

This review suggests that the full potential of 6-thioguanine (6-TG) in cancer therapy may not have been reached. The authors contrast 6-TG and the more widely used 6-mercaptopurine; discuss 6-TG metabolism, pharmacokinetics, dosage and schedule; and summarize many of the early studies that have shown infrequent but nevertheless positive results with 6-TG treatment. The combination of 6-TG and a natural compound, methylthioadenosine (MTA), may provide selective treatment of cancers that have lost the gene methylthioadenosine phosphorylase (MTAP). Administration of MTA decreases 6-TG toxicity to normal host tissues, thus permitting use of increased therapeutic doses of 6-TG. Combinations of 6-TG with other agents, such as methotrexate or pralatrexate, may also enhance therapeutic effects in MTAP-deficient tumors.
Keywords: Chemotherapy, Denovo purine synthesis, 6-thioguanine, Folate antagonists
Abstract
Sixty years ago, 6-thioguanine (6-TG) was introduced into the clinic. We suggest its full potential in therapy may not have been reached. In this paper, we contrast 6-TG and the more widely used 6-mercaptopurine; discuss 6-TG metabolism, pharmacokinetics, dosage and schedule; and summarize many of the early studies that have shown infrequent but nevertheless positive results with 6-TG treatment of cancers. We also consider studies that suggest that combinations of 6-TG with other agents may enhance antitumor effects. Although not yet tested in man, 6-TG has recently been proposed to treat a wide variety of cancers with a high frequency of homozygous deletion of the gene for methylthioadenosine phosphorylase (MTAP), often codeleted with the adjacent tumor suppressor CDKN2A (p16). Among the cancers with a high frequency of MTAP deficiency are leukemias, lymphomas, mesothelioma, melanoma, biliary tract cancer, glioblastoma, osteosarcoma, soft tissue sarcoma, neuroendocrine tumors, and lung, pancreatic, and squamous cell carcinomas. The method involves pretreatment with the naturally occurring nucleoside methylthioadenosine (MTA), the substrate for the enzyme MTAP. MTA pretreatment protects normal host tissues, but not MTAP-deficient cancers, from 6-TG toxicity and permits administration of doses of 6-TG that are much higher than can now be safely administered. The combination of MTA/6-TG has produced substantial shrinkage or slowing of growth in two different xenograft human tumor models: lymphoblastic leukemia and metastatic prostate carcinoma with neuroendocrine features. Further development and a clinical trial of the proposed MTA/6-TG treatment of MTAP-deficient cancers seem warranted.
Implications for Practice:
This review suggests that the full potential of 6-thioguanine (6-TG) in cancer therapy may not have been reached. The authors contrast 6-TG and the more widely used 6-mercaptopurine; discuss 6-TG metabolism, pharmacokinetics, dosage and schedule; and summarize many of the early studies that have shown infrequent but nevertheless positive results with 6-TG treatment. The combination of 6-TG and a natural compound, methylthioadenosine (MTA), may provide selective treatment of cancers that have lost the gene methylthioadenosine phosphorylase (MTAP). Administration of MTA decreases 6-TG toxicity to normal host tissues, thus permitting use of increased therapeutic doses of 6-TG. Combinations of 6-TG with other agents, such as methotrexate or pralatrexate, may also enhance therapeutic effects in MTAP-deficient tumors.
Introduction
6-Mercaptopurine (6-MP) and 6-thioguanine (6-TG), introduced into the clinic in the early 1950s, are two well-studied purine analogs that have both anticancer and immune-suppressive activities. Although used earlier for the treatment of acute and chronic myeloid leukemia [1], use of these agents in myeloid malignancies has been supplanted by newer and more effective agents. 6-MP, however, remains a key drug in curative regimens for children with acute lymphoblastic leukemia (ALL). Although studies in children with ALL did not show any difference in outcome between the use of 6-TG and 6-MP [2], a large body of work on the mechanism of action of 6-TG and 6-MP has indicated that these drugs differ in their mechanism of action (vide infra). Based on limited data, 6-TG has not been shown to be an effective drug for the treatment of solid tumors, and except for use in combinations to treat childhood lymphoma, almost no information is available for single-agent activity in various lymphomas. Emphasis has been placed on host differences in absorption and metabolism of these purine analogs that affect antitumor activity and toxicity, whereas little attention has been paid to the role of methylthioadenosine phosphorylase (MTAP), an enzyme known to be absent in many hematologic and solid tumors (Table 1). Although in vitro and in vivo experimental data predict that patients with tumors that lack this enzyme would be more sensitive to thiopurines [3], the relationship between MTAP deficiency and response to 6-TG (and 6-MP) has not been tested in the clinic. In this review, we discuss reasons for evaluating different dose schedules for this drug; MTAP deficiency as a biomarker for response; and a novel strategy using the substrate of this enzyme, methylthioadenosine (MTA), to protect normal cells but not MTAP-deficient tumor cells from the toxic effects of 6-TG.
Table 1.
