

Rice is a unique crop plant since it can survive under anoxia condition by flooding and also germinate and grow up to coleoptile through its distinctive adaptations. Despite several decades of research on this topic, the current knowledge on the molecular machinery of rice under anoxia is very limited. Therefore, we unraveled the possible regulatory mechanisms by resorting to systems biology approach which combines the metabolic modeling and transcriptome analysis. Such integrative analysis highlight the critical role of MYB, bZIP, ERF and ZnF transcription factors in up-regulating the fermentation and sucrose metabolism genes to generate sufficient energy for cellular growth.
Keywords: Anoxia, cis-elements, flux sampling, rice, systems biology, transcription factors, transcriptional regulation.
Abstract
The ability of rice to germinate under anoxia by extending the coleoptile is a highly unusual characteristic and a key feature underpinning the ability of rice seeds to establish in such a stressful environment. The process has been a focal point for research for many years. However, the molecular mechanisms underlying the anoxic growth of the coleoptile still remain largely unknown. To unravel the key regulatory mechanisms of rice germination under anoxic stress, we combined in silico modelling with gene expression data analysis. Our initial modelling analysis via random flux sampling revealed numerous changes in rice primary metabolism in the absence of oxygen. In particular, several reactions associated with sucrose metabolism and fermentation showed a significant increase in flux levels, whereas reaction fluxes across oxidative phosphorylation, the tricarboxylic acid cycle and the pentose phosphate pathway were down-regulated. The subsequent comparative analysis of the differences in calculated fluxes with previously published gene expression data under air and anoxia identified at least 37 reactions from rice central metabolism that are transcriptionally regulated. Additionally, cis-regulatory content analyses of these transcriptionally controlled enzymes indicate a regulatory role for transcription factors such as MYB, bZIP, ERF and ZnF in transcriptional control of genes that are up-regulated during rice germination and coleoptile elongation under anoxia.
Introduction
Rice is one of the few plant species that can survive submerged conditions. In particular, it can successfully germinate by extending a coleoptile underwater in the complete absence of oxygen (Magneschi and Perata 2009). Most of the early biochemical studies reported that even under anoxia, rice is able to metabolize reserve carbohydrates via glycolysis and fermentation to generate sufficient cellular energy, i.e. ATP needed for plant growth (Mocquot et al. 1981; Alpi and Beevers 1983; Mohanty et al. 1993; Guglielminetti et al. 1995). Microarray data show that this is associated with a strong up-regulation of sucrose and starch metabolism, glycolysis and fermentation genes (Lasanthi-Kudahettige et al. 2007). This work also showed many other significantly up- and down-regulated genes associated with various metabolic and signalling pathways. These findings highlight that rice globally reconfigures its molecular machinery under anoxic stress by selectively synthesizing the necessary transcripts coding for enzymes that produce and conserve energy.
Despite past efforts to characterize the biochemical adaptations of rice under anoxia, current knowledge on how oxygen deficiency is sensed and on the regulatory cascade that fine-tunes the transcriptional and translational changes is still very limited. To date, only the induction of GA-response-free RAmy3D under anoxic conditions (Loreti et al. 2003a, b) is the notable trait unravelled at the molecular level. Even with the availability of abundant high-throughput data, such limitations still exist mainly due to the lack of appropriate systematic frameworks to analyse and derive a valid hypothesis from them. In this regard, constraint-based in silico metabolic modelling and analysis can be helpful. Not only do they allow predictions concerning the physiological behaviour and metabolic states of an organism exposed to various environmental or genetic changes but they also serve as a scaffold to contextualize multiple ‘-omics’ data, thereby enabling biologically meaningful correlations to be identified (Lewis et al. 2012). As a result, several frameworks are now available to integrate metabolic models and high-throughput data such as transcriptomics (Becker and Palsson 2008; Colijn et al. 2009; Bordel et al. 2010), metabolomics (Mo et al. 2009; Selvarasu et al. 2012) and proteomics (Shlomi et al. 2008) within the context of systems biology. One such approach compares the flux levels simulated by the metabolic model with corresponding gene expression data resulting from environmental or genetic changes. This has, for example, highlighted key transcriptional mechanisms in Saccharomyces cerevisiae (Famili et al. 2003; Bordel et al. 2010; Osterlund et al. 2013). Therefore, following a similar method, here, we identified the transcriptionally regulated reactions in rice during its anoxic germination. Our approach used initial random flux sampling using the recently published rice central metabolic network (Lakshmanan et al. 2013), and subsequently compared the differences in flux levels with previously published gene expression data (Lasanthi-Kudahettige et al. 2007) between air and anoxia. Furthermore, to gain a deeper insight into the key regulatory mechanisms during anoxic adaptation, we also found the possible transcription factors (TFs) of transcriptionally regulated enzymes by analysing the distribution of putative cis-elements in their promoter regions.
Methods
Rice central metabolic network
We used the recently published rice central metabolic model (Lakshmanan et al. 2013) to characterize the metabolic states of germinating rice seeds under air and anoxia. The gene–protein reaction (GPR) mappings were updated for all reactions since the original model did not include the unique genes associated with enzymes having multiple isozymes across different cellular compartments. The updated model is provided in Supporting Information.
Microarray data
The raw transcriptome data generated by Lasanthi-Kudahettige et al. (2007) were first downloaded from the Gene Expression Omnibus (accession no. GSE6908) and normalized using the quantile method (Bolstad et al. 2003). Differentially expressed genes were then identified by a linear model (Wettenhall and Smyth 2004). The resulting P values were also corrected for multiple testing using Benjamini–Hochberg correction.
Random flux sampling
The artificial centring hit-and-run (ACHR) Monte Carlo sampling (Thiele et al. 2005; Mo et al. 2009) was utilized to sample uniformly the metabolic flux solution space under aerobic and anaerobic conditions with appropriate flux constraints. Under both conditions, we constrained sucrose uptake rate, oxygen uptake rate and cell growth rate with experimentally measured values (Lakshmanan et al. 2013). To make a fair comparison between both conditions, oxygen uptake and growth rates were normalized with respect to sucrose uptake rate. The solution space was sampled with 100 000 randomly distributed points for 10 000 iterations in each simulation. In this study, the COBRA toolbox (Schellenberger et al. 2011) was utilized to implement the random flux sampling.
