

Abstract
In prostate cancer, bone is a frequent site of metastasis; however, the molecular mechanisms of this tumor tropism remain unclear. Here, we integrate a microfluidic coculture platform with multi-photon imaging based techniques to assess both phenotypic cell behavior and FAD fluorescence intensity and fluorescence lifetime in the same cell. This platform combines two independent assays normally performed with two different cell populations into a single device, allowing us to simultaneously assess both phenotypic cell behavior and enzyme activity. We observed that the osteotropic prostate cancer cell line (C4-2B), when in coculture with bone marrow stromal cells (MC3T3-E1) have increased protrusive phenotype and increased total and protein-bound FAD as compared to their parent cell line (LNCaP). We hypothesized that an increase in ROS-generating APAO activity may be responsible for these effects, and found that the effects were decreased in the presence of the antioxidant N-Acetyl Cysteine (NAC). This suggests that an ROS-related signaling mechanism at the bone metastatic site may be correlated with and play a role in increased invasion of metastasizing prostate cancer cells. The studies enabled with this combined platform will lead to new insight into the mechanisms that drive prostate cancer metastasis.
Introduction
In prostate cancer (PCa), bone is a frequent site of metastasis, with 90% of patients with metastatic prostate cancer displaying lesions in the bone upon autopsy [1]. While there has been much investigation into the role of biologic, genetic, epigenetic, and tissue microenvironmental changes, the molecular mechanism(s) of this tropism remains unknown [2]. Accumulated evidence has shown that excess reactive oxygen species (ROS) often observed in human and animal PCa cells and tissues play a key role in PCa recurrence and progression to castrate-resistant PCa (CRPC). ROS-induced hydroxylation and nitrosylation of DNA and proteins in normal prostatic epithelia and PCa tissues have been shown in various studies [3, 4, 5, 6]. Pairs of cancer and normal tissues from the same PCa patient [7] or from the same transgenic animal developing spontaneous PCa [8] were analyzed and showed that ROS-induced macromolecular modifications are significantly higher in the PCa cells as compared to their normal epithelial counterparts both in mice and men. ROS levels are higher in invading adenocarcinomas as compared to the normal prostatic epithelia [4, 5], with hydroxyl and nitric oxide radicals related to cellular oxidative stress as a putative compound responsible for PCa cell invasion and migration [9, 10]. ROS may activate more than one mechanism to help androgen-dependent PCa (ADPC) cell survival and proliferation in the absence of androgen as well as its metastasis to distant organs leading to PCa progression to CRPC.
The JunD-androgen receptor (AR) complex initiates a metabolic pathway in PCa, which is a likely mechanism for ROS production [11, 12, 13, 14, 15]. Within this pathway, acetyl derivatives of spermidine and spermine are oxidized by flavin adenine dinucleotide (FAD)H2-bound enzyme acetyl polyamine oxidase (APAO), which releases FAD along with the production of excess ROS H2O2 in highly polyamine enriched PCa cells [16, 17, 18]. Increased APAO activity in cancer cells will result in an increase in the FAD concentration within the cell due to enhanced FADH2 to FAD interconversion [14, 15, 19, 20]. Recently, the incorporation of multiphoton excitation [21] and photon-counting techniques [22] have made it possible to estimate FAD in cancer cells and tissues through intrinsic fluorescence of the molecule. This method can be used to estimate total FAD as well as bound/free FAD related to increased APAO activity on a single cell-level.
The ability to couple mechanistic or enzymatic endpoints such as western blots, mRNA analysis, ELISA, etc. with phenotypic or functional assays (typically the gold standard Transwell platform [23]) have enabled researchers to discern which of many mechanisms of action may be responsible for the general invasive phenotype observed in cancer cells. However, the integration of multiphoton microscopy with traditional invasion assays can be limited by the need for high-resolution microscopy compatible glass-bottom trays with analytes in close proximity to the bottom surface. There has been increasing development of microfluidic platforms to look at cellular invasion [24, 25, 26] and the invasion of cancer cells in the bone microenvironment [27], but their adaptation for use with multiphoton imaging technology is limited.
Here, we assessed prostate cancer cell behavior and FAD fluorescence as a marker for ROS-producing APAO activity in response to bone marrow stromal cells. This was enabled by integrating multi-photon imaging techniques with a microscopy compatible and novel microfluidic coculture platform to provide a multiplexed functional and enzymatic activity assay (Figure 1). This platform is simple to set up and operate, as all steps during set up and use require only a pipette to operate compared to many closed microfluidic systems. Additionally, this platform offers several advantages over traditional assays. First, the integration of two independent assays normally performed with two different cell populations into a single device and assay. Second, the need for limited cell numbers and reagents enables one to screen more conditions than would be possible using traditional assays. Third, the matrix used can be customized, in a way that is easier and most cost-effective than traditional platforms. These advantages enable the potential for new mechanistic insight into the role of ROS in prostate cancer metastasis to bone.
Figure 1.