Incidence of MTAP deficiency in human malignancies

Evidence that 6-TG and 6-MP Differ in Their Mechanisms of Action
6-TG and 6-MP were codeveloped by Gertrude Elion and George Hitchings, who received the Nobel Prize for development of these and other anticancer agents [4]. 6-TG and 6-MP were produced by substitution of oxygen by sulfur at carbon 6 of guanine and hypoxanthine, respectively. The remarkable ability of 6-MP to produce remissions in childhood ALL led to its speedy approval by the U.S. Food and Drug Administration (FDA) in 1953. Azathioprine followed in 1963, as a modification of 6-MP, and is used today as one of the standard immunosuppressants in organ transplant patients and in inflammatory diseases [4]. 6-TG was also studied in ALL, both for induction therapy and maintenance. Although studies in ALL did not show any difference in outcome between 6-TG and its sister antimetabolite 6-MP [2], several reports indicate that these drugs differ in their mechanisms of action [5–7]. All thiopurines are prodrugs and are converted into their active metabolic forms in vivo. 6-TG and 6-MP are enzymatically converted by hypoxanthine-guanine phosphoribosyl transferase (HGPRT) into cytotoxic nucleotides. These are incorporated into DNA as fraudulent bases, an important step that results in damage and arrest of replication by cross-linkage, single-strand breaks, interstrand cross-links, and sister chromatid exchange [8]. The incorporation of thioguanine nucleotides (TGNs) into DNA requires the mismatch repair (MMR) system, which, when defective, confers resistance to thioguanines. In cells defective in MMR, the major contributor of cytotoxicity is purine starvation [9]. Morgan et al. reported that 6-TG causes growth arrest of leukemic cell lines that are resistant to 6-MP by mechanisms not involving HGPRT, suggesting an additional mechanism of 6-TG-induced arrest, probably involving incorporation of 6-TG into queuine-containing transfer RNAs (tRNAs) [7].
Metabolism
6-MP conversion to TGNs is less direct than 6-TG in that it involves two additional enzymes: inosine monophosphate dehydrogenase and guanosine monophosphate synthetase. The first intermediate in the pathway, 6-thioinosine 5′ monophosphate, can also be converted by thiopurine methyltransferase (TPMT) into methyl-thioinosine 5′ monophosphate (MeTIMP), a metabolite that strongly inhibits de novo purine synthesis and likely contributes to the cytotoxic effect of 6-MP in addition to formation of toxic nucleotides [8]. Other enzymes competing for initial metabolism of 6-TG and 6-MP are xanthine oxidase and aldehyde oxidase, both producing metabolites that are believed to have few cytotoxic effects (Fig. 1).
Figure 1.

Simplified schematic diagram of the metabolism of thiopurines.
Abbreviations: 6-MP, 6-mercaptopurine; 6-TG, 6-thioguanine; 8-OHTG, 8-hydroxythioguanine; AO, aldehyde oxidase; GMPS, guanosine monophosphate synthase; HGPRT, hypoxanthine guanine phosphoribosyltransferase; IMPD, inosine monophosphate dehydrogenase; MeMP, methylmercaptopurine; MeTG, methylthioguanine; MeTGMP, methylthioguanosine monophosphate; MeTIMP, methylthioinosine monophosphate; PRPP, 5′-phosphoribosyl-1-pyrophosphate; SAM, S-adenosyl-L-methionine; TGMP, thioguanosine monophosphate; TGNs, thioguanine nucleotides; TIMP, thioinosine 5′-monophosphate; TPMT, thiopurine methyltransferase; TXMP, thioxanthine monophosphate; XO, xanthine oxidase.
Although cell differences in metabolic pathways affect the cytotoxic effects of 6-MP and 6-TG, in most tumor cells, 6-MP is believed to exert its action mainly through the inhibition of purine biosynthesis, whereas 6-TG kills cells mainly through incorporation of 6-thioguanine nucleotides into DNA [8].
Although cell differences in metabolic pathways affect the cytotoxic effects of 6-MP and 6-TG, in most tumor cells, 6-MP is believed to exert its action mainly through the inhibition of purine biosynthesis, whereas 6-TG kills cells mainly through incorporation of 6-thioguanine nucleotides into DNA.
Although more than one of the thiopurine metabolites can inhibit de novo purine synthesis, MeTIMP, the product of the enzyme TPMT, is a much more potent inhibitor of purine biosynthesis than methylthioguanosine monophosphate and accounts in part for the difference in the modes of action of the two thiopurines because it is unique to 6-MP.
Approximately 0.3% of people are homozygous for nonfunctional TPMT alleles. These patients are susceptible to severe and potentially fatal 6-MP-induced hematologic toxicity, presumably because more of this drug is converted to thioguanosine monophosphate and incorporated into DNA [8, 9].
The two drugs differ further in catabolism. 6-TG is first acted on by guanase before oxidation by xanthine oxidase to thiouric acid, whereas 6-MP is converted by xanthine oxidase directly to thiouric acid. In an early interesting observation, the combination of 6-TG and 6-MP, over a limited range of doses, has shown potentiation against tumors in vivo, further implying different mechanisms of action of the two drugs [10]. In addition, as has been mentioned, Morgan et al. reported that 6-TG, specifically, can be incorporated into certain tRNAs, causing growth arrest of leukemic cell lines that are resistant to 6-MP [7].