The differences in flux samples between aerobic and anaerobic conditions were quantified using a Z-score approach as described previously (Mo et al. 2009). In this approach, two random flux vectors, vj, one from each sample, i.e. aerobic and anaerobic, were chosen and the difference is calculated as follows:
This approach was repeated 10 000 times to create new flux differences, vj,diff, sampled with 10 000 points. From this flux difference sample, the sample mean, μj, and standard deviation, σj, were computed to calculate the Z-score as follows:
Finally, the absolute Z-scores were translated to P values using the normal cumulative distribution function and the reactions with P values <0.05 were deemed as statistically different between aerobic and anaerobic conditions.
Identification of transcriptionally regulated enzymes
Using the P values of transcriptome data and flux sampling, we identified the reactions that are transcriptionally regulated (Bordel et al. 2010). Briefly, if the flux and gene expression (for both up- and down-regulated) significantly changes in the same direction, the corresponding enzyme is classified as ‘transcriptionally regulated’. On the other hand, if the values significantly change in the opposite direction, the enzyme is classified as ‘metabolically regulated’. In the case of reactions with multiple isozymes, the gene expression was considered to be up- or down-regulated based on the expression values of the majority of the transcripts. For example, if a gene has more up-regulated transcripts than down-regulated ones, then it is considered as up-regulated.
Motif detection and identification of putative TFs
Promoter sequences [−1000, +200 nt relative to the transcription start site (TSS)] for transcriptionally up- and down-regulated genes related to anaerobic central metabolism were extracted from our in-house rice promoter sequence database. Known and novel promoter motifs were detected using the Dragon Motif Builder program with EM2 option (Huang et al. 2005). Thirty motifs were detected each time having a length of 8–10 nucleotides per detection at a threshold value of 0.875. Motif occurrences in over 50 % of the sequences at a threshold e value of ≤10−3 were considered as statistically overrepresented. Motif classes were identified by their matches in different plant Transcription Factor Binding databases such as TRANSFAC (Matys et al. 2003; http://www.gene-regulation.com), PLACE (Higo et al. 1999; http://www.dna.affrc.go.jp/htdocs/PLACE), AGRIS (Yilmaz et al. 2011; http://arabidopsis.med.ohio-state.edu/) and Osiris (Morris et al. 2008; http://www.bioinformatics2.wsu.edu/cgi-bin/Osiris/cgi/home.pl). A similar method was used for the extraction and detection of motifs in the negative sets.
Results
Random sampling reveals significant differences in rice central metabolism between aerobic and anaerobic conditions
We sampled the plausible metabolic states of rice coleoptile in air and under anoxia using ACHR Monte Carlo sampling to estimate the range of possible steady-state flux values through each of the reactions in the rice model (see Methods). The resulting probability distributions of individual reaction fluxes revealed significant differences in central metabolic pathways such as glycolysis, the tricarboxylic acid (TCA) cycle, the pentose phosphate pathway and oxidative phosphorylation between aerobic and anaerobic conditions [see Supporting Information].
Under anoxia, the TCA cycle, the pentose phosphate pathway and oxidative phosphorylation had only a small range of fluxes, with some even having zero flux due to the imposed capacity constraints, i.e. the absence of oxygen exchange [see Supporting Information]. As a result, the fluxes across various amino acid and lipid synthetic pathways were also severely restricted under anoxic conditions (data not shown). On the other hand, random sampling allowed us to observe the possibility of high fluxes through cytosolic glycolysis and fermentation, mainly to produce all the ATP required for anaerobic cell growth. Other energy-producing pathways such as oxidative phosphorylation and the TCA cycle are grossly impaired under such conditions (Mohanty et al. 1993; Lakshmanan et al. 2013).
Transcriptionally regulated reactions during anaerobic adaptation
We compared the differences in flux samples and gene expression data between air and anoxia, thereby identifying the reactions that are likely to be transcriptionally regulated (see Methods). Overall, among the 63 reactions in rice central metabolism, 37 and 5 exhibit transcriptional and metabolic regulation, respectively. The remaining 38 reactions could not be classified in any of these categories as they exhibited an insignificant change in either flux or gene expression. The complete lists of transcriptionally and metabolically regulated reactions are provided in Supporting Information.
Most of the reactions in the TCA cycle, oxidative phosphorylation and the pentose phosphate pathway show down-regulation in both flux and gene expression under anaerobic conditions while several reactions of sucrose metabolism including sucrose synthase (SUS), fructokinase and nucleoside diphosphate kinase are significantly up-regulated at the transcriptional level (Fig. 1). Only invertase in sucrose metabolism shows down-regulation in both flux and gene expression, confirming that rice preferably utilizes SUS to metabolize sucrose under anaerobic conditions. The use of SUS conserves ATP usage (Guglielminetti et al. 1995; Lakshmanan et al. 2013).
Figure 1.