A) This platform enables integration of invasion and enzyme activity assays to study the role of ROS in prostate cancer invasion. The osteoblasts interact with the prostate cancer cells with an observed increase in protrusive cells. The increased activity of ROS-producing APAO, as assessed by the detection of bound FAD via FLIM, suggests one possible mechanism for this response due to ROS. B) Diagram of microfluidic invasion assay set up. 1-The microfluidic device is seeded with MC3T3-E1s and cultured at a 90 degree angle overnight to allow MC3T3-E1s to adhere to the side of the device. 2-The channel is filled with a collagen I solution. 3-Media is flowed through the device via passive pumping (Walker2002) leaving behind a thin coating of collagen I. 4-The collagen solution is polymerized at 37 degrees C and prostate cancer cells (LNCaPs or C4-2Bs) are seeded into the channel. C) Schematic and volume-rendered cross-sections of the device are shown at bottom right. Shown in the volume-rendered cross section: 24 hours after loading LNCaPs into the channels, cells were stained with calcein AM and imaged using multi-photon microscopy. The collagen coating is shown in red and the cells (MC3T3-E1s on the side and LNCaPs in the center channel) are shown in yellow. Scale bar represents 100μm.
Results and Discussion
Characterization of prostate cancer cell line behavior in response to MC3T3-E1s using a microfluidic coculture assay
To investigate mechanisms by which the bone specific microenvironment influences prostate cancer cells, we developed a microfluidic coculture assay and characterized the assay using two prostate cancer cell lines, LNCaP, a prostate cancer cell line isolated from a patient’s lymph node and C4-2B a variant of LNCaP cells isolated from animal bone-metastatic lesion [28]. We used the assay to examine the effects of MC3T3-E1 cells, a cell line that can be differentiated toward an osteoblast-like cell type that is representative of the bone microenvironment.
In initial experiments, MC3T3-E1 cells were differentiated toward an osteoblast lineage and seeded on the sidewall of a microchannel. A collagen I hydrogel coating was then added and LNCaPs or C4-2Bs were seeded into the channels (Figure 1B). The hydrogel coating method, viscous finger patterning, has been previously characterized and it was found that the hydrogel layer has a thickness of approximately 87+/−8μm [29]. After a period of 24 hours, the percentage of protrusive versus rounded cells in each channel were counted and compared to control channels in which MC3T3-E1s were not added prior to collagen coating (Figure 2). We observed that C4-2Bs show a statistically significant increase in percentage of cells with protrusions, when in coculture with MC3T3-E1s as compared to monoculture. This difference was not seen for LNCaP monoculture and coculture with MC3T3-E1s. Each channel had approximately 30 cells within the imaging region (i.e. excluding ports). A minimum of three channels were used for each experiment, with three independent experiments analyzed for each data set.
Figure 2.

A) The percentage of protrusive cells under different conditions was determined by counting the number of LNCaP or C4-2Bs that showed protrusions into the collagen I hydrogel after 24 hours. Scale bars represent 50μm. B) C4-2Bs in coculture with MC3T3-E1s showed a statistically significant increase in protrusive response compared to C4-2Bs in monoculture. This effect was not observed in LNCaP cells. Data was collected from 3 independent experiments with 8 channels per condition. Error bars represent standard deviation.
Using the number of cells with protrusive phenotypes as the assay readout enables researchers to quickly perform and analyze experiments as a clear cellular response is usually observed in as little as 24 hours (using our chosen model), and does not require advanced imaging techniques. However, as we have shown below, more advanced imaging and analysis techniques can be easily integrated with the platform for more in-depth mechanistic studies. While a protrusive phenotype alone does not prove invasion, the formation of cellular protrusions is often a first phenotypically observed step in cellular invasion and can indicate an invasive capacity [30, 31, 32].
The cells used for this investigative study were LNCaPs, an androgen-dependent invasive cell line isolated from a patient’s lymph node, and the bone-metastatic, androgen-independent invasive C4-2B cell line derived from LNCaP cells injected into animals along with a bone marrow cell line. As medium containing 2% FBS was used in experiments limited androgen was added to the system. Thus, the androgen-dependent LNCaPs were not expected to have invasive characteristics towards the bone stromal cell model [11, 33, 34]. Due to the specific ability of the C4-2Bs to be androgen-independent and invasive to bone, the effect of the bone stromal cells was of interest. Even though, MC3T3-E1 cells are heterologous, these cells have been used to mimic human pre-osteoblastic cell lines due to similarities in secretions [35, 36]. The increased percentage of C4-2Bs with cellular protrusions, when placed in coculture with MC3T3-E1s demonstrates the influence of this cell line on the behavior of the C4-2B cell line.
C4-2B response to an added matrix factor
As a proof-of-principle experiment, we investigated the effects of incorporating a matrix protein (osteopontin) into the collagen I hydrogel coating. Osteopontin is a matrix protein that is secreted by osteoblasts and osteoblast-like cells such as MC3T3-E1s [37]. As shown in Figure 3, when osteopontin was added to the matrix, the percentage of C4-2B cells with protrusions in monoculture was increased to similar levels as seen in coculture with MC3T3-E1s. Adding osteopontin to the matrix in coculture experiments did not significantly affect the percentage of protrusive C4-2Bs. The ability to easily customize the matrix by incorporating other factors into a collagen I gel is a significant advantage of this assay. In comparison, traditional coculture assays such as Transwell assays usually use a standardized hydrogel coating that is not user-defined and would be more expensive to modify due to the amount of reagents needed.
Figure 3.