6-TG pharmacokinetics have been extensively studied in patients with acute leukemia or with solid tumors after oral and intravenous use. Blood levels of 6-TG were higher after intravenous administration than after equal doses of oral drug [11–13]. Blood levels and incorporation into the bone marrow DNA were higher with the higher doses. After five daily doses of 6-TG, the guanine of the DNA in the bone marrow was largely replaced by 6-TG [11, 12]. These drugs are often referred to as “self-limiting” because incorporation into DNA decreases when total DNA synthesis is inhibited by purine starvation. The extent of this effect varies with the tissue type or cell line being studied and the concentration of the drug.
Do Dose and Schedule Matter?
6-MP and 6-TG were first studied in patients with acute leukemia in the early 1950s [1], and daily oral administration was found to produce complete albeit transient remission in some children with ALL. Pharmacokinetic studies showed that both drugs were poorly absorbed, and interpatient variability was large. The metabolic enzyme TPMT also showed considerable interpatient variability [8]. Oral daily 6-MP, following induction therapy, continues to be used in combination with weekly methotrexate (MTX) for maintenance therapy in patients with ALL.
Because a few studies did not show promising results with oral continuous dosing of 6-TG in solid tumors, parenteral dosing was investigated in patients with hematologic malignancies and with solid tumors. Kovach et al. [13] evaluated a schedule of intravenous 6-TG and found that a dose of 55–65 mg/m2 given daily for 5 days caused dose-limiting myelosuppression without other significant toxicity. This dose schedule was then used over the next decade to treat patients with various solid tumors [14–23] (Table 2). Minimal antitumor effects were observed, except possibly for the treatment of breast cancer. Noteworthy is the lack of published studies of this or other dose schedules for the treatment of other solid tumors (e.g., gynecologic malignancies, bladder cancer, sarcomas) or of information on tumor MTAP status. Because studies indicated that 6-TG crosses the blood-brain barrier, combinations containing 6-TG have been used to treat both recurrent glioblastomas [24] and low-grade gliomas in children [25]. Because these combinations also contained a nitrosourea and the studies were phase II, the contribution of 6-TG to the effectiveness of the treatment is not clear.
Table 2.
Single-agent data with 6-TG treatment in solid tumors
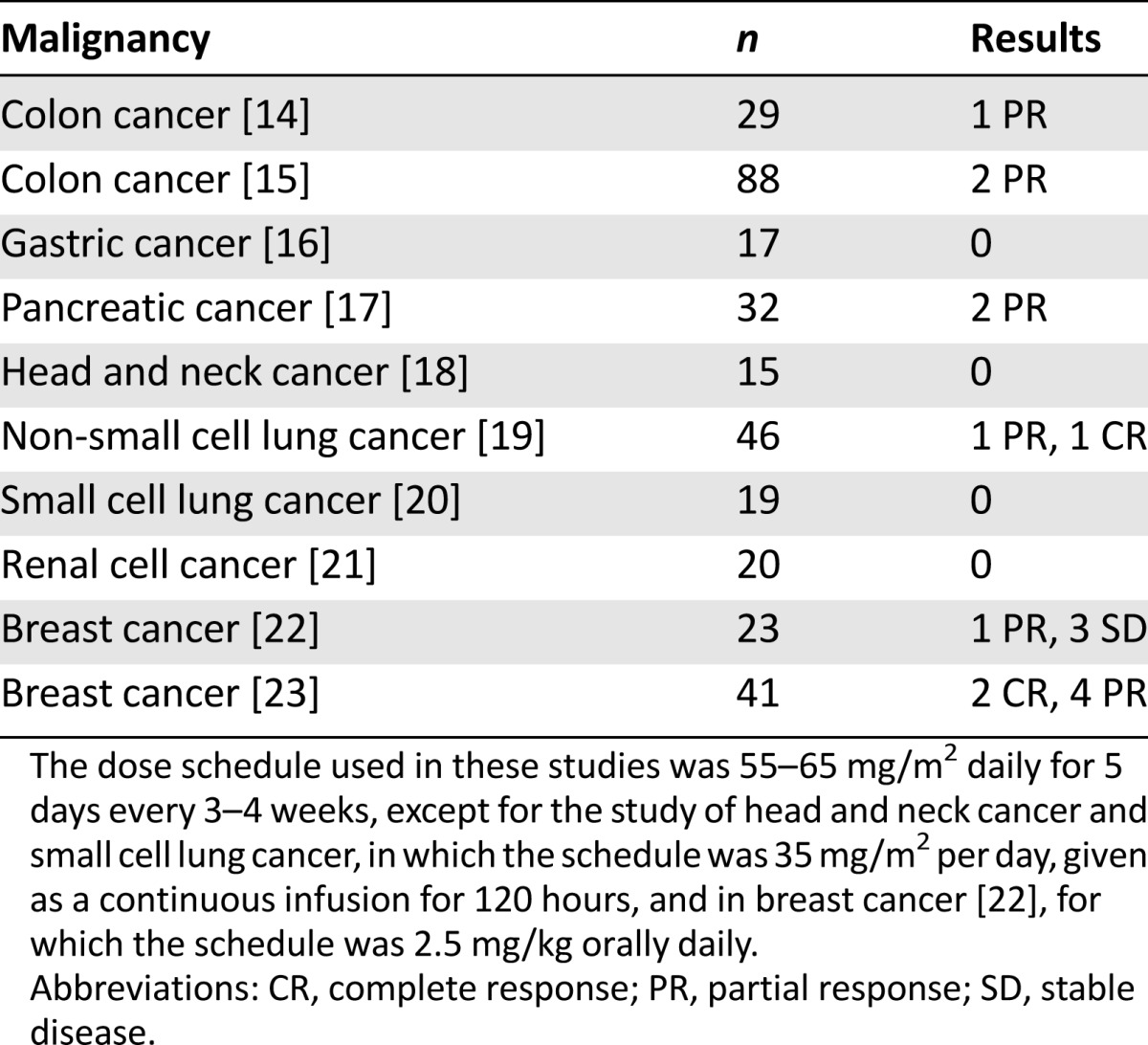
For hematologic malignancies, there were two studies of single-agent 6-TG in multiple myeloma. One was a study of oral continuous 6-TG [26], and a later study used a high-dose intermittent schedule (1,000 mg/m2 every 3 weeks) [27]. In the high-dose study, only 3 of 32 patients were clinically improved, with one partial response [27]. Myelotoxicity was again dose limiting. 6-TG and 6-MP were also used extensively in combination with other agents for the treatment of aggressive lymphomas, especially in children, with a substantial number of cures reported [28]. Interestingly, we could find no reports of single-agent activity in lymphoma. The finding that a high percent of T-cell lymphomas lack MTAP [29] may explain the 30% response rate seen in peripheral T-cell and cutaneous T-cell lymphoma to pralatrexate, which, when polyglutamylated intracellularly, is also an inhibitor of de novo purine synthesis. 6-Thioguanine has not been tested in other lymphomas, in particular, mantle cell lymphoma, in which deficiency of MTAP occurs in 20%–30% of patients and is associated with poor prognosis [30].
The optimal dose schedule of 6-TG used either alone or in combination has not been adequately defined. Such a schedule may depend on whether MTA pretreatment is used to protect normal cells but not MTAP deficient tumor cells from 6-TG toxicity (vide infra). When MTA and 6-TG are both used, based on animal-model studies [31], intermittent high-dose treatment (i.e., every 4–7 days) may be optimal—a dose schedule of MTA and 6-TG not yet tested in humans.
