

Abstract
Intestinal epithelial cells that line the mucosal surface of the gastrointestinal tract are positioned between an anaerobic lumen and a highly metabolic lamina propria. As a result of this unique anatomy, intestinal epithelial cells function within a steep physiologic oxygen gradient relative to other cell types. Furthermore, during active inflammatory disease such as IBD, metabolic shifts toward hypoxia are severe. Studies in vitro and in vivo have shown that the activation of hypoxia-inducible factor (HIF) serves as an alarm signal for the resolution of inflammation in various murine disease models. Amelioration of disease occurs, at least in part, through transcriptional up-regulation of non-classical epithelial barrier genes. There is much recent interest in harnessing hypoxia-inducible pathways, including targeting the hypoxia-inducible factor (HIF) and the proyl-hydroxylase enzyme (which stabilizes HIF), for therapy of IBD. Here, we review the signaling pathways involved and define how hypoxia may serve as an endogenous alarm signal for mucosal inflammatory disease. We also discuss the upside and potential downsides of targeting these pathways to treat patients with IBD.
Introduction
The intestinal epithelium lines the entire gastrointestinal tract, covering a surface area of approximately 300 m2 in the adult human and forming an essential barrier to the outside world. This intestinal epithelial barrier consists of a monolayer of cells with intercellular tight junctions, a complex three dimensional structure and a thick mucous gel layer, and provides a dynamic and regulated barrier to the flux of the luminal contents to the lamina propria1,2. As well as having an important role in nutrient uptake and development of oral tolerance to nonpathogenic antigens, the intestinal epithelial barrier drives the daily absorption of at least 9 l of fluid. Both the absorptive and barrier functions of the intestinal epithelium are regulated by the availability of O23.
It is widely understood that the gastrointestinal tract functions in a state of ‘low grade inflammation’. Such a state results from the constant processing of luminal antigenic material during the development of oral tolerance and the priming of the mucosal immune system for rapid and effective responses to antigens or microbes that may penetrate the barrier.
The anatomy and function of the intestine provide a fascinating oxygenation profile as, even under physiologic conditions, the intestinal mucosa experiences profound fluctuations in blood flow and metabolism. For example, less than 5% of total blood volume is present in the gut during fasting, but, following ingestion of a meal, approximately 30% of total blood volume is present in the gastrointestinal tract. Such changes in blood flow also result in marked shifts in local pO2. Notably, there is a steep oxygen gradient from the anaerobic lumen of the intestine across the epithelium into the highly vascularized sub-epithelium. From this perspective, it is perhaps not surprising that the epithelium has evolved a number of features to cope with this metabolic setting. In fact, studies comparing functional responses between epithelial cells from different tissues have revealed that intestinal epithelial cells seem to be uniquely resistant to hypoxia and that an extremely low level of oxygenation within the normal intestinal epithelial barrier (so-called “physiologic hypoxia”) may be a regulatory adaptation mechanism to the steep oxygen gradient4.
Loss of epithelial barrier function with the resultant unrestricted flux of luminal antigens to the mucosal immune system underlies the pathology of IBD, and results in hypoxia within the chronically inflamed mucosa, particularly within the epithelial cell layer. This loss of epithelial barrier, together with hypoxia and inflammation underlie the pathology of IBD. Ongoing studies suggest that hypoxia-regulated pathways are highly associated with IBD and contribute particularly to the resolution of ongoing inflammation. In this review we discuss the signaling pathways involved in these processes and the possibility of developing therapies to modify the hypoxic state to treat IBD.]
Hypoxia and the immune response
Sites of mucosal inflammation are characterized by profound changes in tissue metabolism, including local depletion of nutrients, imbalances in tissue oxygen supply and demand, and the generation of large quantities of reactive nitrogen and oxygen intermediates3. In part, these changes can be attributed to recruitment of inflammatory cells, including myeloid cells such as neutrophils (polymorphonuclear cells; PMNs) and monocytes (Figure 1). PMNs are recruited by chemical signals, such as the chemokine interleukin 8, complement factor C5a, N-formylated peptides, platelet-activating factor and leukotriene B4, which are generated at sites of active inflammation as part of the innate host immune response to microorganisms. In transit, these cells expend tremendous amounts of energy. For instance, large amounts of ATP are needed for the high actin turnover required for cell migration5. Once at the sites of inflammation, the nutrient, energy and oxygen demands of the PMNs increase to accomplish the processes of phagocytosis and microbial killing. It has long been known that PMNs are primarily glycolytic cells, with few mitochondria and little energy produced from respiration6. A predominantly glycolytic metabolism ensures that PMN can function at the low oxygen concentrations (even anoxia) associated with inflammatory lesions.
Figure 1. Potential sources of hypoxia in mucosal inflammation.