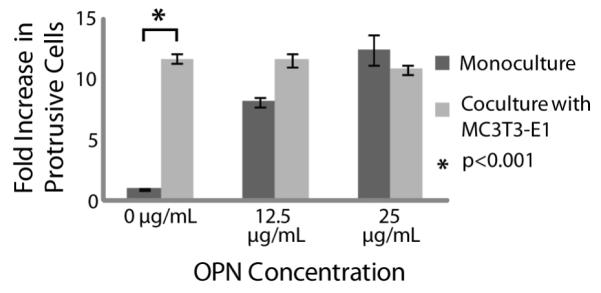
By adding osteopontin into the collagen I hydrogel coating we were able to demonstrate the ability to customize the invasion assay matrix. When osteopontin was added the percentage of C4-2Bs in monoculture with protrusions increased to the level typically observed by C4-2Bs in coculture with MC3T3-E1s. Data was collected from 3 independent experiments with 8 channels per condition. Errors bars represent standard deviation.
Interactions between C4-2B and MC3T3-E1 influence behavior of C4-2Bs
Media containing 2% FBS was conditioned by and collected from cultures of C4-2Bs, MC3T3-E1s, or a mixture of the two cell types after 48 hours. Using the microfluidic coculture assay, C4-2Bs in monoculture were treated for 24 hours with the different conditioned media and the percentage of protrusive cells was assessed (Figure 4). While no significant difference was observed in negative control channels (regular culture media) and C4-2Bs treated with C4-2B conditioned media (C4-2B CM), the percentage of protrusive C4-2Bs was significantly increased, when C4-2Bs were treated with MC3T3-E1 conditioned media (MC3T3-E1 CM). It was increased further when C4-2Bs were treated with conditioned media from a mixed culture of C4-2Bs and MC3T3-E1s (C4-2B/MC3T3-E1 Mix CM). A positive control with C4-2Bs and MC3T3s cocultured within the device (denoted C4-2B/MC3T3-E1 Coculture) is included for comparison. These results indicate that secreted factors from the MC3T3-E1s affect the behavior of C4-2B cells, and that cross-talk interactions between C4-2Bs and MC3T3-E1s provide the maximum effect. Further screening experiments may elucidate the specific secreted factors and cross-talk interactions responsible for the observed increase of protrusive C4-2Bs in coculture with MC3T3-E1s.
Figure 4.
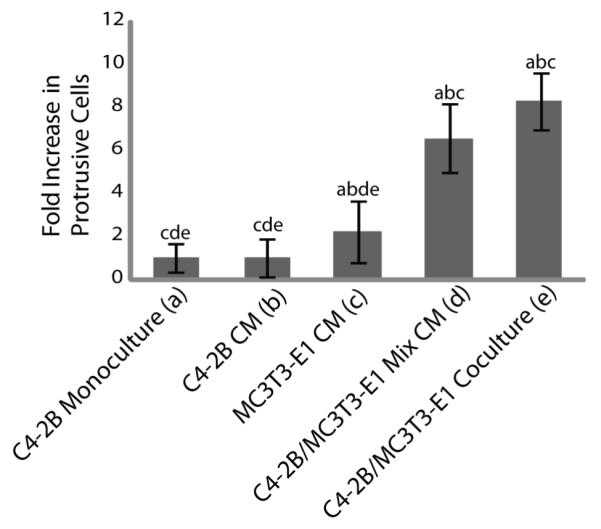
C4-2Bs in monoculture were cultured in conditioned media (CM) collected from a number of different conditions: C4-2B alone (C4-2B CM), MC3T3-E1 alone (MC3T3-E1 CM), and media from a mixed culture of C4-2Bs and MC3T3-E1s (C4-2B/MC3T3-E1 Mix CM). The percentage of C4-2Bs with protrusions after culture under these conditions was compared to C4-2Bs in monoculture (C4-2B Monoculture) as a negative control and in coculture with MC3T3-E1s (C4-2B/MC3T3-E1 Coculture) as a positive control. The letters above each bar indicate a p value less than 0.05 when compared to the bar represented by each corresponding letter. Data were collected from 3 independent experiments with 8 channels per condition. Error bars represent standard deviation.
FAD intensity and percentage of bound FAD detected in C4-2Bs is increased in coculture with MC3T3-E1s
To investigate a potential mechanism for the observed response of C4-2Bs to coculture with MC3T3-e1s, we used multi-photon imaging to detect the levels of the intrinsic fluorescent metabolites FAD and nicotinamind adenine dinuclotide (NAD)H (or NAD(P)H) in C4-2Bs or LNCaPs in coculture with MC3T3-E1s. The results are shown in Figure 5. Using an excitation source tuned to 890 nm with a 562/40 filter for FAD intensity imaging or 780 nm with a 457/50 filter for NADH intensity imaging we are able to specifically distinguish autofluorescence due to these two molecules. We observed a statistically significant increase in the level of total FAD intensity per cell in C4-2Bs cocultured with MC3T3-E1s compared to monoculture (Figure 5A). Using Fluorescence Lifetime Imaging Microscopy (FLIM) we were able to detect the percentage of bound or free FAD or NADH (NAD(P)H) in LNCaPs or C42-2Bs under different conditions. We observed that the percentage of bound FAD in C4-2Bs was significantly increased in coculture. This effect was not observed when comparing LNCaP cells in monoculture and coculture with MC3T3-E1s. No differences were observed in NADH (NAD(P)H) intensity or the percentage of bound NADH (NAD(P)H) between these conditions.
Figure 5.