Rationale for MTA Selective Protection of Normal Tissues and Not MTAP-Deficient Tumors
The high frequency of homozygous deletions of MTAP in many tumors (Table 1) suggested an approach to treatment, involving 6-TG, that could be highly selective [28, 29, 32–41]. The treatment takes advantage of the metabolic pathway involving MTA, the natural substrate of the enzyme MTAP.
In this pathway, MTA is derived from S-adenosyl-L-methionine as a byproduct of polyamine biosynthesis (Fig. 2a). MTAP cleaves MTA to adenine and a precursor of methionine. Adenine is then converted by adenine phosphoribosyl transferase (APRT) to AMP, with phosphoribosyl pyrophosphate (PRPP) serving as donor of the phosphoribosyl moiety.
Figure 2.

MTAP. (A): MTAP metabolic pathway in normal cells. (B): MTAP-deficient cells are unable to salvage adenine and rely on de novo biosynthesis of purines.
Abbreviations: 5-FU, 5-fluorouracil; 6-TG, 6-thioguanine; MTA, methylthioadenosine; MTAP, methylthioadenosine phosphorylase; MTR-1-P, methylthioribose-1-phosphate; PRPP, 5′-phosphoribosyl-1-pyrophosphate.
Remarkably, salvage of purines and an amino acid converge at the branch point where MTA sits. Equally remarkable, this is the only known place in metabolism where adenine exists as a free base. The strategy of treatment is indirect. It does not involve drug and target, as in so many chemotherapeutic applications. Indeed, in a general proposal to kill cells with homozygous deletions, Varshavsky coined the phrase, “targeting the absence” [42].
In the proposed strategy for treatment of MTAP-deficient tumors, two drugs are administered sequentially: MTA and then high-dose 6-TG. In normal cells, which ubiquitously contain MTAP, administration of exogenous MTA produces adenine. The phosphoribosylation of adenine by APRT consumes PRPP [43] and prevents the phosphoribosylation of 6-TG, by HGPRT, to its toxic nucleotide. Hence normal cells are protected from the toxic effects of 6-TG. In contrast, in those cancer cells that lack MTAP, MTA cannot be cleaved to form adenine, and PRPP levels remain high. 6-TG is thus converted by phosphoribosylation to its toxic nucleotide, which inhibits or kills the cancer cells (Fig. 2b). Specificity of treatment is ensured by the absolute difference between normal host cells, which contain the enzyme MTAP, and those cancer cells that contain no MTAP [44].
Five studies provide support for this proposed method of treatment. First, MTA, given intraperitoneally, protected mice from lethal, even supralethal, doses of 6-TG [31]. Second, limited in vitro experiments showed that an analog of MTA, 5′-deoxyadenosine—which, like MTA, gives rise to adenine when acted on by MTAP—protected normal human fibroblasts from 6-TG toxicity, whereas MTAP-deficient tumor cells were not protected [44]. Third, in mouse xenograft experiments, the combination of MTA and 6-TG, given i.p. in three injections 4 days apart, caused complete regression of the MTAP-deficient CCRF-CEM human lymphoma/leukemia tumors, although the tumors subsequently regrew [31]. Fourth, in xenograft experiments similar to those just described, MTA and 6-TG produced marked inhibition of growth of a metastatic prostate neuroendocrine tumor [45]. In both of these in vivo experiments, the dose of 6-TG, given alone, was sufficient to kill the mice, yet in the presence of MTA, the mice were protected, with little or no weight loss. Last, in a thorough in vitro study, Kruger and colleagues showed that the combination of MTA and 6-TG produced an extraordinary increase in the therapeutic index of 6-TG. Their results covered several cell types, namely, pairs of cell lines isogenic for presence or absence of MTAP, including mesothelioma, osteosarcoma, and mouse embryo fibroblasts [46].
