H). Given that (i) Pax2 is associated with coloboma in humans and mice and (ii) PAX6 and PAX2 are mutually antagonistic in the developing retina, we examined the expression of Pax2 in Sox2CONT and Sox2HYP embryos (Sanyanusin et al., 1995; Torres et al., 1996; Schwarz et al., 2000). Whereas Pax2 is strongly expressed in the ventral OC of Sox2CONT embryos, it is strongly downregulated in the Sox2HYP OC (Fig. 6I and J). To determine whether the ciliary margin and RPE are correctly specified in Sox2HYP embryos, we examined the expression of Otx1, which is normally expressed in the distal ciliary margin and the RPE. In Sox2CONT embryos at E10.5, Otx1 expression is restricted to the distal retina and the RPE (Fig. 6K). In E10.5 Sox2HYP OCs, Otx1 expression is normal in the RPE but is expanded into the central neural retina (Fig. 6L).
Fig. 6.
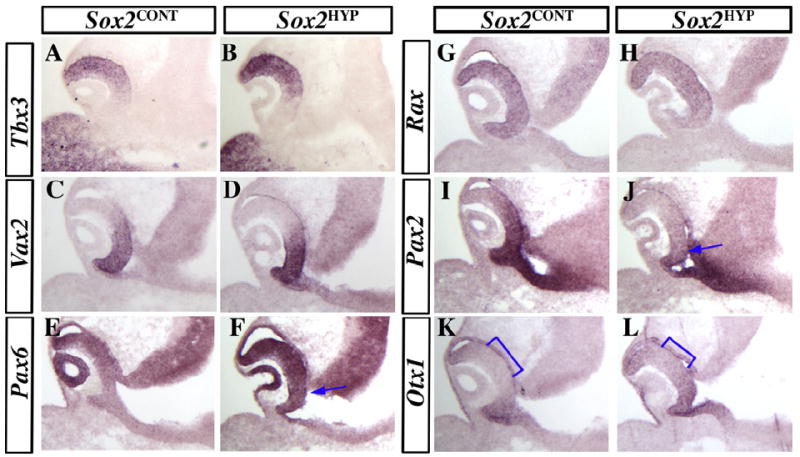
In situ analyses of patterning markers in E10.5 Sox2CONT and Sox2HYP embryos. (A–D) The expression of the dorsal/ventral patterning markers Tbx3 (A and B) and Vax2 (C and D) are unaffected in Sox2HYP embryos. Pax6 (E and F) expression is upregulated in Sox2HYP OCs (arrow F). (G and H) The expression of the retinal progenitor marker Rax is observed in all neural progenitors of the central retina in both Sox2CONT (G) and Sox2HYP (H) OCs. (I and J) Pax2 is expressed throughout the ventral retina in Sox2CONT embryos (I) but is strongly downregulated in the Sox2HYP OC (J, arrow). The expression of the early ciliary margin marker Otx1 is observed in the retinal pigmented epithelium and distal retina in Sox2CONT embryos (K) but is expanded centrally in Sox2HYP OCs (L, brackets). A minimum of n = 3 Sox2CONT and Sox2HYP embryos were analyzed for each in situ probe.
To determine the fate of the distalized, Otx1-expressing cell population observed in E10.5 Sox2HYP OCs, we examined marker expression in the E16.5 neural retina and ciliary margin. In Sox2CONT eyes at E16.5, the neural progenitor marker Notch1 is expressed in the neuroblast layer of the central retina but is excluded from the ciliary margin (Fig. 7A and B) (de la Pompa et al., 1997). In Sox2HYP eyes, however, a greater extent of the distal retina is Notch1-negative (Fig. 7C and D). Instead, this tissue is positive for the ciliary margin marker Otx1 and exhibits an epithelial morphology that is consistent with a ciliary margin identity (Fig. 7E, F vs. G and H).
Fig. 7.