A-B) Fluorescence lifetime imaging of C4-2Bs in monoculture (A) and coculture with MC3T3-E1s (B) in the microfluidic coculture assay. Images show the percentage of protein-bound FAD. Scale bar represents 50μm. C-D) Quantification of FAD (C) and NAD(P)H (D) fluorescence intensity in LNCaPs or C4-2Bs in monoculture or coculture with MC3T3-E1s using multi-photon microscopy. Data was collected from 3 independent experiments with 3 channels per condition. Error bars represent standard deviation. E-F) Quantification of the percentage of bound FAD (E) and NAD(P)H (F) in LNCaPs or C4-2Bs in monoculture or coculture with MC3T3-E1s using fluorescence lifetime imaging microscopy. Data were collected from 3 independent experiments with 3 channels per condition. Error bars represent standard deviation.
The analysis of FAD involves two values, the total FAD fluorescence intensity, and the ratio of protein-bound FAD to free FAD. Importantly, the concentration of FAD and, therefore, its fluorescence intensity will increase with the activation of APAO. While mitochondrial energy metabolism also produces free FAD, the amount of protein-bound FAD increases with APAO and other cytoplasmic FADH2-linked enzyme activity specifically [15]. Polyamines are produced in millimolar concentrations in the prostate cells and thus, FAD produced due to activation of a FAD-linked enzyme involved in polyamine oxidation pathway is likely to predominate the cellular FAD autofluorescence over all other enzymes producing FAD. The data presented here show an increase in both the FAD fluorescence intensity and the protein-bound/free FAD ratio. This suggests that the increase in FAD intensity is likely not due to an increase in the mitochondrial metabolism, but instead due cytoplasmic enzyme activity such as APAO, which is particularly active in aggressive PCa cells [14].
Similarly to FAD, NAD(P)H intensity and the ratio of protein-bound/free NAD(P)H was analyzed using FLIM. NAD(P)-mediated signaling plays an important role in many biological processes such as metabolism and has been suggested as a potential therapeutic target for cancer treatment [38]. In the present study no significant differences between in the fluorescence intensity of NAD(P)H or the percentage of protein-bound NAD(P)H was observed between the LNCaPs or C4-2Bs in monoculture or coculture with MC3T3-E1s. These observations further support the possibility that changes in mitochondrial function between the different conditions in this particular study are not involved in the changes in FAD previously discussed. This observation is from a cell-based assay using a heterogeneous system and further investigation into these findings is needed.
Although not exclusive to APAO, the increased FAD intensity suggests one potential mechanism of ROS production. The hypothesis that the changes in FAD intensity are not due to mitochondrial effects, is further supported by recent data showing ROS by LNCaP and C4-2 cells are coming from cytoplasmic and extracellular H2O2 and not from mitochondrial superoxide (Basu et al, unpublished data). Importantly, this effect was seen in the cocultured conditions, suggesting a correlation and potential role for ROS involvement in the progression of metastatic disease and the initiation of metastases at secondary sites, such as bone.
Protrusive response of C4-2Bs in coculture with MC3T3-E1s decreased in presence of antioxidant N-Acetyl Cysteine
To test whether ROS were playing a role in the protrusive phenotype observed when C4-2Bs are in coculture with MC3T3-E1s, we included an antioxidant, N-Acetyl Cysteine (NAC) into the culture medium (Figure 6). It was found that levels of protrusive C4-2Bs in coculture with at least 0.5mM NAC were decreased to levels similar to monoculture. The effect of ROS on the progression of prostate metastatic disease has been postulated as one of the mechanisms for PCa invasion [4, 5]. In prostate cancer patient tissues, increased protein-bound FAD to free FAD ratios have been observed in metastases, however this pathway has not been explored on the single-cell level [39].
Figure 6.
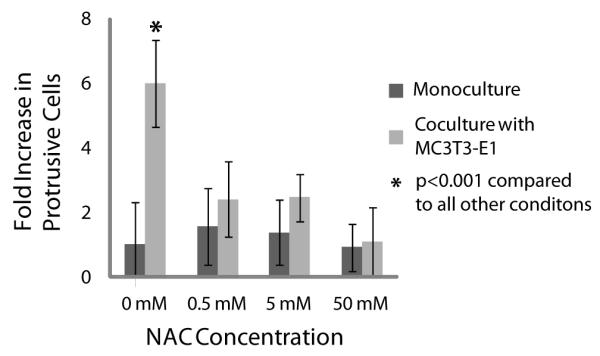
The antioxidant N Acetyl-Cysteine (NAC) was added to culture medium of C4-2Bs in monoculture or in coculture with MC3T3-E1s. The percentage of C4-2Bs showing a protrusive response to coculture with MC3T3-E1s was significantly reduced in the presence of NAC. Data were collected from 3 independent experiments with 8 channels per condition. Error bars represent standard deviation.
Conclusions
Using a microfluidic coculture assay, we assessed the influence of bone stromal cells on prostate cancer cells. Together our results demonstrate that cross-talk between a bone metastatic prostate cancer cell line, C4-2B, and bone stromal cells, MC3T3-E1, increases the protrusive phenotype, and possibly invasive capacity, of the C4-2Bs and that effect may be mediated through an increase in ROS-producing APAO activity as assessed by an increase in total and protein-bound FAD. While further investigation is required, this suggests a potential link between an ROS-related signaling mechanism and increased invasion in prostate cancer cells specifically at the secondary metastatic site of the bone.