The method described in this paper merits three comments. First, Batova et al. [47] described a method in which MTA and other substrates of MTAP rescued MTAP-negative human T-cell lines from inhibition by L-alanosine, an inhibitor of IMP to AMP synthesis. L-alanosine neither consumes nor requires PRPP for its activity, whereas 6-TG does. Competition for PRPP by 6-TG and adenine, derived from MTA, is at the heart of the method described. Hence the methods involving MTA/L-alanosine and MTA/ 6-TG are distinctly different. We note that a clinical trial of L-alanosine designed to treat MTAP-negative cancers did not result in any objective responses [48].
Second, it seems possible that administration of high-dose MTA might result in release of adenine from MTAP-positive host tissues sufficient to rescue MTAP-negative tumors cells from 6-TG toxicity. The positive results obtained, however, with CCRF-CEM and prostate cancer xenografts suggest that excess adenine release does not interfere with the MTA/6-TG method.
Third, although MTA/6-MP might be expected to substitute for MTA/6-TG in the method described, it does not, perhaps because 6-MP is not as potent as is 6-TG or for other reasons that are not yet known (unpublished data).The optimal dosage and schedule for in vivo experiments with the MTA/6-TG method have barely been explored. Refinements would undoubtedly produce even more striking results. In addition, the combination of 6-TG with 5-fluorouracil, which, like 6-TG, competes with adenine for the common phosphoribosyl donor PRPP, may result in synergistic efficacy. Such experiments have not yet been carried out but, in principle, are expected to produce promising results.
Will Combinations of 6-TG With Other Inhibitors of De Novo Purine Synthesis Enhance Antitumor Effects?
Based on a study showing enhanced antitumor effects when mice bearing a transplanted sarcoma-180 tumor were treated with the combination of 6-TG and azaserine, a potent inhibitor of the first step in purine biosynthesis [49], two clinical studies tested this combination, one in multiple myeloma [50] and the other in patients with advanced solid tumors [51]. Results were disappointing. It would be worth reinvestigating this combination in tumors known to be MTAP deficient and with MTA protection of normal tissues.
Although 6-TG or 6-MP have been part of many clinically effective combinations (the first curative regimen for some children with ALL was POMP: prednisone, oncovin, 6-MP, methotrexate), the role of 6-TG or 6-MP in the treatment of cancer has diminished as other effective drugs have become available. Presently, 6-MP in combination with MTX is used as treatment following induction for patients with ALL. The explanation for the antileukemia synergy noted was shown to be associated with an increase in conversion of 6-MP to 6-TG nucleotides, associated with an increase in long-chain MTX polyglutamates. MTX polyglutamates, in addition to inhibiting dihydrofolate reductase, also inhibit de novo purine synthesis; therefore, the synergy noted may be due to the combined effects on purine biosynthesis [52]. By inhibiting de novo purine synthesis, MTX increases levels of PRPP, thus increasing the conversion of 6-thiopurines into nucleotides [53].
The explanation for the selective action of the MTX/6-MP combination as treatment during remission for children with ALL cannot be explained entirely by MTAP deficiency because only 30% of childhood ALL patients lack MTAP (Table 1). The dose schedule used currently (daily oral 6-MP, weekly oral or intravenous MTX) has been one of convenience, and although improved dose schedules are possible, the ease of administration and the lack of serious side effects, together with the high rates of cure for childhood ALL, make it unlikely that further changes in this schedule will occur without new information. In other malignancies that are not curable, optimal dose schedules for the combination of MTX (or newer antifolates, including pralatrexate and pemetrexed) with 6-TG are worthy of investigation for the treatment of MTAP-deficient tumors, either alone or together with MTA protection of normal tissues. We have shown, for example, that 6-TG alone or in combination with pralatrexate is much more effective against xenografts of T-cell lymphoma lacking MTAP (CCRF-CEM) than with a T-cell lymphoma (MOLT-4) that expresses MTAP (unpublished data). In addition, combining 6-TG and MTA would allow the use of higher doses of 6-TG, leading to greater tumor kill without increasing toxicity [31].
Prospects for a Clinical Trial
A clinical trial of the MTA/6-TG method for therapy of MTAP-deficient cancers would require additional studies in mouse xenografts of MTAP-deficient human tumors to optimize dosage and schedule. Although MTA has been widely studied in rodent models and, as a natural substance, appears to be without significant toxicity, MTA must be obtained that meets good manufacturing practice standards, and toxicity studies must be repeated to meet FDA requirements. MTA is readily derived from S-adenosyl-L-methionine.
The National Cancer Institute or a major foundation is the likely source of funding. MTA and 6-TG are both known compounds and cannot receive the patent protection required by the pharmaceutical industry. The method described, however, can be protected and hence could attract funding from industry.