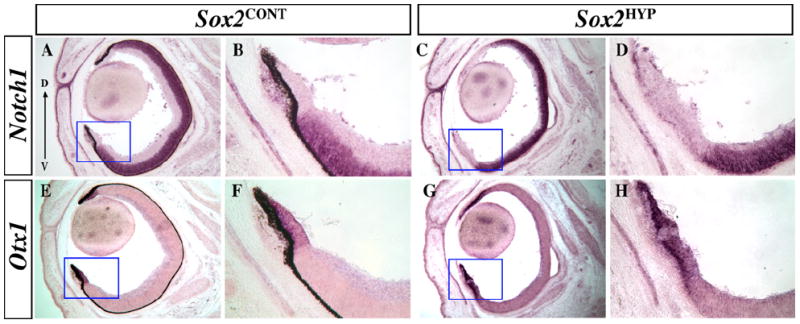
In situ analyses of Notch1 and Otx1 expression in E16.5 Sox2CONT and Sox2HYP eyes. (A–D) Notch1 expression in E16.5 Sox2CONT (A and B) and Sox2HYP (C and D) eyes. Notch1 expression is notably absent from a wide portion of the distal retina in Sox2HYP eyes. (B) and (D) are magnified images of the indicated areas in (A) and (C), respectively. (E–H) Otx1 expression in E16.5 Sox2CONT (I) and Sox2HYP (K) eyes. Otx1 expression is expanded in Sox2HYP eyes. (F) and (H) are magnified images of the indicated areas in (E) and (G), respectively. D: dorsal, V: ventral.
These molecular data suggest that the Sox2HYP retinal primordium is patterned normally at E10.5, but that (i) Pax6 expression is expanded at the expense of Pax2; and (ii) at both E10.5 and E16.5, cells in the distal retina have transitioned from a neural to a ciliary margin identity.
4. Discussion
Here, we present the first analyses of early hypothalamic and ocular development in a mouse model of human SOX2 haploinsufficiency, a genetic disorder characterized by severe diencephalic and ocular defects. These findings indicate that SOX2 is required for the proper morphogenesis and molecular patterning of the posterior prechordal plate, optic stalk and OC. Furthermore, these results (i) define the morphological and temporal onset of the defects observed in Sox2HYP embryos, and (ii) identify Shh signaling as a potential mediator of these abnormalities.
In humans, SOX2 haploinsufficiency causes abnormalities in several brain regions, including the hypothalamus, optic nerve, and eyes (Kelberman et al., 2006). The nature and timing of the onset of these defects are unknown, as are the downstream pathways that are involved in mediating their development. We generated Sox2HYP embryos to directly address these questions, and thereby identified novel functions for SOX2 in the morphogenesis and molecular patterning of the diencephalon and the eye. The generation and detailed molecular analysis of Sox2HYP embryos provide an understanding of the molecular nature and temporal onset of the defects observed in SOX2 haploinsufficient humans.