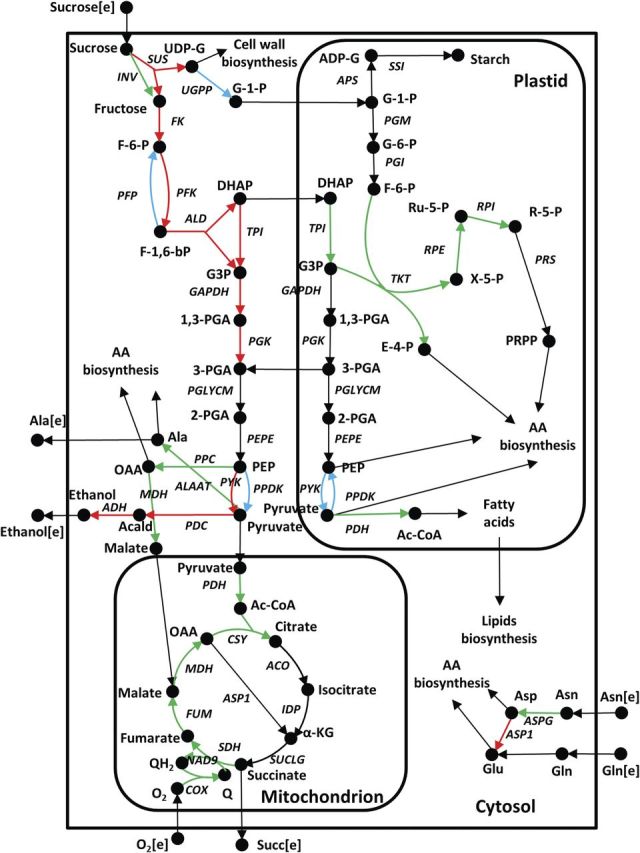
Central metabolic reactions of rice showing transcriptional and metabolic regulation under anoxia. Green, red and blue colours indicate the transcriptionally down-, up- and metabolically regulated enzymes, respectively. Reactions with black arrows represent enzymes whose regulation mechanism has not been investigated or identified in this study. Metabolite abbreviations are as follows: α-KG, α-ketoglutarate; 1,3-PGA, 1,3-diphosphoglycerate; 2-PGA, 2-phosphoglycerate; 3-PGA, 3-phosphoglycerate; AA, amino acids; Acald, acetaldehyde; Ac-CoA, acetyl-coenzyme A; ADP-G, ADP-glucose; Ala, alanine; Asn, asparagine; Asp, aspartate; DHAP, dihydroxyacetone phosphate; E-4-P, erythrose-4-phosphate; F-6-P, fructose-6-phosphate; F-1,6-bP, fructose-1,6-bisphosphate; G-1-P, glucose-1-phosphate; G3P, glyceraldehyde-3-phosphate; G-6-P, glucose-6-phosphate; Gln, glutamine; Glu, glutamate; OAA, oxaloacetate; PEP, phosphoenolpyruvate; PRPP, phosphoribosyl pyrophosphate; Q, ubiquinone; QH2, ubiquinol; R-5-P, ribose-5-phosphate; Ru-5-P, ribulose-5-phosphate; UDP-G, UDP-glucose; X-5-P, d-xylulose-5-phosphate. Enzyme abbreviations are as follows: ACO, aconitase; ADH, alcohol dehydrogenase; ALAAT, alanine aminotransferase; ALD, aldolase; APS, glucose-1-phosphate adenylyltransferase; ASP1, aspartate aminotransferase; ASPG, asparaginase; COX, cytochrome c oxidase; CSY, citrate synthase; FK, fructokinase; FUM, fumarase; GAPDH, glyceraldehyde phosphate dehydrogenase; IDP, isocitrate dehydrogenase (NADP-dependent); INV, invertase; MDH, malate dehydrogenase; NAD9, NADH dehydrogenase; PDC, pyruvate decarboxylase; PDH, pyruvate dehydrogenase; PEPE, phosphoenolpyruvate enolase; PFK, 6-phosphofructokinase; PFP, PPi-dependent phosphofructokinase; PGI, phosphoglucoisomerase; PGK, phosphoglycerate kinase; PGLYCM, phosphoglucomutase; PGM, phosphoglucomutase; PPC, phosphoenolpyruvate carboxylase; PPDK, pyruvate orthophosphate dikinase; PRS, ribose-phosphate diphosphokinase; PYK, pyruvate kinase; RPE, ribose-5-phosphate epimerase; TKT, transketolase; TPI, triose phosphate isomerase; SDH, succinate dehydrogenase; SSI, starch synthase; SUCLG, succinyl-CoA ligase; SUS, sucrose synthase; UGPP, UDP-glucose pyrophosphorylase.
All the reactions in the fermentation pathway including pyruvate decarboxylase (PDC), alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH) were up-regulated (Fig. 1). In rice, both alcohol and aldehyde dehydrogenases have multiple isozymes and a few of them such as ADH1, ADH2 and ALDH2a showed a drastic increase in gene expression (Lasanthi-Kudahettige et al. 2007). Interestingly, the up-regulation of ALDH at both flux and transcriptional level is in very good agreement with earlier experimental reports on both tolerant and intolerant lines of rice under hypoxic conditions during submergence (Nakazono et al. 2000; Ismail et al. 2012; Miro et al. 2013), indicating that ALDH is likely to play a key role in detoxifying the excess acetaldehyde from PDC that cannot be metabolized via ADH. In this regard, Lasanthi-Kudahettige et al. (2007) hypothesized that the acetate resulting from ALDH can enter the TCA cycle to further fuel amino acid synthesis pathways. However, our flux analysis revealed that the acetate from ALDH primarily fuels lipid biosynthesis in plastids, as fatty acid synthesis needs sufficient carbon flux to synthesize the required saturated fatty acids under anoxia.
Unlike other central metabolic pathways, the glycolytic reactions did not show an overall up- or down-regulation in gene transcription and fluxes (Fig. 1). Out of a total of 25 reactions, only 11 showed transcriptional regulation. Among the remaining 14, 5 were oppositely correlated between flux and gene expression, indicating that these enzymes are most likely to be metabolically regulated. Plastidic pyruvate kinase, UDP-glucose pyrophosphorylase, PPi-dependent phosphofructokinase (PFP), and cytosolic and plastidic pyruvate phosphate dikinase (PPDK) are the five enzymes with metabolic regulation. Such observations are in good agreement with the earlier hypothesis by Plaxton (1996), who suggested that PK, PPDK, PFK and PFP are involved in the fine control of plant glycolysis and are most likely to be regulated based on the variation in substrate(s) and cofactor concentrations, pH and other metabolite effectors.