These studies were enabled by the integration of a novel microfluidic coculture assay with multiphoton imaging. Importantly, the platform offers several key advantages that have the potential to enable insight into a variety of biological studies involving interactions between multiple cell types. This platform combines two independent assays normally performed with two different cell populations into a single device, allowing us to simultaneously assess both cellular behavior and enzyme activity. These integrated experiments require only small numbers of cells, which is a key advantage over traditional well-plate assays and is a necessary step towards translating this technology for analysis of human tissues. As demonstrated with the doped osteopontin assay, various matrix factors or other microenvironmental factors can be added and as enabled by microfluidics, can be done in a highly arrayable technique. These techniques could be integrated with other methods of analysis or cancer models to provide insight to other mechanisms of action for various cancers and disease sites.
Materials and Methods
Cell Lines and Maintenance
MC-3T3-E1 cells were obtained from Dr. S H Lin’s lab at MD Anderson and maintained in MEM-alpha (no ascorbic acid, Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin (PS) (Invitrogen, Carlsbad, CA, USA). LNCaPs and C4-2Bs were obtained from Dr. Bushman’s lab at the UW-Madison and maintained in MEM-alpha (no ascorbic acid, Invitrogen, Carlsbad, CA, USA) with 10% FBS and 1% PS. Cell Check services were performed at IDEXX RADIL (Columbia, MO, USA) and showed that the LNCaP and C4-2B cell lines were confirmed to be of human origin and no mammalian inter-species contamination was detected. The LNCaP cell line used had an extra allele at markers TH01 and vWA, but the genetic profile is otherwise consistent with the genetic profile reported for this cell line. The C4-2B cell line contains an extra allele at markers CSF1PO and D16S539 as well as a different allele at marker D13S317 and is missing the Y allele marker when compared to the LNCaP cell line genetic profile. The MC3T3-E1 cell line was confirmed to be of mouse origin and no mammalian inter-species contamination was detected.
Device Fabrication
Polydimethylsiloxane (PDMS, Sylgard 184 Silicone Elastomer Kit, Dow Corning, Midland, MI) elastomer base and curing agent were mixed at a 10:1 ratio and degassed for 45 min under vacuum at room temperature. The degassed PDMS was then poured over SU-8 master molds that were generated using standard soft lithography methods [40]. PDMS was cured at 80°C for 4 h. Prior to use, devices were oxygen-plasma-treated to bond the PDMS channels to a glass surface (the inside of a glass-bottom Petri dish, MatTek, Ashland, ME, USA). All devices consisted of single straight microchannels 5 cm in length 500 μm wide, 500 μm tall, with input and output port diameters of 1mm and 1.5 mm respectively.
Assay Setup
Following device preparation, microchannels were coated with 100 μg/mL fibronectin (FN) (Sigma-Aldrich, St. Louis, MO, USA) to facilitate adhesion of MC3T3-E1s and collagen l coating to the channel walls. After at least 20 min incubation at room temperature, the FN solution was aspirated from the channel. For coculture conditions with osteoblasts on the side of the channels, 2×105 MC3T3-E1 cells/mL were seeded into the channels in MEM-alpha (no ascorbic acid) medium with 2% FBS, 1% PS. The channels were subsequently cultured at a 90 degree angle at 37°C overnight. This is to enable a separation between cell types and clearer imaging as MC3T3-E1s are kept out of the light path. Channels were then coated with a layer of 4.28 mg/mL type I collagen (8.56 mg/ml, rat tail, BD Biosciences, Bedford, MA, USA). The collagen I hydrogel solution was prepared by neutralizing the collagen I stock solution in a 1:1 ratio with HEPES buffer in 2× PBS. The collagen I solution was incubated on ice for 5-10 minutes prior to use as an additional nucleation phase [41]. The hydrogel solution was then loaded into the microchannels and subsequently rinsed with media via passive pumping [42], leaving a thin coating of collagen I along the edges of the microchannel [29]. Following polymerization for 10 minutes at 37°C, LNCaPs or C4-2Bs cells were added to the channels at a density of 1×105 cells/ml which resulted in ~30 cells per channel (excluding ports). This process is outlined in Figure 5. Cells were cultured in Minimum Essential Medium (MEM) α, without ascorbic acid containing 2% FBS and 1% PS.
To test the effects of secreted factors on the invasion of C4-2Bs, conditioned media was collected from C4-2Bs, MC3T3-E1s, or a mixed culture of C4-2Bs and MC3T3-E1s (in a ratio of 1:2 in order to more closely replicate ratio found in the device coculture conditions). Cells were plated in regular tissue culture flasks at a concentration of 250,000 cells/ml and incubated with MEM-α medium supplemented with 2% FBS and 1% PS. Media was collected after 48 h. To test the effects of incorporating other microenvironmental factors into the matrix, 0, 12.5, or 25 ng/ml osteopontin (OPN) (Sigma-Aldrich, St. Louis, MO, USA) was added to the collagen solution during preparation. To test the effects of reducing ROS, an antioxidant NAcetyl Cysteine (Sigma-Aldrich, St. Louis, MO, USA) was included in the culture media at 0mM, 0.5mM, 5mM, and 50mM. The pH of the media was adjusted as needed by adding NaOH to the media until the appropriate pH was reached as detected by phenol red in the media.