Hypothalamic hamartomas are benign overgrowths of the ventral hypothalamus and are associated with a variety of developmental problems, cognitive deficiencies, and seizures (Freeman, 2003). In this study, we used SEM to obtain three-dimensional, high-resolution images of the Sox2HYP hypothalamus, and observed that E10.5 Sox2HYP embryos exhibit a broad diencephalic prechordal floor from the mammillary area to the infundibulum. This region encompasses the tuberal region of the hypothalamus, which is associated with HHs in humans (Booth et al., 2004; Schneider et al., 2009). In E12.5 Sox2HYP embryos, this expansion manifests as a protuberance into the lumen of the neural tube. The morphogen Shh acts as a powerful patterning agent along the length of the mouse neural tube, including the hypothalamus (Briscoe et al., 1999; Szabó et al., 2009). However, in contrast to much of the ventral midline, Shh expression is normally excluded from the posterior prechordal floor (Shimamura et al., 1995). Gli3, a member of the Gli family of transcription factors, has been demonstrated in several contexts to antagonize Shh signaling; a loss of Gli3 in mouse leads to an upregulation of Shh in the developing spinal cord (Ruiz i Altaba, 1998), and Shh and Gli3 have been shown to be functionally antagonistic in the mouse forebrain (Rallu et al., 2002). Consistent with these findings, Gli3 is normally expressed in the Shh-negative hypothalamic prechordal floor, and a loss of GLI3 expression in humans has been shown to result in HH. Molecular analysis of the protuberance observed in Sox2HYP embryos indicates that the cells at its apex ectopically express Shh and abnormally downregulate Gli3. These results raise the intriguing possibility that the hypothalamic defects that are observed in SOX2 haploinsufficient humans result from a dysregulation of Shh signaling. Sox2HYP embryos recover normal molecular patterning in the hypothalamus by late gestational stages, consistent with the observation that Gli3-deficient mice, despite exhibiting nearly all of the defects that are associated with human GLI3 mutations, are not reported to exhibit HH (Böse et al., 2002). These data suggest a murine-specific compensatory mechanism in the hypothalamus. Our results also address the question of why the ventral hypothalamus is more severely affected by SOX2 deficiency than are other regions of the diencephalon. We find that that Sox2 expression is nearly eliminated in the prechordal floor of Sox2HYP embryos, which may reflect Sox2 autoregulation and explain the apparent hypersensitivity of this tissue to SOX2 deficiency. Consistent with this interpretation, evidence for SOX2 autoregulation has been observed in mouse and chick neural stems cells (Miyagi et al., 2004: Miyagi et al., 2006).
Ocular defects are by far the most commonly described phenotypes in SOX2 haploinsufficient humans and range from coloboma to bilateral anophthalmia (Ragge et al., 2005; Wang et al., 2008; Schneider et al., 2009). However, there is currently no description of the effects of germline Sox2 deficiency on oculogenesis earlier than E13.5 in mouse, by which point mature retinal morphology has been established. Specifically, it is unknown what role SOX2 plays in the development of the OC or the optic stalk. We report that the ventral OCs of E10.5 Sox2HYP embryos are abnormally separated from the surface ectoderm but do not exhibit decreased cell number or proliferation or increased cell death. These data suggest that a hypoplastic ventral optic stalk leads to abnormal OC morphology. Indeed, we find that Sox2HYP optic stalks are shorter and exhibit increased cell death. These results are the first to define a role for SOX2 in optic stalk morphogenesis and indicate that germline Sox2 deficiency can disrupt OC morphogenesis by causing optic stalk hypoplasia.
The role of SOX2 in the maintenance of neural competency is well established, although it is unknown how germline Sox2 hypomorphism affects early retinal development (i.e., prior to E13.5) (Avilion et al., 2003; Favaro et al., 2009). Recent work from our laboratory demonstrated that conditional ablation of Sox2 from the neural retina at E10.5 using the Pax6α enhancer-driven Cre results in the conversion of the retinal neuroepithelium to non-neural ciliary epithelium (Marquardt et al., 2001; Taranova et al., 2006; Matsushima et al., 2011). These data also demonstrate that SOX2 and PAX6 are mutually antagonistic in the retina, respectively promoting neural and ciliary fates. Distal (i.e., ciliary) identity is indicated by Otx1 expression and high expression levels of Pax6 (Martinez-Morales et al., 2001; Matsushima et al., 2011). We demonstrate that although overall patterning of the neural retina is maintained at E10.5, the expression of Otx1 is centrally expanded, and Pax6 is upregulated in the Sox2HYP retina at this stage. These results indicate that retinal neural progenitors exhibit the first molecular indications of a loss of neuronal competency at E10.5 in the absence of the full complement of SOX2 dosage, defining a temporal requirement for SOX2 in the establishment of neural fate in the mouse OC. Consistent with the results of the E10.5 expression analyses, E16.5 Sox2HYP eyes lack expression of the neural progenitor marker Notch1 in the distal retina. Instead, this region expresses Otx1 and exhibits an epithelial morphology consistent with a ciliary margin identity. The ciliary fate transition that is observed in Sox2HYP retinas is intriguing in that it suggests a mechanism by which microphthalmia develops, i.e., the expansion of the more slowly proliferating, non-neural, ciliary epithelium would be expected to result in a smaller retina; however, this hypothesis will have to be directly addressed.