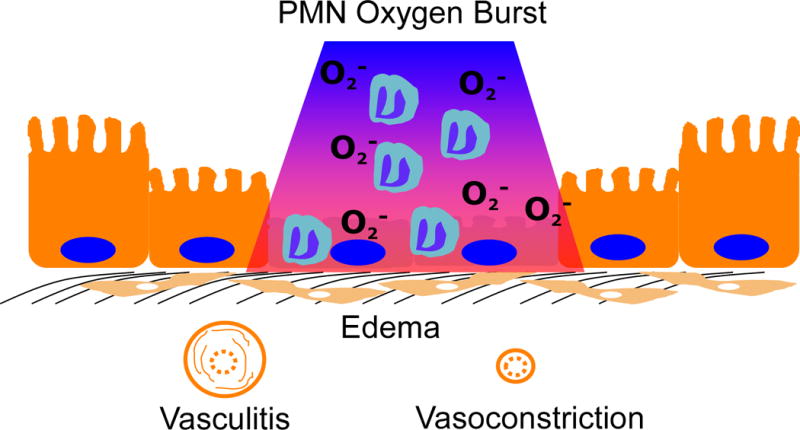
During episodes of inflammation, a number of factors influence the supply and demand of oxygen to the tissues, as well as influencing oxygen delivery. Noted here are edema, vasculitis and vasoconstriction, which separate epithelial cells from the blood supply and limit oxygen availability. In addition, local depletion of oxygen through processes such as the polymorphonuclear cells oxygen burst can usee large quantities of oxygen and result in localized hypoxia, where red depicts normoxia and blue represents hypoxia.
Once at the inflammatory site, PMNs recognize and engulf pathogens and activate the release of antibacterial peptides, proteases and reactive oxygen species (ROS; superoxide anion, hydrogen peroxide, hydroxyl radical and hypochlorous acid) into the vacuole, which together kill the invading microbes (Figure 1)7. ROS are produced by phagocytes in a powerful oxidative burst, driven by a rapid increase in oxygen uptake and glucose consumption, which in turn triggers further generation of ROS. When activated, it is estimated that PMN can consume up to 10 times more O2 than any other cell in the body. Notably, the PMN oxidative burst is not hindered by even relatively low O2 (as low as 4.5% O2)8, which is important, since it means that ROS can be generated in the relatively low O2 environments of inflamed intestinal mucosa3.
In contrast to the innate immune response associated with PMNs, adaptive immune responses to inflammation in the intestine are characterized by a combination recruitment and high rates of local T and B cell proliferation. As a result, adaptive immune responses have markedly different demands for glucose, oxygen and ATP.9,10. In the past 10 years we have begun to understand the nature of interactions between microenvironmental metabolic changes and the generation of recruitment signals and molecular mechanisms of leukocyte migration into these areas. The metabolic changes that occur as a result of the recruitment and activation of leukocytes during inflammation provide information about the potential sources of hypoxia at the intestinal epithelial barrier (Figure 1).
IBD is an interesting disease for studying the metabolic changes associated with inflammation, particularly the development of hypoxia within inflammatory lesions3. Some microvascular abnormalities that occur in patients with IBD have been associated with abnormal blood flow to the intestine, including increased production of tissue vasoconstrictor molecules and a reduced generation of nitric oxide by endothelial cels11, as well as vascular endothelial growth factor-dependent angiogenesis12. In addition, studies of active inflammation in mouse models of IBD have shown the intestinal epithelial cell to be a primary target for hypoxia13.
Hypoxia in murine models of IBD was revealed using 2-nitroimidazole dyes, a class of compounds known to undergo intracellular metabolism depending tissue oxygenation (Figure 2)14. Tissue staining with these nitroimidazole dyes revealed two profound observations. First, in the normal intestinal epithelial cells, especially in the colon, physiologic hypoxia predominates (Figure 2). Whether such low oxygen levels function to regulate basal gene expression in intestinal epithelial cells is not known. Second, the inflammatory lesions seen in these mouse models are profoundly hypoxic or even anoxic, similar to that seen in some large tumors, and penetrate deep into the mucosal tissue. It is likely that there are multiple contributing factors, such as vasculitis, vasoconstriction, edema, increased O2 consumption; Figure 1), which predispose the inflamed intestinal epithelia to decreased oxygen delivery and hypoxia13. While these 2-niroimidazole compounds have not been used to image inflammatory lesions, they have shown significant clinical utility in tumor imaging and in the identification of stroke regions within the brain of patients15. As opposed to other imaging techniques, these compounds have the advantages that they image only viable tissue and are not active within apoptoptic or necrotice regions16. Likewise, studies are underway to use these compounds as adjunct radiosensitizers for enhancing chemotherapy targeting17.
Figure 2. Detection of hypoxia in the mucosa.