TFs associated with transcriptionally regulated genes
To identify the potential TFs involved in the transcriptional control of the transcriptionally regulated reactions, we performed promoter analysis of the corresponding genes. In total, promoter sequences for 26 transcriptionally up-regulated genes and 27 down-regulated genes were used for cis-element detection [see Supporting Information]. This analysis identified several highly enriched putative cis-elements associated with different potential TFs for both up- and down-regulated transcriptionally controlled genes (Tables 1 and 2). A high enrichment of putative cis-elements such as AT-hook/PE-1-like, GT element-like, pyrimidine-box-like, GARE-like and MYb-box-like associated with MYB TFs was observed among both up- and down-regulated genes. However, the high enrichment of these cis-elements in the up-regulated genes signifies that MYB TFs especially play an important role in the transcriptional control of up-regulated genes under anoxia. Similarly, putative cis-elements such as AS-1/ocs-like and ABRE-like associated with bZIP TFs were also found in both up- and down-regulated genes but with a high percentage of occurrences only in up-regulated genes. Besides, a number of other putative cis-elements such as ERE-like/GCC-box-like and zinc finger binding element-like associated with ERF and ZnF TFs were also more overrepresented in the up-regulated genes albeit being present in both up- and down-regulated genes. Furthermore, we noticed a moderate enrichment of other ERE like-elements such as JA response element-like elements among the up-regulated genes (Table 1). Collectively, these results indicate that all these TFs, i.e. MYB, bZIP, ERF and ZnF, may work together in the transcriptional control of the up-regulated genes in rice central metabolism. Interestingly, we also noticed the enrichment of MBF1C binding element and CRT/DRE-like elements associated with MBF1C and CBF/DREB TFs only in down-regulated genes. This highlights the possibility that this TF could be involved in transcriptional control of down-regulated genes under anoxia (Table 2).
Table 1.
Potential cis-elements identified in the promoters of transcriptionally controlled up-regulated genes of rice seeds germinated under anoxia.
| Cis-elements | Motifs | Associated TFs | % (TIC), e value |
|---|---|---|---|
| AT-hook/PE1-like | TTTTTTCA | MYB (PF1) | 73 (12.78), 2e−004 |
| AATTTTTTT | MYB (PF1) | 65 (16.05), 3e−005 | |
| ATAAAAAAAA | MYB (PF1) | 58 (16.67), 0e+000 | |
| AAGAAAAAG | MYB (PF1) | 58 (13.91), 1e−004 | |
| AAAAATAC | MYB (PF1) | 54 (13.32), 1e−004 | |
| TTTTTTCTTT | MYB (PF1) | 50 (16.74), 3e−005 | |
| GT-element-like | TGGTTTGT | MYB (GT-1/GT-3b) | 81 (12.15), 1e−004 |
| TTTTTTCA | MYB (GT-1/GT-3b) | 73 (12.78), 2e−004 | |
| GGCTTGTG | MYB (GT-1/GT-3b) | 69 (11.62), 2e−000 | |
| AGGAAAAAG | MYB (GT-1/GT-3b) | 58 (13.91), 1e−004 | |
| AAATCATA | MYB (GT-1) | 62 (12.80), 8e−005 | |
| AAATCAAAT | MYB (GT-1) | 62 (13.69), 1e−004 | |
| TTTTTTCTTT | MYB (GT-1) | 50 (16.74), 3e−005 | |
| Pyrimidine-box-like | TTTTTTCA | MYB (R1, R2R3) | 73 (12.78), 2e−004 |
| CTTTTGCT | MYB (R1, R2R3) | 65 (12.33), 9e−005 | |
| GARE-like | AAAACAAA | MYB (R1, R2R3) | 58 (12.66), 2e−004 |
| MYB-box-like | TGGTTTAT | MYB (R2R3) | 81 (12.15), 1e−004 |
| TGGTTTGT | MYB (R2R3) | 81 (12.15), 1e−004 | |
| AACTTGTT | MYB (R2R3) | 54 (13.18), 3e−005 | |
| As-1/ocs-like | TTTTTTCA | bZIP (Gr. D, I, S) | 73 (12.78), 2e−004 |
| AAATCATA | bZIP (Gr. D, I, S) | 62 (12.80), 8e−005 | |
| ATGAAAAAG | bZIP (Gr. D, I, S) | 58 (13.91), 1e−004 | |
| ABRE-like | AAATCAAAT | bZIP (Gr. A) | 62 (12.80), 8e−005 |
| CTTTGCCA | bZIP (Gr. A) | 58 (13.43), 1e−004 | |
| GAGCGCCA | bZIP (Gr. A) | 54 (12.18), 3e−004 | |
| RSG binding element-like | AACTTGTT | bZIP | 54 (13.18), 3e−005 |
| CAMTA3 binding site-like | GAAGAAAA | bZIP | 63 (14.30), 2e−004 |
| RISbZ1 binding site-like | AAAACAAA | bZIP (RISbZ1) | 58 (12.66), 2e−004 |
| CAMTA3 binding site-like | GAGAAAGAA | bZIP | 58 (14.52), 1e−004 |
| AAGAAGAG | bZIP | 50 (13.41), 2e–004 | |
| Zinc finger binding element-like | AAGAAGAG | ZnF (ZCT1, ZCT2, ZCT3) | 62 (13.69), 1e−004 |
| AAATCATA | ZnF (ZCT1, ZCT2, ZCT3) | 62 (12.80), 8e−005 | |
| AAATCAAAT | ZnF (ZCT1, ZCT2, ZCT3) | 50 (13.41), 2e−004 | |
| ERE-like (JA response element-like) | AAATCATA | ERF (Gr. VI, VIII, IX) | 62 (12.80), 8e−005 |
| AAATCAAAT | ERF (Gr. VI, VIII, IX) | 50 (13.41), 2e−004 | |
| GCC-box-like | CTCCGCCGC | ERF (I, IV, VII, X) | 50 (15.38), 2e−005 |
| AuxRe-like | CTTTTGCT | ARF | 65 (12.33), 9e−005 |
| CTTTGCCA | ARF | 58 (13.43), 1e−004 | |
| AAAAG/element-like | CTTTTGCT | DOF (Dof1/4/11/22) | 65 (12.33), 9e−005 |
| AAGAAAAAG | DOF (Dof1/4/11/22) | 58 (13.43), 1e−004 | |
| MYC-box-like | TGCTACTC | bHLH (JAMYC2) | 65 (11.96), 1e−004 |
| ARR10 binding element-like | AAATCATA | ARR-B (ARR10) | 62 (13.69), 1e−004 |
| TATA-box-like | TATAAATT | TBP | 96 (12.32), 3e−005 |
| DBP element-like | AAAAATAC | DBP | 54 (13.32), 1e−004 |
| DBP1 element-like | AATATATTA | DBP1 | 50 (15.09), 8e−005 |
Table 2.