For a clinical trial, immunohistochemistry can identify MTAP-deficient tumors and be further validated with a polymerase chain reaction DNA assay [48]. Alternatively, MTAP deficiency may be determined by direct sequencing of tumor samples.
MTAP deficiency is found so frequently in many tumors with a dismal prognosis that a novel method for therapy with a reasonable chance of success surely deserves a clinical trial.
MTAP deficiency is found so frequently in many tumors with a dismal prognosis that a novel method for therapy with a reasonable chance of success surely deserves a clinical trial.
Author Contributions
Conception/Design: Joseph R. Bertino, Martin Lubin
Provision of study material or patients: Joseph R. Bertino, Pashna N. Munshi
Collection and/or assembly of data: Joseph R. Bertino, Pashna N. Munshi
Data analysis and interpretation: Joseph R. Bertino, Martin Lubin
Manuscript writing: Joseph R. Bertino, Pashna N. Munshi, Martin Lubin
Final approval of manuscript: Joseph R. Bertino, Martin Lubin
Disclosures
Martin Lubin: Applied for a patent on the MTA/TG method described (IP). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Burchenal JH, Murphy ML, Ellison RR, et al. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of acute leukemia and allied diseases. Blood. 1953;8:965–999. [PubMed] [Google Scholar]
- 2.Stork LC, Matloub Y, Broxson E, et al. Oral 6-mercaptopurine versus oral 6-thioguanine and veno-occlusive disease in children with standard-risk acute lymphoblastic leukemia: Report of the Children’s Oncology Group CCG-1952 clinical trial. Blood. 2010;115:2740–2748. doi: 10.1182/blood-2009-07-230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coulthard SA, Redfern CP, Vikingsson S, et al. Increased sensitivity to thiopurines in methylthioadenosine phosphorylase-deleted cancers. Mol Cancer Ther. 2011;10:495–504. doi: 10.1158/1535-7163.MCT-10-0798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hitchings GH, Elion GB. The chemistry and biochemistry of purine analogs. Ann N Y Acad Sci. 1954;60:195–199. doi: 10.1111/j.1749-6632.1954.tb40008.x. [DOI] [PubMed] [Google Scholar]
- 5.Maybaum J, Hink LA, Roethel WM, et al. Dissimilar actions of 6-mercaptopurine and 6-thioguanine in Chinese hamster ovary cells. Biochem Pharmacol. 1985;34:3677–3682. doi: 10.1016/0006-2952(85)90230-8. [DOI] [PubMed] [Google Scholar]
- 6.Nelson JA, Carpenter JW, Rose LM, et al. Mechanisms of action of 6-thioguanine, 6-mercaptopurine, and 8-azaguanine. Cancer Res. 1975;35:2872–2878. [PubMed] [Google Scholar]
- 7.Morgan CJ, Chawdry RN, Smith AR, et al. 6-Thioguanine-induced growth arrest in 6-mercaptopurine-resistant human leukemia cells. Cancer Res. 1994;54:5387–5393. [PubMed] [Google Scholar]
- 8.Coulthard S, Hogarth L. The thiopurines: An update. Invest New Drugs. 2005;23:523–532. doi: 10.1007/s10637-005-4020-8. [DOI] [PubMed] [Google Scholar]
- 9.Karran P. Thiopurines, DNA damage, DNA repair and therapy-related cancer. Br Med Bull. 2006;79-80:153–170. doi: 10.1093/bmb/ldl020. [DOI] [PubMed] [Google Scholar]
- 10.Henderson JF, Junga IG. Potentiation of carcinostasis by combinations of thioguanine and 6-mercaptopurine. Biochem Pharmacol. 1960;5:167–168. doi: 10.1016/0006-2952(60)90020-4. [DOI] [PubMed] [Google Scholar]
- 11.Lepage GA, Jones M. Further studies on the mechanism of action of 6-thioguanine. Cancer Res. 1961;21:1590–1594. [PubMed] [Google Scholar]
- 12.LePage GA, Whitecar JP., Jr Pharmacology of 6-thioguanine in man. Cancer Res. 1971;31:1627–1631. [PubMed] [Google Scholar]
- 13.Kovach JS, Rubin J, Creagan ET, et al. Phase I trial of parenteral 6-thioguanine given on 5 consecutive days. Cancer Res. 1986;46:5959–5962. [PubMed] [Google Scholar]
- 14.Britell JC, Moertel CG, Kvols LK, et al. Phase II trial of iv 6-thioguanine in advanced colorectal carcinoma. Cancer Treat Rep. 1981;65:909–910. [PubMed] [Google Scholar]
- 15.Presant CA, Denes AE, Liu C, et al. Prospective randomized reappraisal of 5-fluorouracil in metastatic colorectal carcinoma. A comparative trial with 6-thioguanine. Cancer. 1984;53:2610–2614. doi: 10.1002/1097-0142(19840615)53:12<2610::aid-cncr2820531207>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 16.Ajani JA, Pazdur R, Winn RJ, et al. Phase II study of intravenous 6-thioguanine in patients with advanced gastric carcinoma. Invest New Drugs. 1991;9:257–259. doi: 10.1007/BF00176979. [DOI] [PubMed] [Google Scholar]