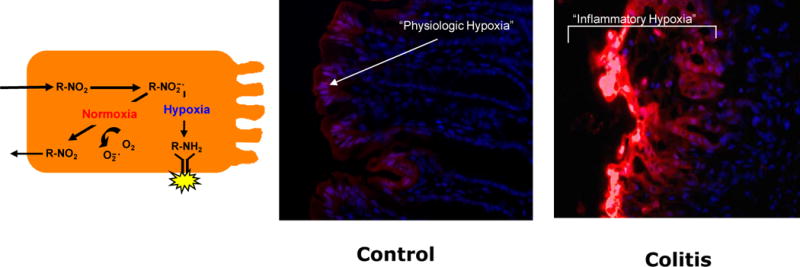
a) The in vivo evidence for hypoxia associated with inflammation (so called “inflammatory hypoxia”) are provided using nitroimidazole-based dye retention in vitro and in mice. These molecules (R-NO2) are taken into cells passively and reduced to highly reactive nitrogen intermediates (R-NO2-[dot]). In the absence of adequate oxygen to regenerate the native compound, these intermediates react with cellular proteins to form adducts (R-NH2), which can be visualized using labeled antibodies. b) In the colons of mice with no inflammation (control) small amounts of nitroimidazole adduct is detected along the luminal aspect of the colon (red), suggesting a degree of physiological hypoxia in the normal colon. c) During episodes of inflammation, such as seen here in a mouse model of colitis, intense and deep tissue hypoxia is prevalent, particularly in areas overlying lesions.
Hypoxia-inducible factor
The same series of studies which identified inflammation-related hypoxia also showed that expression of HIF-1 is induced in the inflammatory lesions13. Many cell types, including intestinal epithelial cells18, express both HIF-1 and HIF-2 and murine knockout studies suggest that these proteins have non-redundant roles19. Some have suggested that distinct transcriptional responses mediated by HIF-1 and HIF-2 may be integrated in ways that support particular adaptations to hypoxia. For example, the transcriptional responses that coordinate the glycolytic pathways include more than 11 target genes and seem to be more selective for the HIF-1α than for the HIF-2α isoform19. Studies addressing the selectivity of the two isoforms for erythropoietin induction have suggested a more important role for the HIF-2α isoform19. Currently, this specificity is not well understood. Some have suggested that binding of HIF-1α or HIF-2α to other transcription factors at the site of DNA binding could determine such specificity19, but this is not conclusive.
Several studies have shown that HIF-1 triggers the transcription of many genes that enable intestinal epithelial cells to act as an effective barrier4,20–22. Originally shown by microarray analysis of hypoxic intestinal epithelial cells21, these studies have been validated in animal models of intestinal inflammation13,23–27 and in human intestinal inflammation tissues28–30. The functional proteins encoded by hypoxia-induced, HIF-dependent mRNAs localize primarily to the most luminal aspect of polarized epithelia. Molecular studies of these hypoxia-elicited pathway(s) have shown a dependence on HIF-mediated transcriptional responses. Notably, epithelial barrier protective pathways driven by HIF tend not to be the classical regulators of barrier function, such as the tight junction proteins occludin or claudin. Rather, the HIF-dependent pathways are more to do with overall tissue integrity, ranging from increased mucin production,31 including molecules that modify mucins, such as, intestinal trefoil factor4, to xenobiotic clearance by P-glycoprotein,20 to nucleotide metabolism (by ecto-5′-nucleotidase and CD73)21,22 and nucleotide signaling through the adenosine A2B receptor22.
As an extension of the original studies identifying HIF induction within the intestinal mucosa, Karhausen, et al. generated transgenic miceexpressing either mutant Hif1-α (causing constitutive repression of Hif1-α) or mutant von Hippel-Lindau (causing constitutive overexpression of HIF) targeted to the intestinal epithelial cells13. Loss of epithelial HIF-1α resulted in a more severe colitic phenotype than wild-type animals, with increased weight loss, decreased colon length and increased epithelial permeability, whereas constitutively active intestinal epithelial HIF was protective for these parameters. These findings may be somewhat model-dependent, since epithelial HIF-based signaling has also been shown to promote inflammation in another study27. However, the findings confirmed that intestinal epithelial cells can adapt to hypoxia and that HIF may contribute to this.
Non-epithelial cell types within the gastrointestinal mucosa have also been studied for HIF expression and response to hypoxia. Activated T cells show increased expression of HIF-1α which prevents them from undergoing activation-induced cell death in hypoxic settings. T-cell survival in hypoxia is, at least in part, mediated by the vasoactive peptide adrenomedullin32.
Other studies using chimeric mice bearing HIF-1α-deficient T and B cells have revealed lineage-specific defects that result in increased autoimmunity, including autoantibodies, increased rheumatoid factor and kidney damage10. HIF function has also been studied in some detail in myeloid cells. Cre-LoxP-based elimination of HIF-1α in cells of the myeloid lineage (lysozyme M promoter) have revealed multiple features which importantly implicate metabolic control of myeloid function33. In particular, these studies have shown that PMN and macrophage bacterial killing capacities are severely limited in the absence of HIF-1α, as HIF-1α is central to production of antimicrobial peptides and granule proteases. These findings are explained, at least in part, by the inability of myeloid cells to mount appropriate metabolic responses to diminished O2 characteristic of infectious sites33. Finally, compelling evidence have revealed that HIF-1α transcriptionally controls the critical integrin important in all myeloid cell adhesion and transmigration, namely the β2 integrin (CD18)34,35. Such findings are important for our current understanding of the role of functional PMN in IBD. A recent study, for example, used PMN depletion techniques to document a central role for PMN in the resolution of inflammation in several murine IBD models36.