Potential cis-elements identified in the promoters of transcriptionally controlled down-regulated genes of rice seeds germinated under anoxia.
| Cis-elements | Motifs | Associated TFs | % (TIC), e value |
|---|---|---|---|
| AT-hook/PE1-like | ATATTTTTAT | MYB (PF1) | 59 (16.39), 6e−005 |
| TTTAAAAAA | MYB (PF1) | 59 (16.42), 2e−005 | |
| GT-element-like | ATTGGCTA | MYB (GT-1) | 56 (12.42), 2e−004 |
| MYB-box-like | AAAATCCA | MYB (R2R3, MCB1/2) | 70 (13.11), 2e−004 |
| As-1/ocs-like | TCGTCGCG | bZIP (Gr. D, I, S) | 63 (13.21), 0e+000 |
| ACGTGTCA | bZIP (Gr. D, I, S) | 59 (11.79), 3e−004 | |
| AGACGTTG | bZIP (Gr. D, I, S) | 56 (11.91), 3e−005 | |
| ABRE-like | ACGTGACA | bZIP (Gr. A) | 59 (11.79), 3e−004 |
| TCGCCGGC | bZIP (Gr. A) | 59 (13.20), 5e−005 | |
| ER stress RE-like | ATTGGCTA | bZIP (Gr. D) | 56 (12.42), 2e−004 |
| RISbZ1 binding site-like | AAAACAAA | bZIP (RISbZ1) | 58 (12.66), 2e−004 |
| AuxRe-like | ACTACTAT | ARF1 | 67 (12.28), 9e−005 |
| TCGTCGCG | ARF1 | 63 (13.21), 0e+000 | |
| ACGTGACA | ARF1 | 59 (11.79), 3e−004 | |
| AATCCTTT | ARF1 | 56 (13.21), 9e−005 | |
| GAGA element-like | CTCCTCTC | GAGA-binding factor BBR/BPC2 | 63 (14.39), 7e−004 |
| TCCTCTAT | GAGA-binding factor BBR | 52 (13.62), 4e−004 | |
| GGGAGAGGG | GAGA-binding factor BBR | 52 (15.63), 3e−005 | |
| DBP1 element-like | TTTATTTT | DBP1 | 85 (13.63), 2e−004 |
| ACATTAAA | DBP1 | 78 (12.88), 2e−004 | |
| AAATAATA | DBP1 | 62 (13.44), 9e−005 | |
| GCC-box-like | GGCGGCGGC | ERF (I, IV, VII, X) | 70 (15.92), 1e−004 |
| CCGCCGCC | ERF (I, IV, VII, X) | 56 (13.95), 3e−004 | |
| ARR10 binding element-like | AAAATCCA | ARR-B (ARR10) | 70 (13.11), 2e−004 |
| AATCCTTT | ARR-B (ARR10, ARR5, ARR1) | 56 (13.21), 9e−005 | |
| CRT/DRE-like | TCGTCGCG | CBF1/DREB | 63 (13.21), 0e+000 |
| AAAGG element-like | AATCCTTT | DOF | 56 (13.21), 9e−005 |
| ATTTAAAGA | DOF (Dof1/4/11/22) | 52 (14.11), 9e−005 | |
| Zinc finger binding element-like | GAGGAGGAG | ZnF | 56 (16.04), 6e−005 |
| MBF1C binding element-like | GAGGAGGAG | MBF1C | 56 (16.04), 6e−005 |
| TATA-box-like | TTTTATATA | TBP | 63 (15.28), 2e−004 |
| DBP element-like | ATATTTTTAT | DBP | 59 (16.39), 6e−005 |
Motif detection in negative sets
To ensure that our cis-element analysis did not identify an excessive number of false positives, we sought motifs from the promoters of a similar number of genes that are not anoxia specific using the same protocol (see Methods). For this purpose, we used two negative datasets: (i) randomly selected genes from the non-differentially expressed gene list under anoxia (Lasanthi-Kudahettige et al. 2007) (negative Set 1) and (ii) up-regulated genes associated with drought response in rice (Zhang et al. 2012) (negative Set 2). The list of genes from the negative sets and the results of this analysis are provided in Supplementary Information. Using the negative control Set 1, we identified a few common motifs such as MYB, bZIP and DOF, and their total enrichment was much lower compared with the motifs detected in transcriptionally up- or down-regulated genes of anoxia (Tables 1, 2 and Supplementary Information). Similarly, the motif analysis of negative Set 2 also revealed a significantly different cis-element enrichment pattern where most of the identified TFs such as MYB, bZIP, ERF, NAC and MYC have been experimentally confirmed to play a regulatory role in drought stress response (Shinozaki and Yamaguchi-Shinozaki 2007; Zhang et al. 2012). Collectively, these results clearly demonstrate that our cis-element method identifies reasonably precise motifs that are specific to anoxic stress.
Discussion
Our study analysed the plausible metabolic states of rice under aerobic and anaerobic conditions using random sampling and compared their differences. We also compared the changes in flux levels of individual enzymes in the central metabolism of rice grown in the two conditions with that of gene expression and identified the transcriptionally controlled reactions. Accordingly, the major contribution of the current work is the demonstration of the utility of random flux sampling and gene expression data for elucidating metabolic differences and the identification of corresponding putative TFs responsible for such changes under the stress conditions.