- 17.Ajani JA, Pazdur R, Winn RJ, et al. Phase II study of intravenous 6-thioguanine in patients with advanced carcinoma of the pancreas. Invest New Drugs. 1991;9:369–371. doi: 10.1007/BF00183584. [DOI] [PubMed] [Google Scholar]
- 18.Kruter F, Eisenberger M, Sinibaldi V, et al. Phase II trial of 5 day continuous intravenous infusion of 6-thioguanine in patients with recurrent and metastatic squamous cell carcinoma of the head and neck. Invest New Drugs. 1992;10:89–91. doi: 10.1007/BF00873122. [DOI] [PubMed] [Google Scholar]
- 19.Vokes EE, Lyss AP, Herndon JE, II, et al. Intravenous 6-thioguanine or cisplatin, fluorouracil and leucovorin for advanced non-small cell lung cancer: A randomized phase II study of the Cancer and Leukemia Group B. Ann Oncol. 1992;3:727–732. doi: 10.1093/oxfordjournals.annonc.a058328. [DOI] [PubMed] [Google Scholar]
- 20.Williamson SK, Crowley J, Livingston RB, et al. Phase II trial of 6-thioguanine administered as 120 hour continuous infusion for refractory or recurrent small cell lung cancer. A Southwest Oncology Group study. Invest New Drugs. 1993;11:81–83. doi: 10.1007/BF00873917. [DOI] [PubMed] [Google Scholar]
- 21.Witte RS, Elson P, Stewart JA, et al. A phase II trial of melphalan or thioguanine in the treatment of patients with advanced renal cell cancer. Urol Oncol. 1996;2:96–98. doi: 10.1016/s1078-1439(96)00073-7. [DOI] [PubMed] [Google Scholar]
- 22.Pandya KJ, Tormey DC, Davis TE, et al. Phase II trial of 6-thioguanine in metastatic breast cancer. Cancer Treat Rep. 1980;64:191–192. [PubMed] [Google Scholar]
- 23.Ingle JN, Twito DI, Suman VJ, et al. Evaluation of intravenous 6-thioguanine as first-line chemotherapy in women with metastatic breast cancer. Am J Clin Oncol. 1997;20:69–72. doi: 10.1097/00000421-199702000-00015. [DOI] [PubMed] [Google Scholar]
- 24.Kyritsis AP, Yung WK, Jaeckle KA, et al. Combination of 6-thioguanine, procarbazine, lomustine, and hydroxyurea for patients with recurrent malignant gliomas. Neurosurgery. 1996;39:921–926. doi: 10.1097/00006123-199611000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Heath JA, Turner CD, Poussaint TY, et al. Chemotherapy for progressive low-grade gliomas in children older than ten years: The Dana-Farber experience. Pediatr Hematol Oncol. 2003;20:497–504. doi: 10.1080/08880010390232709. [DOI] [PubMed] [Google Scholar]
- 26.Carbone PP, Frei E, III, Owens AH, Jr, et al. 6-Thioguanine (NSC-752)-1 therapy in patients with multiple myeloma. Cancer Chemotherapy Rep. 1964;36:59–62. [PubMed] [Google Scholar]
- 27.Edelstein MB, Crowley JJ, Valeriote FA, et al. A-phase II study of intravenous 6-thioguanine (NSC-752) in multiple myeloma. A Southwest Oncology Group study. Invest New Drugs. 1990;8(suppl 1):S83–S86. doi: 10.1007/BF00171990. [DOI] [PubMed] [Google Scholar]
- 28.Sullivan MP, Boyett J, Pullen J, et al. Pediatric Oncology Group experience with modified LSA2-L2 therapy in 107 children with non-Hodgkin’s lymphoma (Burkitt’s lymphoma excluded) Cancer. 1985;55:323–336. doi: 10.1002/1097-0142(19850115)55:2<323::aid-cncr2820550204>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 29.Bertino JR, Lubin M, Johnson-Farley N, et al. Lack of expression of MTAP in uncommon T-cell lymphomas. Clin Lymphoma Myeloma Leuk. 2012;12:306–309. doi: 10.1016/j.clml.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Marcé S, Balagué O, Colomo L, et al. Lack of methylthioadenosine phosphorylase expression in mantle cell lymphoma is associated with shorter survival: Implications for a potential targeted therapy. Clin Cancer Res. 2006;12:3754–3761. doi: 10.1158/1078-0432.CCR-05-2780. [DOI] [PubMed] [Google Scholar]
- 31.Bertino JR, Waud WR, Parker WB, et al. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: Current strategies. Cancer Biol Ther. 2011;11:627–632. doi: 10.4161/cbt.11.7.14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Illei PB, Rusch VW, Zakowski MF, et al. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesothelioma. Clin Can Res. 2003;9:2108–2113. [PubMed] [Google Scholar]
- 33.Hustinx SR, Hruban RH, Leoni LM, et al. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: A potential new target for therapy. Cancer Biol Ther. 2005;4:83–86. doi: 10.4161/cbt.4.1.1380. [DOI] [PubMed] [Google Scholar]
- 34.Nobori T, Szinai I, Amox D, et al. Methylthioadenosine phosphorylase deficiency in human non-small cell lung cancers. Cancer Res. 1993;53:1098–1101. [PubMed] [Google Scholar]
- 35.García-Castellano JM, Villanueva A, Healey JH, et al. Methylthioadenosine phosphorylase gene deletions are common in osteosarcoma. Clin Cancer Res. 2002;8:782–787. [PubMed] [Google Scholar]