Prolyl-hydroxylases and HIF Expression
In the past 10 years, the molecular mechanisms of HIF stabilization have been clarified. Four HIF-hydroxylases termed PHD1-3 and Functional Inhibitor of HIF (FIH) have been demonstrated to be important in the hypoxic regulation of the HIF pathway37. As depicted in Figure 3, each of these hydroxylases are encoded by different genes and their gene product enzymes demonstrate tissue specific expression patterns37. All three PHD’s and FIH are found in the intestinal epithelium23,26,38. Significantly different phenotypes in mice genetically lacking individual isoforms of the hydroxylases exist. For instance, PHD1−/− mice demonstrate a reprogrammed basal metabolic profile in normal tissue which decreases exercise performance but these animals are protected against acute liver and muscle ischemia39,40. PHD2 homozygous knockout is embryonic lethal due to developmental angiogenesis dysfunction41,42. PHD2 heterozygous knockout animals demonstrate enhanced tumor angiogenesis but decreased metastasis41. PHD3 homozygous knockout mice demonstrate reduced neuronal apoptosis, abnormal sympathoadrenal development and reduced blood pressure43. These diverse phenotypes strongly suggests distinct isoform-specific functions in vivo.
Figure 3. Structural features of hypoxia-inducible factor (HIF) and mechanism of HIF stabilization.
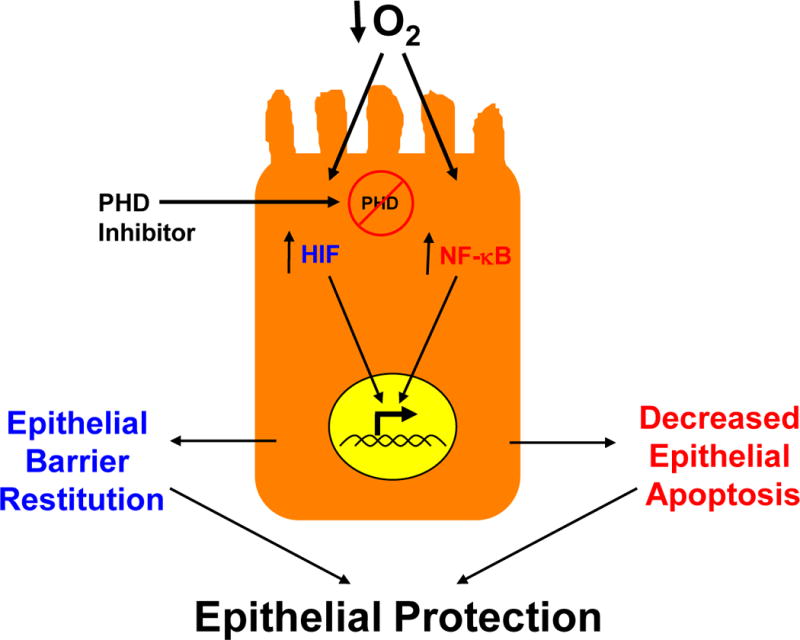
A) The alpha subunit of HIF (HIF-1, HIF-2 and HIF-3) consist of a C-terminal basic helix-loop-helix domain (bHLH, gray), a Per-Arnt-SIM (PAS, purple), a C- and N-terminal oxygen-dependent degradation domain (ODD) region (orange) and a N-terminal transactivation domain (TAD, blue). Also shown are the proline (Pro) and asparagines (Asn) sites for hydroxylation by proly-hydroxylases (PHD) 1–3 and functional inhibitor of HIF (FIH), respectively. B) Depiction of HIF hydroxylation by the combination of alpha-ketoglutarate (αKG), molecular oxygen (O2) and the PHD enzyme in normoxia. When O2 becomes limiting (hypoxia), the alpha subunit is stabilized, binds to the HIF-1 beta subunit within the nucleus where it becomes transcriptionally active upon binding to the hypoxia-response element (HRE) consensus sequence on DNA.
In the presence of 2-oxoglutarate, Fe2+ and molecular oxygen, these enzymes hydroxylate HIF alpha subunits leading to directed ubiquitination and subsequent degradation. Hypoxia or pharmacologic agents (such as DMOG) inhibit HIF-hydroxylases leading to derepression of HIF. The impact of HIF hydroxylase inhibitors on epithelial cell gene expression is not restricted to regulation via HIF. For example, NF-κB has recently been reported to be regulated by HIF hydroxylases in a number of studies37,44. The transcriptional targets of HIF hydroxylases can impact upon epithelial barrier function in a number of ways. For example, HIF regulates the expression of a family of barrier protective factors including intestinal trefoil factor4, the mucins31 and actin cytoskeletal crosslinkers45. Likewise, NF-κB is thought to be largely protective in the intestinal epithelium via the inhibition of enterocyte apoptosis leading to enhanced intestinal barrier function23.