Coloboma is a ventral eye deformity that frequently afflicts human carriers of SOX2 mutations but which has not been described in a mouse model of SOX2 deficiency (Schneider et al., 2009). PAX2 is also associated with coloboma in humans and mouse, and its expression is strongly downregulated in the ventral Sox2HYP OC (Sanyanusin et al., 1995; Torres et al., 1996), suggesting a mechanism by which SOX2 deficiency results in coloboma.
The question naturally arises of why certain regions of the neuroepithelium are more severely affected by SOX2 deficiency than others. One possible explanation involves the other SOXB1 family members, SOX1 and SOX3, which are also broadly expressed in the developing neural tube, where their functions are thought to be partially redundant (Wood and Episkopou, 1999; Bylund et al., 2003; Graham et al., 2003). However, SOX1 and SOX3 are not expressed in the neural retina following OC invagination, and SOX1 is excluded from the ventral midline (Kamachi et al., 1998; Aubert et al., 2003). Given that these two tissues are the most severely affected in Sox2HYP embryos, these expression profiles fit a model in which (i) a certain degree of SOXB1 dosage is required in all regions of the neural epithelium; and (ii) the neural retina and the ventral midline are the most sensitive tissues to SOX2-deficiency. The phenotypes of SOX3 and SOX1-deficient mice are consistent with this hypothesis (Malas et al., 2003; Rizzoti et al., 2004; Donner et al., 2007).
In summary, this is the first study to describe early diencephalic and optic development in a mouse model of human SOX2 haploinsufficiency. Our results demonstrate that SOX2 is required for proper formation and patterning of the prechordal floor, optic stalk, and the OC. All of the phenotypes of Sox2HYP embryos recapitulate those described in SOX2 haploinsufficient humans. Furthermore, this work provides significant insights into the temporal and molecular requirements for SOX2 in the morphogenesis and patterning of the mammalian CNS.
Supplementary Material
Acknowledgments
We would like to thank Megumi Aita of the UNC In Situ Core Facility, Dr. Cynthia Andoniadou for insights into data interpretation and manuscript preparation, Deborah Dehart for sample processing expertise, Hal Mekeel for electron microscopy expertise, and Connie Wang for lab assistance. This work was supported by the National Institute of Mental Health and the National Eye Institute (Grant numbers 1RO1 MH071822 and 1R01 EY018261-06A1 to L.H.P.) at the National Institutes of Health.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.mod.2012.04.001.
References
- Anchan RM, Drake DP, Haines CF, Gerwe EA, LaMantia AS. Disruption of local retinoid-mediated gene expression accompanies abnormal development in the mammalian olfactory pathway. J Comp Neurol. 1997;379:171–184. [PubMed] [Google Scholar]
- Aubert J, Stavridis MP, Tweedie S, O’Reilly M, Vierlinger K, Li M, Ghazal P, Pratt T, Mason JO, Roy D, Smith A. Screening for mammalian neural genes via fluorescence-activated cell sorter purification of neural precursors from Sox1-gfp knock-in mice. Proc Natl Acad Sci USA. 2003;100(Suppl. 1):11836–11841. doi: 10.1073/pnas.1734197100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakrania P, Robinson DO, Bunyan DJ, Salt A, Martin A, Crolla JA, Wyatt A, Fielder A, Ainsworth J, Moore A, Read S, Uddin J, Laws D, Pascuel-Salcedo D, Ayuso C, Allen L, Collin JR, Ragge NK. SOX2 anophthalmia syndrome: 12 new cases demonstrating broader phenotype and high frequency of large gene deletions. Br J Ophthalmol. 