- 36.Nobori T, Karras JG, Della Ragione F, et al. Absence of methylthioadenosine phosphorylase in human gliomas. Cancer Res. 1991;51:3193–3197. [PubMed] [Google Scholar]
- 37.Huang HY, Li SH, Yu SC, et al. Homozygous deletion of MTAP gene as a poor prognosticator in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:6963–6972. doi: 10.1158/1078-0432.CCR-09-1511. [DOI] [PubMed] [Google Scholar]
- 38.Batova A, Diccianni MB, Nobori T, et al. Frequent deletion in the methylthioadenosine phosphorylase gene in T-cell acute lymphoblastic leukemia: Strategies for enzyme-targeted therapy. Blood. 1996;88:3083–3090. [PubMed] [Google Scholar]
- 39.Mirebeau D, Acquaviva C, Suciu S, et al. The prognostic significance of CDKN2A, CDKN2B and MTAP inactivation in B-lineage acute lymphoblastic leukemia of childhood. Results of the EORTC studies 58881 and 58951. Haematologica. 2006;91:881–885. [PubMed] [Google Scholar]
- 40.Traweek ST, Riscoe MK, Ferro AJ, et al. Methylthioadenosine phosphorylase deficiency in acute leukemia: Pathologic, cytogenetic, and clinical features. Blood. 1988;71:1568–1573. [PubMed] [Google Scholar]
- 41.Hori Y, Hori H, Yamada Y, et al. The methylthioadenosine phosphorylase gene is frequently co-deleted with the p16INK4a gene in acute type adult T-cell leukemia. Int J Cancer. 1998;75:51–56. doi: 10.1002/(sici)1097-0215(19980105)75:1<51::aid-ijc9>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 42.Varshavsky A. Targeting the absence: Homozygous DNA deletions as immutable signposts for cancer therapy. Proc Natl Acad Sci USA. 2007;104:14935–14940. doi: 10.1073/pnas.0706546104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Savarese TM, Chu SH, Chu MY, et al. 5′-Deoxy-5′-methylthioadenosine phosphorylase—III. Role of the enzyme in the metabolism and action of 5′-halogenated adenosine analogs. Biochem Pharmacol. 1985;34:361–367. doi: 10.1016/0006-2952(85)90044-9. [DOI] [PubMed] [Google Scholar]
- 44.Lubin M, Lubin A. Selective killing of tumors deficient in methylthioadenosine phosphorylase: A novel strategy. PLoS One. 2009;4:e5735. doi: 10.1371/journal.pone.0005735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins CC, Volik SV, Lapuk AV, et al. Next generation sequencing of prostate cancer from a patient identifies a deficiency of methylthioadenosine phosphorylase, an exploitable tumor target. Mol Cancer Ther. 2012;11:775–786. doi: 10.1158/1535-7163.MCT-11-0826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang B, Testa JR, Kruger WD. Increasing the therapeutic index of 5-fluorouracil and 6-thioguanine by targeting loss of MTAP in tumor cells. Cancer Biol Ther. 2012;13:1082–1090. doi: 10.4161/cbt.21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Batova A, Cottam H, Yu J, et al. EFA (9-beta-D-erythrofuranosyladenine) is an effective salvage agent for methylthioadenosine phosphorylase-selective therapy of T-cell acute lymphoblastic leukemia with L-alanosine. Blood. 2006;107:898–903. doi: 10.1182/blood-2005-06-2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kindler HL, Burris HA, III, Sandler AB, et al. A phase II multicenter study of L-alanosine, a potent inhibitor of adenine biosynthesis, in patients with MTAP-deficient cancer. Invest New Drugs. 2009;27:75–81. doi: 10.1007/s10637-008-9160-1. [DOI] [PubMed] [Google Scholar]
- 49.Sartorelli AC, Lepage GA. Inhibition of ascites cell growth by combinations of 6-thioguanine and azaserine. Cancer Res. 1958;18:938–942. [PubMed] [Google Scholar]
- 50.Hayes DM, Costa J, Moon JH, et al. Combination therapy with thioguanine (NSC-752) and azaserine (NSC-742) for multiple myeloma. Cancer Chemother Rep. 1967;51:235–238. [PubMed] [Google Scholar]
- 51.Schroeder JM, Ansfield AR, Curreri AR, et al. Toxicity and clinical trial of azaserine and 6-thioguanine in advanced solid malignant neoplasms. Br J Cancer. 1964;13:449–458. doi: 10.1038/bjc.1964.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allegra CJ, Drake JC, Jolivet J, et al. Inhibition of phosphoribosylaminoimidazolecarboxamide transformylase by methotrexate and dihydrofolic acid polyglutamates. Proc Natl Acad Sci USA. 1985;82:4881–4885. doi: 10.1073/pnas.82.15.4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tan CT, Wollner N, Trippett T, et al. Pharmacologic-guided trial of sequential methotrexate and thioguanine in children with advanced malignancies. J Clin Oncol. 1994;12:1955–1962. doi: 10.1200/JCO.1994.12.9.1955. [DOI] [PubMed] [Google Scholar]