Convergence of inflammation and hypoxia
The oxygen-dependent regulatory role of PHDs may not be restricted to the HIF pathway and may provide a means to better understand how hypoxia contributes to other aspects of inflammation. For instance, NF-κB may interact with with the HIF pathway and is activated during inflammation. NF-κB consists of either homodimers or heterodimers and which, on activation, translocates to the nucleus and binds with the transcriptional coactivator CBP/p300 to begin transcription or repression of various genes. The activity of NF-κB is regulated by the inhibitory IκB proteins46. The best-studied complex is IκBα bound to the NF-κB p50–p65 dimer46. The interaction with IκBα inhibits NF-κB from binding to DNA and maintains the complex in the cytoplasm. On activation by various extracellular signals, IκB kinase (IKK) is activated, resulting in phosphorylation47 and polyubiquitylation of IκBα48. The polyubiquitinated IκBα is then selectively degraded by the S26 proteasome. Once dissociated from IκBα, NF-κB rapidly enters the nucleus and activates gene expression.
It is now appreciated that NF-κB-dependent pathways are also be regulated by PHDs and that the usefulness PHD inhibitors for murine colitis also target the NF-kB pathway. Indeed, hypoxia has been shown to activate NF-κB and this seems, at least in part to be, to be dependent on PHD-mediated hydroxylation38,49 of IKKβ.38 In normoxia, IKKβ activity is held in check through LXXLAP-dependent hydroxylation by PHD1 and PHD238. It is notable that conditional deletion of the NF-κB pathway in intestinal epithelial cells in mice leads to an increased susceptibility to colitis50, similar to that of the mice expressing homozygous mutant HIF1α13. This implicates epithelial NF-κB in a prominently protective role in colitis, probably through the expression of anti-apoptotic genes in intestinal epithelial cells and through enhanced epithelial barrier function. Some studies have suggested that both the HIF and NF-κB pathways may also be influenced by mediators found within inflammatory sites, including microbial products, cytokines and even intact bacteria33. NF-κB is a classic transcriptional regulator activated by a spectrum of agonists, the activation of which drives a complex series of receptor-mediated signaling pathways. Recent studies indicate that transcription of HIF-1α is activated by NF-κB-mediated signaling51. Inflammation-associated upregulation of HIF-1α mRNA occurs in an NF-κB-dependent manner51. It also seems that increased NF-κB activity in hypoxia can be regulated by HIF-144, thus a cross-regulatory loop may exist between these two pathways and may involve other transcriptional regulators that bear non-redundant PHD sensitivity, including activating transcription factor-4 and Notch52,53, both critical regulators of cell fate (see Figure 4). Given that intestinal epithelial cells are in an environment with constant exposure to potentially inflammatory stimuli, the cross regulation of HIF and NF-κB may have profound implications for intestinal epithelial cell function and survival under both homeostatic and disease conditions.
Figure 4. Activation of hypoxia-inducible factor (HIF) and NF-κB in IEC’s during inflammation.
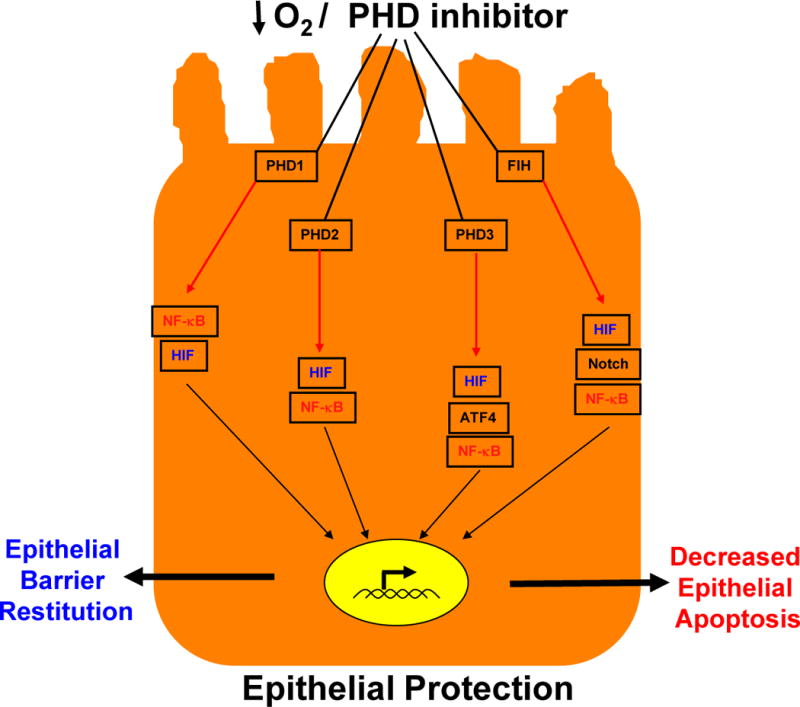
Episodes of inflammation and hypoxia activate a number of pathways in intestinal epithelial cells. The transcriptional activation of both HIF (blue) and NF-κB (red) is stabilized by proly-hydoxylase enzymes (PHDs) and functional inhibitor of HIF (FIH). Activation of these pathways has been shown to promote epithelial barrier function and to decrease epithelial apoptosis, with an overall epithelial protective phenotype. As noted, the various PHD enzymes cross-regulate both HIF and NF-κB in a non-redundant manner, including the regulation of activating transcription factor-4 (ATF-4) and Notch (see text). Green arrows indicate hydroxylation reactions and black arrows indicate transcriptional activation. Activation of HIF through these hydroxylation reactions (blue) promotes barrier function while activation of NF-κB (red) diminishes epithelial apoptosis.