Phenotypic Invasion Imaging and Analysis
Samples were imaged with an Olympus IX70 microscope (Center Valley, PA, USA) and images were acquired using MetaMorph 7.5 (Molecular Devices, LLC, Sunnyvale, CA, USA). The high-throughput data analysis platform, JeXperiment (http://jexperiment.wikidot.com), was used to perform custom image processing and analysis as previously described [43]. [31]Protrusive and rounded cells in each sample channel were counted and JeXperimet was used to calculate the average increase in the protrusive phenotype observed in each condition. Fold increase in protrusive cells was calculated by normalizing this number to the experimental control (C4-2B cells in monoculture). A student T-test was used to determine significant differences between samples. P values less than 0.05 were considered to be significant.
Multiphoton Intensity and Fluorescence Lifetime Imaging and Analysis
Multiphoton laser-scanning microscopy (MPLSM) imaging was performed on an Optical Workstation that was constructed around a Nikon Eclipse TE300 (Nikon, Mehlville, NY). A MaiTai Deepsee Ti:sapphire laser (Spectra Physics, Mountain View, CA) excitation source tuned to 890 nm with a 562/40 filter for FAD intensity imaging or 780 nm with a 457/50 filter for NAD(P)H intensity imaging. The beam was focused onto the sample with a Nikon 20× Plan Apo VC air immersion lens (numerical aperture [NA] = 0.75). Image acquisition was performed with WiscScan (http://www.loci.wisc.edu/software/wiscscan), a laser-scanning software acquisition package developed at LOCI (Laboratory for Optical and Computational Instrumentation, University of Wisconsin, Madison, WI) described elsewhere [44, 45]. For volume-rendered images, stacks were acquired and volume-rendered images were generated using OsiriX imaging software. For three independent experiments FAD and NAD(P)H intensity images were collected from three samples each of LNCaPs and C4-2B in monoculture or coculture with MC3T3-E1s in the microfluidic invasion assay. FAD and NAD(P)H intensity images were imported into JeXperiment to enable the quantification of the mean intensity of the images. This was divided by the number of cells in the image to yield the average intensity per cell. A student T-test was used to determine significant differences between samples. P values less than 0.05 were considered to be significant.
Fluorescence Lifetime Imaging Microscopy (FLIM) was performed as described in Skala et al [44] but using a newer electronic system for recording fast light signals by time correlated single photon counting (SPC-830, Becker & Hickl). Single photon counting for FLIM measurements was performed at a rate of ≈1 × 105 photons per s (by adjusting the power incident on the sample). The integration time for the FLIM images was 0 to 120 s. The instrument response function (IRF) measured with a second harmonic signal from a urea crystal was used in the lifetime fit model. For three independent experiments FLIM images were collected and analyzed from three samples each of LNCaPs and C4-2B in monoculture or coculture with MC3T3-E1s in the microfluidic invasion assay. SPCImage software (Becker and Hickl) was used to analyze the fluorescence lifetime decay curves. The lifetime decay curve of each pixel was fit to a double-exponential decay model, and the mean relative contributions of the lifetime components were measured. A student T-test was used to determine significant differences between samples. P values less than 0.05 were considered to be significant.
Acknowledgements
This work was supported by National Institute of Health (NIH) grants R33 CA137673 (B.P. Casavant & D.J. Beebe), T32HL007889 (L.L. Bischel), R43 -01A1 (H.S. Basu), R21CA176218-01 (H.S. Basu), R01 CA136590 (K.E. Eliceri), R01EB10039 (D.J. Beebe), and R33CA160244 (D.J. Beebe) and Department of Defense (DOD) grants W81XWH-09-1-0192 (B.P. Casavant & D.J. Beebe), W81XWH-12-1-0025 (P.A. Young), W81XWH-12-1-0085 (H.S. Basu), W81XWH-10-1-0169 (H.S. Basu), and W81XWH-12-1-0367 (H.S. Basu). David J. Beebe has an ownership interest in Bellbrook Labs LLC, which has licensed technology reported in this publication. Hirak S. Basu has ownership interest in Colby Pharmaceutical Company that is involved in drug development targeting invasion and metastasis of prostate cancer. The authors would like to thank Ashleigh Theberge for specific help with comparing fluid motion. Also Jay Warrick, Erwin Berthier, and Edmond Young provided specific guidance when developing the project.