2007;91:1471–1476. doi: 10.1136/bjo.2007.117929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri AM, Lupo G, Bulfone A, Andreazzoli M, Mariani M, Fougerousse F, Consalez GG, Borsani G, Beckmann JS, Barsacchi G, Ballabio A, Banfi S. A homeobox gene, vax2, controls the patterning of the eye dorsoventral axis. Proc Natl Acad Sci USA. 1999;96:10729–10734. doi: 10.1073/pnas.96.19.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumer N, Marquardt T, Stoykova A, Ashery-Padan R, Chowdhury K, Gruss P. Pax6 is required for establishing naso–temporal and dorsal characteristics of the optic vesicle. Development. 2002;129:4535–4545. doi: 10.1242/dev.129.19.4535. [DOI] [PubMed] [Google Scholar]
- Booth TN, Timmons C, Shapiro K, Rollins NK. Pre- and postnatal MR imaging of hypothalamic hamartomas associated with arachnoid cysts. Am J Neuroradiol. 2004;25:1283–1285. [PMC free article] [PubMed] [Google Scholar]
- Böse J, Grotewold L, Rüther U. Pallister–Hall Syndrome phenotype in mice mutant for Gli3. Hum Mol Genet. 2002;11:1129–1135. doi: 10.1093/hmg/11.9.1129. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Sussel L, Serup P, Hartigan-O’Connor D, Jessell TM, Rubenstein JL, Ericson J. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature. 1999;398:622–627. doi: 10.1038/19315. [DOI] [PubMed] [Google Scholar]
- Bylund M, Andersson E, Novitch BG, Muhr J. Vertebrate neurogenesis is counteracted by So1–3 activity. Nat Neurosci. 2003;6:1162–1168. doi: 10.1038/nn1131. [DOI] [PubMed] [Google Scholar]
- Chan AOK, Dong M, Wang L, Chan WY. Somite as a morphological reference for staging and axial levels of developing structures in mouse embryos. Neuroembryol Aging. 2004;3:102–110. [Google Scholar]
- de la Pompa JL, Wakeham A, Correia KM, Samper E, Brown S, Aguilera RJ, Nakano T, Honjo T, Mak TW, Rossant J, Conlon RA. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development. 1997;124:1139–1148. doi: 10.1242/dev.124.6.1139. [DOI] [PubMed] [Google Scholar]
- Domyan ET, Ferretti E, Throckmorton K, Mishina Y, Nicolis SK, Sun X. Signaling through BMP receptors promotes respiratory identity in the foregut via repression of Sox2. Development. 2011;138:971–981. doi: 10.1242/dev.053694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S, Leung KK, Pelling AL, Lee PY, Tang AS, Heng HH, Tsui LC, Tease C, Fisher G, Steel KP, Cheah KS. Circling, deafness, and yellow coat displayed by yellow submarine (Ysb) and light coat and circling (Lcc) mice with mutations on chromosome 3. Genomics. 2002;79:777–784. doi: 10.1006/geno.2002.6783. [DOI] [PubMed] [Google Scholar]
- Donner AL, Ko F, Episkopou V, Maas RL. Pax6 is misexpressed in Sox1 null lens fiber cells. Gene Expr Patterns. 2007;7:606–613. doi: 10.1016/j.modgep.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis P, Fagan BM, Magness ST, Hutton S, Taranova O, Hayashi S, McMahon A, Rao M, Pevny L. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev Neurosci. 2004;26:148–165. doi: 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M, Szumska D, Kockx C, van Ijcken W, Dekkers DH, Demmers J, Rijkers EJ, Bhattacharya S, Philipsen S, Pevny LH, Grosveld FG, Rottier RJ, Lenhard B, Poot RA. Sox2 cooperates with Chd7 to regulate genes that mutated in human syndromes. Nat Genet. 2011;43:607–611. doi: 10.1038/ng.825. [DOI] [PubMed] [Google Scholar]