Monte Carlo flux sampling has been shown to be an effective tool to analyse and compare the plausible solution space of a metabolic network across different environmental/genetic conditions (Famili et al. 2003; Bordel et al. 2010; Osterlund et al. 2013). When compared with the commonly used constraint-based analysis methods such as flux balance analysis (FBA) and minimization of metabolic adjustment, which predict just a single flux distribution based on a particular objective function, random sampling allows us to account for uncertainty in the flux distributions as it uniformly samples the possible solution space in an unbiased manner and, thus, potentially minimizes the possibility of overestimating the actual flux differences between the two conditions. This can be clearly exemplified by comparing the results of previous work (Lakshmanan et al. 2013), which utilized FBA for simulating flux differences between air and anoxia, with our current results. Although both studies show that most of the central metabolic reactions possess different flux levels in aerobic and anaerobic conditions, flux sampling can provide confidence scores to the reactions, thus allowing us to consider the reactions with the most plausible fluxes. Furthermore, by comparing the significant flux differences with gene expression data, we determined the reactions that are most probably regulated at the transcriptional level. Most importantly, we revealed that although the glycolytic reactions are not transcriptionally regulated, genes coding for reactions upstream and downstream of glycolysis, i.e. sucrose metabolism and fermentation, respectively, are regulated transcriptionally. It will be important now to compare and contrast such findings with anoxia-intolerant higher plants such as Arabidopsis thaliana, wheat and maize to analyse whether the corresponding gene orthologues are commonly regulated at a similar level.
In recent years, in silico promoter analysis of differentially expressed genes from microarray data has improved our understanding of the transcriptional regulation of gene expression markedly. Several in silico analyses on the promoter cis-elements and their cognate TFs have been experimentally verified for predicted functions, highlighting its reliability (Meier et al. 2008; Yun et al. 2010; Stamm et al. 2012). Therefore, in this study, we analysed the cis-regulatory content of transcriptionally controlled reactions through such a method and unravelled the possible involvement of a number of TFs in transcriptional control of sucrose metabolism and fermentation under anoxia. Among all the putative cis-elements identified, the elements associated with MYB family TFs were found to be highly enriched in transcriptionally up-regulated genes. Coincident with these observations a number of MYB family genes are also up-regulated under anoxia (Table 3). MYB proteins represent important plant TFs and are found to be involved in various developmental and physiological processes including transcriptional activation, kinase activity, protein binding and transcription repressor activation under abiotic and biotic stresses (Dubos et al. 2010). In rice, a recent analysis highlighted that 98.70 % of total MYB proteins are fully involved in transcriptional activation (Katiyar et al. 2012). Furthermore, the role of MYB TFs that bind specifically to MYB box/GT element during hypoxic and anoxic responses has already been reported through promoter analysis, both computationally and experimentally (Dolferus et al. 2003; Mohanty et al. 2005, 2012). Specifically, the AtMYB2 in Arabidopsis has been shown to be a key regulator of the ADH1 promoter under low oxygen conditions (Hoeren et al. 1998). When this AtMYB2 was driven by a constitutive promoter, it was able to transactivate ADH1 expression not only in Arabidopsis but also in Nicotiana plumbaginifolia and Pisum sativum. Collectively, these results fully support the hypothesis that MYB TFs play an important role in the up-regulation of sucrose metabolism and fermentation enzymes at the transcriptional level.
Table 3.
List of anoxia-stressed up-regulated TFs with potential significance to the pattern of cis-element enrichment among the up-regulated genes.
| Family | Locus_ID (Annotation) | Fold increase |
|---|---|---|
| MYB/MYB-related | Os02g0706400 (Myb-related, similar to Radialis) | 9.0 |
| Os06g0728700 (Homeodomain-like protein) | 7.0 | |
| Os08g0151000 (Myb-like, SHAQKYF class) | 7 | |
| Os01g0524500 (Myb-like, SHAQKYF class) | 6 | |
| Os01g0863300 (similar to MCB2 protein) | 4 | |
| Os08g0549000 (similar to MybHv5) | 3 | |
| Os05g0459000 (c-Myb protein) | 2 | |
| Os04g0480300 (Myb-like protein) | 2 | |
| bZIP | Os09g0306400 (bZIP-1 domain protein) | 16.0 |
| Os03g0336200 (RF2b transcription factor) | 6.0 | |
| Os06g0662200 (bZIP-1 domain protein) | 4.0 | |
| Os01g0867300 (G-box binding factor) | 3.0 | |
| Os05g0489700 (similar to BZO2H3) | 2.0 | |
| Os05g0129300 (bZIP protein) | 2.0 | |
| Os05g0569300 (G-box binding factor) | 2.0 | |
| ERF | Os03g0341000 (similar to RAP2.2) | 29.0 |
| Os01g0131600 (similar to PTI6, pathogenesis-related) | 3.0 | |
| Os06g0604000 (similar to ERF1 and ERF3) | 3.0 | |
| ZnF | Os05g0525900 (similar to Zinc finger transcription factor PEI1) | 21.0 |
| Os09g0560900 (zinc finger, C2H2-like domain containing protein) | 2.0 | |
| Zinc finger, CCCH-type domain containing protein. (Os04t0663200-01) (similar to OSIGBa0099L20.3 protein) | 2.0 | |
| Os02g0672100 (zinc finger, C2H2-type domain containing protein) | 2.0 | |
| Os09g0560900 (zinc finger, C2H2-like domain containing protein) | 2.0 | |
| ARF | Os04g0671900 (similar to auxin response factor) | 2.0 |
| Os06g0677800 (similar to auxin response factor) | 2.0 | |
| DOF | Os05g0112200 (Dof domain, zinc finger family protein, expressed) | 2.0 |
| bHLH | Os11t0523700 (similar to transcription factor ICE1 (Inducer of CBF expression 1) (basic helix–loop–helix protein 116) (bHLH116) | 3.0 |
| Os02t0433600 (helix–loop–helix DNA-binding domain containing protein) | 2.0 | |
| Pseudo-ARR-B | Os11t0157600 (similar to timing of CAB expression) | 3.0 |