Therapies for IBD
The finding that hypoxia serves as an alarm signal to promote the resolution of inflammation in IBD holds specific promise for the development of therapies targeting hypoxia pathways. While pre-clinical studies in murine models have focused primarily on the use of PHD inhibitors in acute models of colitis, the ultimate goal should be the development of viable therapies for active and chronic intestinal inflammation.
The identification of HIF-selective PHDs has provided unique opportunities for the development of PHD-based therapies54,55. While there is wide interest in developing HIF-1 inhibitors as potential cancer therapies, opportunities also exist to selectively stabilize HIF in an attempt to support mucosal barrier function and promote inflammatory resolution in IBD. One approach that we have taken is to stabilize HIF through the use of PHD inhibitors. For example, 2-OG analogues can stabilize HIF-α54. While this approach is not selective for particular PHD isoforms, some in vitro studies suggest that marked differences in substrate specificity may exist and could be harnessed for selectivity. For example, all PHDs hydroxylate the C-ODD domain more efficiently than the N-ODD domain (see Figure 3), and PHD2 hydroxylates the N-ODD domain less efficiently on HIF-2α than on HIF-1α. In addition, PHD3 does not hydroxylate the N-ODD domain of HIF-1α56,57. It is also likely that the protection afforded by PHD inhibitors (e.g. decreased tissue inflammatory cytokines, increased barrier function, decreased epithelial apoptosis) may involve both HIF and NF-κB activities (Figure 4).
Given the central role of HIF-mediated signaling on erythropoietin production, these drugs have been developed and are in clinical trials for the treatment of anemia58. Several PHD inhibitors have been described, including direct inhibitors59, analogs of naturally occurring cyclic hydroxamates60 and antagonists of alpha-keto-glutarate54. Each of these molecules serve as competitive inhibitors of the PHDs through substitution for alpha-ketoglutarate in the hydroxylation reaction shown in Figure 3. Within the gastrointestinal tract, the PHD inhibitors DMOG and FG-4497 have been used effectively to reduce symptoms in at least two mouse models of colitis23,26. Indeed, these studies have shown that both DMOG and FG-4497 provide an overall beneficial influence on multiple parameters studied in chemically induced (trinitrobenzene sulfonic acid [TNBS] or dextran sodium sulfate [DSS] mouse models of colitis. In these mouse models, the drugs were well tolerated with no significant adverse side effects. In our experience, both FG-4497 and DMOG can be delivered by multiple routes of administration (intraperitoneal, oral and intravenous)., and both FG-4497 are absorbed orally with only a slight loss of efficacy compared to intraperitoneal administration. To date, no trials have been initiated for the treatment of IBD.
Two notes of caution are worthwhile discussing regarding the use of HIF activators (e.g. PHD inhibitors) as potential therapies. First, this class of drugs substantially elevates hematocrit through increased erythropoietin production. In our initial studies with murine colitis, we observed that high doses of FG-4497 (>60 mg/kg) with 5 daily intervals of dosing resulted in occasional vascular occlusion within the intestine. Likely, these side effects were from erythrocyte sludging due to high hematocrits. This problem was rectified by lowering the dose and decreasing the interval of dosing26. As a second precautionary note, chronic HIF-1 and HIF-2 stabilization have been significantly associated with a number of cancers. Whether pharmacological HIF stabilization could initiate or promote tumor development is not currently known but should be considered as a potential serious side effect of these drugs. Until proven otherwise, the safest use of PHD inhibitors may be for acute exposure or as an adjunct therapy with other drugs over a short term period.
Conclusions
The gastrointestinal mucosa is an interesting tissue in which to investigate tissue oxygenation and disease-based metabolism. In this review, we have outlined the evidence for hypoxia as an important alarm signal within the intestinal mucosa. Studies derived from cultured cell systems, animal models and patient-derived materials have documented the that hypoxia is a significant component of the inflammatory microenvironment. Likewise, studies to date in animal models of intestinal inflammation have demonstrated an almost uniformly beneficial influence of HIF stabilization on disease outcomes. It is notable that the increased susceptibility to colitis following the genetic deletion of intestinal epithelial HIF may be somewhat model-dependent and will require additional validatation with PHD inhibitors before clinical studies can be implements. Ongoing studies to define the differences and similarities between innate and adaptive immune responses will continue to teach us important lessons about the complexity of the gastrointestinal tract. Such information will provide new insight into the pathogenesis disease and importantly, will provide new targets as templates for the development of therapies for human disease.
Key points.