- Fantes J, Ragge NK, Lynch SA, McGill NI, Collin JR, Howard-Peebles PN, Hayward C, Vivian AJ, Williamson K, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia. Nat Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- Favaro R, Valotta M, Ferri AL, Latorre E, Mariani J, Giachino C, Lancini C, Tosetti V, Ottolenghi S, Taylor V, Nicolis SK. Hippocampal development and neural stem cell maintenance require Sox2-dependent regulation of Shh. Nat Neurosci. 2009;12:1248–1256. doi: 10.1038/nn.2397. [DOI] [PubMed] [Google Scholar]
- Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, Ottolenghi S, Pandolfi PP, Sala M, DeBiasi S, Nicolis SK. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131:3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- Freeman JL. The anatomy and embryology of the hypothalamus in relation to hypothalamic hamartomas. Epileptic Disord. 2003;5:177–186. [PubMed] [Google Scholar]
- Furukawa T, Kozak CA, Cepko CL. Rax, a novel paired-type homeobox gene, shows expression in the anterior neural fold and developing retina. Proc Natl Acad Sci USA. 1997;94:3088–3093. doi: 10.1073/pnas.94.7.3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Calero E, De Puelles E, Puelles L. EphA7 receptor is expressed differentially at chicken prosomeric boundaries. Neuroscience. 2006;141:1887–1897. doi: 10.1016/j.neuroscience.2006.04.074. [DOI] [PubMed] [Google Scholar]
- Gontan C, de Munck A, Vermeij M, Grosveld F, Tibboel D, Rottier R. Sox2 is important for two crucial processes in lung development: branching morphogenesis and epithelial cell differentiation. Dev Biol. 2008;317:296–309. doi: 10.1016/j.ydbio.2008.02.035. [DOI] [PubMed] [Google Scholar]
- Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- Kamachi Y, Uchikawa M, Collignon J, Lovell-Badge R, Kondoh H. Involvement of Sox1, 2 and 3 in the early and subsequent molecular events of lens induction. Development. 1998;125:2521–2532. doi: 10.1242/dev.125.13.2521. [DOI] [PubMed] [Google Scholar]
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116:2442–2455. doi: 10.1172/JCI28658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan AE, Pelling AL, Leung KK, Tang AS, Bell DM, Tease C, Lovell-Badge R, Steel KP, Cheah KS. Sox2 is required for sensory organ development in the mammalian inner ear. Nature. 2005;434:1031–1035. doi: 10.1038/nature03487. [DOI] [PubMed] [Google Scholar]
- Lai MI, Wendy-Yeo WY, Ramasamy R, Nordin N, Rosli R, Veerakumarasivam A, Abdullah S. Advancements in reprogramming strategies for the generation of induced pluripotent stem cells. J Assist Reprod Genet. 2011;28:291–301. doi: 10.1007/s10815-011-9552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litingtung Y, Chiang C. Specification of ventral neuron types is mediated by an antagonistic interaction between Shh and Gli3. Nat Neurosci. 2000;3:979–985. doi: 10.1038/79916. [DOI] [PubMed] [Google Scholar]
- Malas S, Postlethwaite M, Ekonomou A, Whalley B, Nishiguchi S, Wood H, Meldrum B, Constanti A, Episkopou V. Sox1-deficient mice suffer from epilepsy associated with abnormal ventral forebrain development and olfactory cortex hyperexcitability. Neuroscience. 2003;119:421–432. doi: 10.1016/s0306-4522(03)00158-1. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Martinez-Morales JR, Signore M, Acampora D, Simeone A, Bovolenta P. Otx genes are required for tissue specification in the developing eye. Development. 2001;128:2019–2030. doi: 10.1242/dev.128.11.2019. [DOI] [PubMed] [Google Scholar]