References
- [1].Bubendorf L, Schopfer A, Wagner U, Sauter G, Moch H, Willi N, et al. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Hum Pathol. 2000;31:578–583. doi: 10.1053/hp.2000.6698. [DOI] [PubMed] [Google Scholar]
- [2].Jin JK, Dayyani F, Gallick GE. Steps in prostate cancer progression that lead to bone metastasis. Int J Cancer. 2011;128:2545–2561. doi: 10.1002/ijc.26024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bostwick D, Alexander E, Singh R, Shan A, Qian J, Santella R, et al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer. 2000;89:123–134. [PubMed] [Google Scholar]
- [4].Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68:1777–1785. doi: 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- [5].Gupta-Elera G, Garrett AR, Robison RA, O’Neill KL. The role of oxidative stress in prostate cancer. Eur J Cancer Prev. 2012;21:155–162. doi: 10.1097/CEJ.0b013e32834a8002. [DOI] [PubMed] [Google Scholar]
- [6].Sung SY, Kubo H, Shigemura K, Arnold RS, Logani S, Wang R, et al. Oxidative stress induces ADAM9 protein expression in human prostate cancer cells. Cancer Res. 2006;66:9519–9526. doi: 10.1158/0008-5472.CAN-05-4375. [DOI] [PubMed] [Google Scholar]
- [7].Oberley T, Zhong W, Szweda L, Oberley L. Localization of antioxidant enzymes and oxidative damage products in normal and malignant prostate epithelium. Prostate. 2000;44:144–155. doi: 10.1002/1097-0045(20000701)44:2<144::aid-pros7>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- [8].Tam N, Nyska A, Maronpot R, Kissling G, Lomnitski L, Suttie A, et al. Differential attenuation of oxidative/nitrosative injuries in early prostatic neoplastic lesions in TRAMP mice by dietary antioxidants. Prostate. 2006;66:57–69. doi: 10.1002/pros.20313. [DOI] [PubMed] [Google Scholar]
- [9].Polytarchou C, Hatziapostolou M, Polmenidi E, Mikelis C, Papadopoulou A, Parthymou A, et al. Nitric oxide stimulates migration of human endothelial and prostate cancer cells through up-regulation of pleiotrophin expression and its receptor protein tyrosine phosphatase beta/zeta. Int J Cancer. 2009;124:1785–1793. doi: 10.1002/ijc.24084. [DOI] [PubMed] [Google Scholar]
- [10].Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer A double-edged sword with therapeutic potential. Oxidative Med Cell Lon. 2010;3:23–34. doi: 10.4161/oxim.3.1.10095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mehraein-Ghomi F, Lee E, Church DR, Thompson TA, Basu HS, Wilding G. JunD mediates androgen-induced oxidative stress in androgen dependent LNCaP human prostate cancer cells. Prostate. 2008;68:924–934. doi: 10.1002/pros.20737. [DOI] [PubMed] [Google Scholar]
- [12].Church D, Lee E, Thompson T, Basu H, Ripple M, Ariazi E, et al. Induction of AP-1 activity by androgen activation of the androgen receptor in LNCaP human prostate carcinoma cells. Prostate. 2005;63:155–168. doi: 10.1002/pros.20172. [DOI] [PubMed] [Google Scholar]
- [13].Mehraein-Ghomi F, Lee E, Church D, Thompson T, Basu H, G W. Androgen Receptor Requires JunD as a Co-activator to Switch on an Oxidative Stress Generation Pathway in Prostate Cancer Cells. Cancer Res. 2010;70:4560–4568. doi: 10.1158/0008-5472.CAN-09-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Basu HS, Thompson TA, Church DR, Clower CC, Mehraein-Ghomi F, Amlong CA, et al. A Small Molecule Polyamine Oxidase Inhibitor Blocks Androgen-Induced Oxidative Stress and Delays Prostate Cancer Progression in the Transgenic Adenocarcinoma of the Mouse Prostate Model. Cancer Res. 2009;69:7689–7695. doi: 10.1158/0008-5472.CAN-08-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Holtta E. Oxidation of spermidine and spermine in rat liver - purification and properties of polyamine oxidase. Biochemistry-US. 1977;16:91–100. doi: 10.1021/bi00620a015. [DOI] [PubMed] [Google Scholar]
- [16].Wang Y, Hacker A, Murray-Stewart T, Frydman B, Valasinas A, Fraser A, et al. Properties of recombinant human N1-acetylpolyamine oxidase (hPAO): potential role in determining drug sensitivity. Cancer Chemoth Pharm. 2005;56:83–90. doi: 10.1007/s00280-004-0936-5. [DOI] [PubMed] [Google Scholar]
- [17].Seiler N. Polyamine Metabolism. Digestion. 1990;46:319–330. doi: 10.1159/000200405. [DOI] [PubMed] [Google Scholar]
- [18].Casero RA, Jr, Pegg AE. Polyamine catabolism and disease. Biochem J. 2009;421:323–338. doi: 10.1042/BJ20090598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cohen S. A Guide to the Polyamines. Oxford University Press; 1998. [Google Scholar]
- [20].Henriques B, Olsen R, Bross P, Gomes C. Emerging roles for riboflavin in functional rescue of mitochondrial beta-oxidation flavoenzymes. Curr Med Chem. 2010;17:3842–54. doi: 10.2174/092986710793205462. [DOI] [PubMed] [Google Scholar]