As mentioned above, our promoter analysis also highlighted significant overrepresentation of several other motifs associated with bZIP, ERF and ZnF TFs in the transcriptionally up-regulated genes. In this regard, an increase in transcript levels of bZIP TF (AtbZIP50) in anoxia-exposed root cultures of Arabidopsis (Klok et al. 2002), the up-regulation of an ABRE-binding bZIP TF (OsABF1) in rice shoot and root under anoxic treatment (Amir Hossain et al. 2010) and the up-regulation of a number of bZIP TFs in rice anoxic coleoptile (Table 3) support the view that bZIP could orchestrate the transcription of up-regulated genes upon anoxic stress. The major role of ERF TF has been identified as a positive regulator of Sub1A expression in a flood-tolerant rice variety during hypoxic conditions caused by submergence (Xu et al. 2006). However, the anoxia-tolerant rice variety ‘Nipponbare’ that germinates and elongates under anoxia does not have this gene. Surprisingly, the presence of highly enriched putative GCC-box-like/ERE-like cis-elements in the promoters of all anoxia up-regulated genes in this variety of rice highlights the possiblity that these TFs might also control the transcription of relevant genes as it is in Arabidopsis under hypoxic conditions (Hinz et al. 2010; Licausi et al. 2010, 2011). Unlike the ERF TFs, the role of ZnF TF in response to submergence/anoxia is not yet known. However, matching with our identification of ZnF binding site-like elements in transcriptionally up-regulated genes, the increase in expression of ZnF TF genes (Table 3) in response to hypoxia/anoxia in both rice and Arabidopsis allows us to speculate on its potential regulatory role in transcriptional control under oxygen stress (Loreti et al. 2005; Pandey and Kim 2012).
As mentioned earlier, we utilized the promoter sequences of 26 transcriptionally up-regulated and 27 down-regulated genes for the cis-element detection in this study. When compared with the normal promoter analysis method that considers the whole set of differentially expressed genes, the current study takes into account relatively few genes. Therefore, in order to argue that this small set of genes is sufficient to generate biologically meaningful results, we first compared the TFs identified in this study with previous work that considered the whole list of up-regulated (842) and down-regulated (1794) genes during anoxic adaptation (Mohanty et al. 2012). Interestingly, the comparison result revealed that the TFs associated with up- and down-regulated genes are the same between two studies where the motif enrichment scores of certain TFs such as MYB, bZIP, ZnF and ERF are much higher in the current study, highlighting the possible role of these TFs in anoxic adaptation. To further confirm our findings, we also analysed the individual promoter sequences of four of the key transcriptionally regulated enzymes, i.e. SUS, PDC, ADH and ALDH, and identified the same TFs as fully responsible for their transcription (Fig. 2). Therefore, based on these motif analysis results and experimental evidence from the literature, we hypothesize a positive role of MYB together with bZIP, ERF and ZnF in the transcriptional control of sucrose metabolism and fermentation during germination and coleoptile elongation of rice under anoxia.
Figure 2.
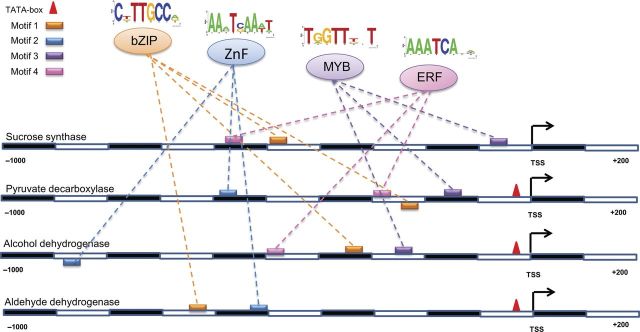
Presence of common putative cis-elements in the key transcriptionally regulated genes. The presence of potential putative cis-elements and their cognate known TFs are shown in different strands of the promoter region (−1000, +200 nt relative to TSS) of the key genes. Each putative cis-element/motif is represented by its consensus logo. TATA boxes are located between 25 and 30 nt upstream from the TSS.
The current methods we used successfully identified several transcriptionally regulated reactions and their related TFs, some of which are experimentally confirmed. However, the overall results of this approach await experimental validation since the current prediction relies on several assumptions concerning model completeness, constraints used during simulation and the statistical cut-off values chosen for comparative analysis. In our work, we utilized the central model to simulate the differences in metabolic fluxes between air and anoxia. It should be noted that although this model predicts the overall cellular phenotype quite accurately (Lakshmanan et al. 2013), it may not capture the global changes in cellular metabolism. This reservation is needed since the model does not take account of the secondary metabolic pathways. Moreover, we have utilized only a few external fluxes, i.e. sucrose uptake, O2 uptake and growth yield, as the sole constraints to identify the internal flux distributions during flux sampling simulations. In such cases, the internal fluxes of some reactions are determined with statistically low confidence scores due to the possibility of multiple flux solutions. Such limitations can be overcome by the use of C13 flux measurements as constraints to the internal reactions. Therefore, the list of transcriptionally regulated reactions and the TFs identified in this study need further confirmation by direct experiments.
Conclusions
In this study, we presented a combined in silico modelling and gene expression data analysis framework to identify transcriptionally regulated reactions in rice central metabolism. Random flux sampling simulations highlighted significant changes in flux levels across oxidative phosphorylation, the TCA cycle, the pentose phosphate pathway, glycolysis, sucrose metabolism and fermentation. The subsequent comparative analysis of changes in flux levels and gene expression between aerobic conditions identified 37 reactions from these pathways that are regulated at the transcriptional level. The motif enrichment analysis of transcriptionally regulated enzymes highlighted the potential involvement of TFs MYB, bZIP, ERF and ZnF in controlling the transcription of sucrose metabolism and fermentation genes under anaerobic conditions. Thus, the current study successfully demonstrated how condition-specific gene expression data can be exploited to narrow down the metabolic changes in rice under anaerobic adaptations and in silico analysis to help us unravel the relevant transcriptional mechanisms.