A steep oxygen gradient exists from the anaerobic lumen of the intestine, across the intestinal epithelial barrier into the highly vascular sub-epithelium
Loss of function of the intestinal epithelial barrier, together with hypoxia and inflammation underlie the pathology of IBD
Hypoxia-inducible factor 1 (HIF-1) is induced in inflammed lesions, and triggers transcription of many genes that allow the intestinal epithelial cells to act as an effective barrier, and is thought to be protective against inflammation and IBD
Proly-hydroxylases (PHDs) are expressed in the intestinal mucosal tissue and regulate the atability of HIF
Targeted therapies to stabilize HIF, for instance by inhibiting PHDs, are in development to treat IBD
Acknowledgments
This work was supported by National Institutes of Health grants DK50189, DE016191, HL60569 and by grant from the Crohn’s and Colitis Foundation of America.
Footnotes
Competing interests
The authors declare no competing interests.
Contributor Information
Sean P. Colgan, Mucosal Inflammation Program, Division of Gastroenterology, Mucosal Inflammation Program, University of Colorado Denver, 12700 E. 19th Ave MS B146, Aurora, CO 80045, USA
Cormac T. Taylor, UCD Conway Institute, School of Medicine and Medical Science, College of Life Sciences, University College Dublin, Belfield, Dublin 4, Ireland
References
- 1.Laukoetter MG, Bruewer M, Nusrat A. Regulation of the intestinal epithelial barrier by the apical junctional complex. Curr Opin Gastroenterol. 2006;22:85–9. doi: 10.1097/01.mog.0000203864.48255.4f. [DOI] [PubMed] [Google Scholar]
- 2.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 3.Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med. 2008;85:1295–1300. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 4.Furuta GT, et al. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Ex Med. 2001;193:1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–65. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 6.Borregaard N, Herlin T. Energy metabolism of human neutrophils during phagocytosis. J Clin Invest. 1982;70:550–7. doi: 10.1172/JCI110647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Benna J, Dang PM, Gougerot-Pocidalo MA. Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin Immunopathol. 2008;30:279–89. doi: 10.1007/s00281-008-0118-3. [DOI] [PubMed] [Google Scholar]
- 8.Gabig TG, Bearman SI, Babior BM. Effects of oxygen tension and pH on the respiratory burst of human neutrophils. Blood. 1979;53:1133–9. [PubMed] [Google Scholar]
- 9.Fox CJ, Hammerman PS, Thompson CB. Fuel feeds function: energy metabolism and the T-cell response. Nat Rev Immunol. 2005;5:844–52. doi: 10.1038/nri1710. [DOI] [PubMed] [Google Scholar]
- 10.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat Rev Immunol. 2005;5:712–21. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 11.Hatoum OA, Heidemann J, Binion DG. The intestinal microvasculature as a therapeutic target in inflammatory bowel disease. Ann N Y Acad Sci. 2006;1072:78–97. doi: 10.1196/annals.1326.003. [DOI] [PubMed] [Google Scholar]
- 12.Danese S, Dejana E, Fiocchi C. Immune regulation by microvascular endothelial cells: directing innate and adaptive immunity, coagulation, and inflammation. J Immunol. 2007;178:6017–22. doi: 10.4049/jimmunol.178.10.6017. [DOI] [PubMed] [Google Scholar]
- 13.Karhausen JO, et al. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114:1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans SM, et al. Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res. 2000;60:2018–24. [PubMed] [Google Scholar]
- 15.Takasawa M, Moustafa RR, Baron JC. Applications of nitroimidazole in vivo hypoxia imaging in ischemic stroke. Stroke. 2008;39:1629–37. doi: 10.1161/STROKEAHA.107.485938. [DOI] [PubMed] [Google Scholar]
- 16.Kizaka-Kondoh S, Konse-Nagasawa H. Significance of nitroimidazole compounds and hypoxia-inducible factor-1 for imaging tumor hypoxia. Cancer Sci. 2009;100:1366–73. doi: 10.1111/j.1349-7006.2009.01195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Overgaard J. Hypoxic radiosensitization: adored and ignored. J Clin Oncol. 2007;25:4066–74. doi: 10.1200/JCO.2007.12.7878. [DOI] [PubMed] [Google Scholar]
- 18.Mastrogiannaki M, et al. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009 doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest. 2007;117:862–5. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Comerford KM, et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–94. [PubMed] [Google Scholar]
- 21.Synnestvedt K, et al. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 (HIF-1) mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eltzschig HK, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Ex Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cummins EP, et al. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134:156–65. doi: 10.1053/j.gastro.2007.10.012. Epub 2007 Oct 10. [DOI] [PubMed] [Google Scholar]
- 24.Han IO, Kim HS, Kim HC, Joe EH, Kim WK. Synergistic expression of inducible nitric oxide synthase by phorbol ester and interferon-gamma is mediated through NF-kappaB and ERK in microglial cells. J Neurosci Res. 2003;73:659–69. doi: 10.1002/jnr.10706. [DOI] [PubMed] [Google Scholar]