- [21].Zipfel W, Williams R, Webb W. Nonlinear magic: multiphoton microscopy in the biosciences. Nat Biotechnol. 2003;21:1368–1376. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- [22].Lakowicz J, Szmacinski H, Nowaczyk K, Johnson M. Flourescence lifetime imaging of free and protein-bound NADH. PNAS. 1992;89:1271–1275. doi: 10.1073/pnas.89.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nabha SM, dos Santos EB, Yamamoto HA, Belizi A, Dong Z, Meng H, et al. Bone marrow stromal cells enhance prostate cancer cell invasion through type I collagen in an MMP-12 dependent manner. Int J Cancer. 2008;122:2482–2490. doi: 10.1002/ijc.23431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shin Y, Kim H, Han S, Won J, Jeong HE, Lee ES, et al. Extracellular Matrix Heterogeneity Regulates Three-Dimensional Morphologies of Breast Adenocarcinoma Cell Invasion. Adv Healthcare Mat. 2013;2:790–794. doi: 10.1002/adhm.201200320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vickerman V, Blundo J, Chung S, Kamm R. Design, fabrication and implementation of a novel multi-parameter control microfluidic platform for three- dimensional cell culture and real-time imaging. Lab Chip. 2008;8:1468–1477. doi: 10.1039/b802395f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Abhyankar VV, Toepke MW, Cortesio CL, Lokuta MA, Huttenlocher A, Beebe DJ. A platform for assessing chemotactic migration within a spatiotemporally defined 3D microenvironment. Lab Chip. 2008;8:1507–1515. doi: 10.1039/b803533d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bersini S, Jeon JS, Dubini G, Arrigoni C, Chung S, Charest JL, et al. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials. 2013;35:2454–2461. doi: 10.1016/j.biomaterials.2013.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Litvinov IV, Vander Griend DJ, Xu Y, Antony L, Dalrymple SL, Isaacs JT. Low-calcium serum-free defined medium selects for growth of normal prostatic epithelial stem cells. Cancer Res. 2006;66:8598–8607. doi: 10.1158/0008-5472.CAN-06-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bischel LL, Lee SH, Beebe DJ. A Practical Method for Patterning Lumens through ECM Hydrogels via Viscous Finger Patterning. JALA. 2012;17:96–103. doi: 10.1177/2211068211426694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kikuchi K, Takahashi K. WAVE2-and microtubule-dependent formation of long protrusions and invasion of cancer cells cultured on three-dimensional extracellular matrices. Cancer Sci. 2008;99:2252–2259. doi: 10.1111/j.1349-7006.2008.00927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yamaguchi H. Pathological roles of invadopodia in cancer invasion and metastasis. Eur J Cell Bio. 2012;91:902–907. doi: 10.1016/j.ejcb.2012.04.005. [DOI] [PubMed] [Google Scholar]
- [32].Jung O, Choi YJ, Kwak TK, Kang M, Lee MS, Ryu J, et al. The COOH-terminus of TM4SF5 in hepatoma cell lines regulates c-Src to form invasive protrusions via EGFR Tyr845 phosphorylation. BBA-Mol Cell Res. 2013;1833:629–642. doi: 10.1016/j.bbamcr.2012.11.026. [DOI] [PubMed] [Google Scholar]
- [33].Thalmann G, Sikes R, Wu T, Degeorges A, Chang S, Ozen M, et al. LNCaP progression model of human prostate cancer: Androgen-independence and osseous metastasis. Prostate. 2000;44:91–103. doi: 10.1002/1097-0045(20000701)44:2<91::aid-pros1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- [34].Bonaccorsi L, Marchiani S, Ferruzzi P, Muratori M, Crescioli C, Forti G, et al. Non-genomic effects of the androgen receptor and vitamin D agonist are involved in suppressing invasive phenotype of prostate cancer cells. Steroids. 2006;71:304–309. doi: 10.1016/j.steroids.2005.09.010. [DOI] [PubMed] [Google Scholar]
- [35].Bodenstine T, Beck B, Cao X, Cook L, Ismail A, Powers S, et al. Pre-osteoblastic MC3T3-E1 cells promote breast cancer growth in bone in a murine xenograft model. Chin J Cancer. 2011;3:189–96. doi: 10.5732/cjc.010.10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Czekanska EM, Stoddart MJ, Richards RG, Hayes JS. In search of an osteoblast cell model for in vitro research. Eur Cells Mater. 2012;24:1–17. doi: 10.22203/ecm.v024a01. [DOI] [PubMed] [Google Scholar]
- [37].Kim SK, Kwon JY, Nam TJ. Involvement of ligand occupancy in Insulin-like growth factor-I (IGF-I) induced cell growth in osteoblast like MC3T3-E1 cells. BioFactors. 2007;29:187–202. doi: 10.1002/biof.5520290403. [DOI] [PubMed] [Google Scholar]
- [38].Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome - a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–752. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- [39].Huang W, Young P, Eliceiri K, Wilding G, Basu Activation of a Specific Metabolic Enzyme Distinguishes Aggressive from Indolent Prostate Cancer. Proc Aacr Mol Targets Cancer Ther. 2012 [Google Scholar]
- [40].Duffy D, McDonald J, Schueller O, Whitesides G. Rapid prototyping of microfluidic systems in poly(dimethylsiloxane) Anal Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- [41].Sung KE, Su G, Pehlke C, Trier SM, Eliceiri KW, Keely PJ, et al. Control of 3-dimensional collagen matrix polymerization for reproducible human mammary fibroblast cell culture in microfluidic devices. Biomaterials. 2009;30:4833–4841. doi: 10.1016/j.biomaterials.2009.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Walker G, Beebe D. A passive pumping method for microfluidic devices. Lab Chip. 2002;2:131–134. doi: 10.1039/b204381e. [DOI] [PubMed] [Google Scholar]
- [43].Montanez-Sauri S, Sung KE, Berthier E, Beebe DJ. Enabling screening in 3D microevnironment probing matrix and stromal effects on the morphology and proliferation of T47D breast carcinoma cells. Integrative Biology. 2013;5:631–640. doi: 10.1039/c3ib20225a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Skala MC, Riching KM, Gendron-Fitzpatrick A, Eickhoff J, Eliceiri KW, White JG, et al. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. PNAS. 2007;104:19494–19499. doi: 10.1073/pnas.0708425104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sung KE, Yang N, Pehlke C, Keely PJ, Eliceiri KW, Friedl A, et al. Transition to invasion in breast cancer: a microfluidic in vitro model enables examination of spatial and temporal effects. Integrative Biology. 2010;3:439–450. doi: 10.1039/c0ib00063a. [DOI] [PMC free article] [PubMed] [Google Scholar]