- Mathers PH, Grinberg A, Mahon KA, Jamrich M. The Rx homeobox gene is essential for vertebrate eye development. Nature. 1997;387:603–607. doi: 10.1038/42475. [DOI] [PubMed] [Google Scholar]
- Matsushima D, Heavner W, Pevny LH. Combinatorial regulation of optic cup progenitor cell fate by SOX2 and PAX6. Development. 2011;138:443–454. doi: 10.1242/dev.055178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi S, Saito T, Mizutani K, Masuyama N, Gotoh Y, Iwama A, Nakauchi H, Masui S, Niwa H, Nishimoto M, Muramatsu M, Okuda A. The Sox-2 regulatory regions display their activities in two distinct types of multipotent stem cells. Mol Cell Biol. 2004;24:4207–4220. doi: 10.1128/MCB.24.10.4207-4220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi S, Nishimoto M, Saito T, Ninomiya M, Sawamoto K, Okano H, Muramatsu M, Oguro H, Iwama A, Okuda A. The Sox2 regulatory region 2 functions as a neural stem cell-specific enhancer in the telencephalon. J Biol Chem. 2006;281:13374–13381. doi: 10.1074/jbc.M512669200. [DOI] [PubMed] [Google Scholar]
- Miyagi S, Masui S, Niwa H, Saito T, Shimazaki T, Okano H, Nishimoto M, Muramatsu M, Iwama A, Okuda A. Consequence of the loss of Sox2 in the developing brain of the mouse. FEBS Lett. 2008;582:2811–2815. doi: 10.1016/j.febslet.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Mui SH, Kim JW, Lemke G, Bertuzzi S. Vax genes ventralize the embryonic eye. Genes Dev. 2005;19:1249–1259. doi: 10.1101/gad.1276605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nornes HO, Dressler GR, Knapik EW, Deutsch U, Gruss P. Spatially and temporally restricted expression of Pax2 during murine neurogenesis. Development. 1990;109:797–809. doi: 10.1242/dev.109.4.797. [DOI] [PubMed] [Google Scholar]
- Okubo T, Pevny LH, Hogan BL. Sox2 is required for development of taste bud sensory cells. Genes Dev. 2006;20:2654–2659. doi: 10.1101/gad.1457106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontecorvi M, Goding CR, Richardson WD, Kessaris N. Expression of Tbx2 and Tbx3 in the developing hypothalamic-pituitary axis. Gene Expr Patterns. 2008;8:411–417. doi: 10.1016/j.gep.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Puelles L, Rubenstein JL. Forebrain gene expression domains and the evolving prosomeric model trends. Neurosci. 2003;26:469–476. doi: 10.1016/S0166-2236(03)00234-0. [DOI] [PubMed] [Google Scholar]
- Ragge NK, Lorenz B, Schneider A, Bushby K, de Sanctis L, de Sanctis U, Salt A, Collin JR, Vivian AJ, Free SL, Thompson P, Williamson KA, Sisodiya SM, van Heyningen V, Fitzpatrick DR. SOX2 Anophthalmia syndrome. Am J Med Genet A. 2005;135:1–7. doi: 10.1002/ajmg.a.30642. discussion 8. [DOI] [PubMed] [Google Scholar]
- Rallu M, Machold R, Gaiano N, Corbin JG, McMahon AP, Fishell G. Dorsoventral patterning is established in the telencephalon of mutants lacking both Gli3 and Hedgehog signaling. Development. 2002;129:4963–4974. doi: 10.1242/dev.129.21.4963. [DOI] [PubMed] [Google Scholar]
- Rizzoti K, Brunelli S, Carmignac D, Thomas PQ, Robinson IC, Lovell-Badge R. SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat Genet. 2004;36:247–255. doi: 10.1038/ng1309. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development. 1998;125:2203–2212. doi: 10.1242/dev.125.12.2203. [DOI] [PubMed] [Google Scholar]