- 25.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136:607–18. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 26.Robinson A, et al. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134:145–55. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah YM, et al. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134:2036–48. 2048 e1–3. doi: 10.1053/j.gastro.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giatromanolaki A, et al. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56:209–13. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mariani F, et al. Cyclooxygenase-2 and Hypoxia-Inducible Factor-1alpha protein expression is related to inflammation, and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett. 2009;279:221–9. doi: 10.1016/j.canlet.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 30.Matthijsen RA, et al. Enterocyte shedding and epithelial lining repair following ischemia of the human small intestine attenuate inflammation. PLoS One. 2009;4:e7045. doi: 10.1371/journal.pone.0007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louis NA, et al. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem. 2006;99:1616–27. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 32.Makino Y, et al. Hypoxia-inducible factor regulates survival of antigen receptor-driven T cells. J Immunol. 2003;171:6534–40. doi: 10.4049/jimmunol.171.12.6534. [DOI] [PubMed] [Google Scholar]
- 33.Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9:609–17. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kong T, Eltzschig HK, Karhausen J, Colgan SP, Shelley CS. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc Natl Acad Sci U S A. 2004;101:10440–5. doi: 10.1073/pnas.0401339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong T, Scully M, Shelley CS, Colgan SP. Identification of Pur alpha as a new hypoxia response factor responsible for coordinated induction of the beta 2 integrin family. J Immunol. 2007;179:1934–41. doi: 10.4049/jimmunol.179.3.1934. [DOI] [PubMed] [Google Scholar]
- 36.Kuhl AA, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133:1882–92. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 37.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 38.Cummins EP, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103:18154–9. doi: 10.1073/pnas.0602235103. Epub 2006 Nov 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aragones J, et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–80. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- 40.Schneider M, et al. Loss or Silencing of the PHD1 Prolyl Hydroxylase Protects Livers of Mice Against Ischemia/Reperfusion Injury. Gastroenterology. 2009 doi: 10.1053/j.gastro.2009.09.057. [DOI] [PubMed] [Google Scholar]
- 41.Mazzone M, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–51. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ozolins TR, et al. Defects in embryonic development of EGLN1/PHD2 knockdown transgenic mice are associated with induction of Igfbp in the placenta. Biochem Biophys Res Commun. 2009;390:372–6. doi: 10.1016/j.bbrc.2009.08.057. [DOI] [PubMed] [Google Scholar]
- 43.Bishop T, et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3−/− mice. Mol Cell Biol. 2008;28:3386–400. doi: 10.1128/MCB.02041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-kappaB in hypoxic inflammation. J Physiol. 2008;586:4055–9. doi: 10.1113/jphysiol.2008.157669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenberger P, et al. Identification of vasodilator-stimulated phosphoprotein (VASP) as an HIF-regulated tissue permeability factor during hypoxia. FASEB J. 2007;21:2613–21. doi: 10.1096/fj.06-8004com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z, et al. Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway. Genes Dev. 1995;9:1586–97. doi: 10.1101/gad.9.13.1586. [DOI] [PubMed] [Google Scholar]
- 47.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death–a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–65. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cockman ME, et al. Posttranslational hydroxylation of ankyrin repeats in IkappaB proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH) Proc Natl Acad Sci U S A. 2006;103:14767–72. doi: 10.1073/pnas.0606877103. Epub 2006 Sep 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zaph C, et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007;446:552–6. doi: 10.1038/nature05590. [DOI] [PubMed] [Google Scholar]
- 51.Rius J, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–11. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Coleman ML, et al. Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem. 2007;282:24027–38. doi: 10.1074/jbc.M704102200. [DOI] [PubMed] [Google Scholar]
- 53.Koditz J, et al. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–7. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- 54.Mole DR, et al. 2-oxoglutarate analogue inhibitors of HIF prolyl hydroxylase. Bioorg Med Chem Lett. 2003;13:2677–80. doi: 10.1016/s0960-894x(03)00539-0. [DOI] [PubMed] [Google Scholar]
- 55.Masson N, Ratcliffe PJ. HIF prolyl and asparaginyl hydroxylases in the biological response to intracellular O(2) levels. J Cell Sci. 2003;116:3041–9. doi: 10.1242/jcs.00655. [DOI] [PubMed] [Google Scholar]
- 56.Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5:343–54. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 57.Bruick RK. Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia-inducible transcription factor. Genes Dev. 2003;17:2614–23. doi: 10.1101/gad.1145503. [DOI] [PubMed] [Google Scholar]
- 58.Jelkmann W. Control of erythropoietin gene expression and its use in medicine. Methods Enzymol. 2007;435:179–97. doi: 10.1016/S0076-6879(07)35010-6. [DOI] [PubMed] [Google Scholar]
- 59.Nwogu JI, et al. Inhibition of collagen synthesis with prolyl 4-hydroxylase inhibitor improves left ventricular function and alters the pattern of left ventricular dilatation after myocardial infarction. Circulation. 2001;104:2216–21. doi: 10.1161/hc4301.097193. [DOI] [PubMed] [Google Scholar]
- 60.Schlemminger I, et al. Analogues of dealanylalahopcin are inhibitors of human HIF prolyl hydroxylases. Bioorg Med Chem Lett. 2003;13:1451–4. doi: 10.1016/s0960-894x(03)00149-5. [DOI] [PubMed] [Google Scholar]