- Sajedi E, Gaston-Massuet C, Signore M, Andoniadou CL, Kelberman D, Castro S, Etchevers HC, Gerrelli D, Dattani MT, Martinez-Barbera JP. Analysis of mouse models carrying the I26T and R160C substitutions in the transcriptional repressor HESX1 as models for septo–optic dysplasia and hypopituitarism. Dis Model Mech. 2008;1:241–254. doi: 10.1242/dmm.000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Eccles MR. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet. 1995;9:358–364. doi: 10.1038/ng0495-358. [DOI] [PubMed] [Google Scholar]
- Schneider A, Bardakjian T, Reis LM, Tyler RC, Semina EV. Novel SOX2 Mutations and genotype–phenotype correlation in anophthalmia and microphthalmia. Am J Med Genet A. 2009;149A:2706–2715. doi: 10.1002/ajmg.a.33098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz M, Cecconi F, Bernier G, Andrejewski N, Kammandel B, Wagner M, Gruss P. Spatial specification of mammalian eye territories by reciprocal transcriptional repression of Pax2 and Pax6. Development. 2000;127:4325–4334. doi: 10.1242/dev.127.20.4325. [DOI] [PubMed] [Google Scholar]
- Shimamura K, Hartigan DJ, Martinez S, Puelles L, Rubenstein JL. Longitudinal organization of the anterior neural plate and neural tube. Development. 1995;121:3923–3933. doi: 10.1242/dev.121.12.3923. [DOI] [PubMed] [Google Scholar]
- Simeone A, Acampora D, Gulisano M, Stornaiuolo A, Boncinelli E. Nested expression domains of four homeobox genes in developing rostral brain. Nature. 1992;358:687–690. doi: 10.1038/358687a0. [DOI] [PubMed] [Google Scholar]
- Sowden JC, Holt JK, Meins M, Smith HK, Bhattacharya SS. Expression of Drosophila omb-related T-box genes in the developing human and mouse neural retina. Invest Ophthalmol Vis Sci. 2001;42:3095–3102. [PubMed] [Google Scholar]
- Sulik K, Dehart DB, Iangaki T, Carson JL, Vrablic T, Gesteland K, Schoenwolf GC. Morphogenesis of the murine node and notochordal plate. Dev Dyn. 1994;201:260–278. doi: 10.1002/aja.1002010309. [DOI] [PubMed] [Google Scholar]
- Szabó NE, Zhao T, Cankaya M, Theil T, Zhou X, Alvarez-Bolado G. Role of neuroepithelial sonic hedgehog in hypothalamic patterning. J Neurosci. 2009;29:6989–6700. doi: 10.1523/JNEUROSCI.1089-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Taranova OV, Magness ST, Fagan BM, Wu Y, Surzenko N, Hutton SR, Pevny LH. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006;20:1187–1202. doi: 10.1101/gad.1407906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M, Gomez-Pardo E, Gruss P. Pax2 contributes to inner ear patterning and optic nerve trajectory. Development. 1996;122:3381–3391. doi: 10.1242/dev.122.11.3381. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Freeman JL, Shouri MR, Izzillo PA, Rosenfeld JV, Mulley JC, Harvey AS, Berkovic SF. Somatic mutations in GLI3 can cause hypothalamic hamartoma and gelastic seizures. Neurology. 2008;70:653–655. doi: 10.1212/01.wnl.0000284607.12906.c5. [DOI] [PubMed] [Google Scholar]
- Wang P, Liang X, Yi J, Zhang Q. Novel SOX2 mutation associated with ocular coloboma in a chinese family. Arch Ophthalmol. 2008;126:709–713. doi: 10.1001/archopht.126.5.709. [DOI] [PubMed] [Google Scholar]
- Wood HB, Episkopou V. Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech Dev. 1999;86:197–201. doi: 10.1016/s0925-4773(99)00116-1. [DOI] [PubMed] [Google Scholar]
- Zhang J, Woodhead GJ, Swaminathan SK, Noles SR, McQuinn ER, Pisarek AJ, Stocker AM, Mutch CA, Funatsu N, Chenn A. Cortical neural precursors inhibit their own differentiation via N-cadherin maintenance of beta-catenin signaling. Dev Cell. 2010;18:472–479. doi: 10.1016/j.devcel.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.