Sources of Funding
This work was supported by the National University of Singapore, the Competitive Research Programme of the National Research Foundation of Singapore (NRF-CRP5-2009-03), the Biomedical Research Council of A*STAR (Agency for Science, Technology and Research), Singapore, and a grant from the Next-Generation BioGreen 21 Program (SSAC, No. PJ009520), Rural Development Administration, Republic of Korea.
Contributions by the Authors
M.L., B.M. and D.Y.L. conceived and designed the study. M.L. carried out flux sampling and gene expression data analysis. B.M. carried out the ab initio analysis of cis-regulatory elements. M.L., B.M. and D.Y.L. wrote the manuscript. S.H.L. and S.H.H. provided critical comments and helped in revising the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest Statement
None declared.
Supporting Information
The following Supporting Information is available in the online version of this article –
File S1. Table. Rice central model with updated GPR associations.
File S2. Diagram. Sampling histogram of glycolysis, pentose phosphate pathway, TCA cycle, oxidative phosphorylation and sucrose metabolism under aerobic and anaerobic conditions.
File S3. Table. List of rice central metabolic reactions and their flux differences with P values. Expression patterns of corresponding gene louses and type of regulation, i.e. transcriptional or metabolic, are also listed.
File S4. Table. List of genes used in negative sets.
File S5. Table. Potential cis-elements identified in the promoters of negative sets.
Acknowledgements
We thank Prof. Vladimir B. Bajic for the use of the Dragon Motif Builder Program. We also thank the associate editor Prof. Michael Jackson and anonymous referees for their useful comments.
Literature Cited
- Alpi A, Beevers H. Effects of O2 concentration on rice seedlings. Plant Physiology. 1983;71:30–34. doi: 10.1104/pp.71.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir Hossain M, Lee Y, Cho JI, Ahn CH, Lee SK, Jeon JS, Kang H, Lee CH, An G, Park PB. The bZIP transcription factor OsABF1 is an ABA responsive element binding factor that enhances abiotic stress signaling in rice. Plant Molecular Biology. 2010;72:557–566. doi: 10.1007/s11103-009-9592-9. [DOI] [PubMed] [Google Scholar]
- Becker SA, Palsson BO. Context-specific metabolic networks are consistent with experiments. PLoS Computational Biology. 2008;4:e1000082. doi: 10.1371/journal.pcbi.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Bordel S, Agren R, Nielsen J. Sampling the solution space in genome-scale metabolic networks reveals transcriptional regulation in key enzymes. PLoS Computational Biology. 2010;6:e1000859. doi: 10.1371/journal.pcbi.1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colijn C, Brandes A, Zucker J, Lun DS, Weiner B, Farhat MR, Cheng TY, Moody DB, Murray M, Galagan JE. Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Computational Biology. 2009;5:e1000489. doi: 10.1371/journal.pcbi.1000489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolferus R, Klok EJ, Delessert C, Wilson S, Ismond KP, Good AG, Peacock WJ, Dennis ES. Enhancing the anaerobic response. Annals of Botany. 2003;91:111–117. doi: 10.1093/aob/mcf048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubos C, Stracke R, Grotewold E, Weisshaar B, Martin C, Lepiniec L. MYB transcription factors in Arabidopsis. Trends in Plant Science. 2010;15:573–581. doi: 10.1016/j.tplants.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Famili I, Forster J, Nielsen J, Palsson BO. Saccharomyces cerevisiae phenotypes can be predicted by using constraint-based analysis of a genome-scale reconstructed metabolic network. Proceedings of the National Academy of Sciences of the USA. 2003;100:13134–13139. doi: 10.1073/pnas.2235812100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielminetti L, Perata P, Alpi A. Effect of anoxia on carbohydrate metabolism in rice seedlings. Plant Physiology. 1995;108:735–741. doi: 10.1104/pp.108.2.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo K, Ugawa Y, Iwamoto M, Korenaga T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Research. 1999;27:297–300. doi: 10.1093/nar/27.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz M, Wilson IW, Yang J, Buerstenbinder K, Llewellyn D, Dennis ES, Sauter M, Dolferus R. Arabidopsis RAP2.2: an ethylene response transcription factor that is important for hypoxia survival. Plant Physiology. 2010;153:757–772. doi: 10.1104/pp.110.155077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeren FU, Dolferus R, Wu Y, Peacock WJ, Dennis ES. Evidence for a role for AtMYB2 in the induction of the Arabidopsis alcohol dehydrogenase gene (ADH1) by low oxygen. Genetics. 1998;149:479–490. doi: 10.1093/genetics/149.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E, Yang L, Chowdhary R, Kassim A, Bajic VB. An algorithm for ab-initio DNA motif detection. In: Bajic VB, Tan TW, editors. Information processing and living system. London: World Scientific, Imperial College Press; 2005. pp. 611–614. [Google Scholar]
- Ismail AM, Johnson DE, Ella ES, Vergara GV, Baltazar AM. Adaptation to flooding during emergence and seedling growth in rice and weeds, and implications for crop establishment. AoB PLANTS. 2012;2012:pls019. doi: 10.1093/aobpla/pls019. doi:10.1093/aobpla/pls019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar A, Smita S, Lenka SK, Rajwanshi R, Chinnusamy V, Bansal KC. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genomics. 2012;13:544. doi: 10.1186/1471-2164-13-544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klok EJ, Wilson IW, Wilson D, Chapman SC, Ewing RM, Somerville SC, Peacock WJ, Dolferus R, Dennis ES. Expression profile analysis of the low-oxygen response in Arabidopsis root cultures. The Plant Cell. 2002;14:2481–2494. doi: 10.1105/tpc.004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmanan M, Zhang Z, Mohanty B, Kwon JY, Choi HY, Nam HJ, Kim DI, Lee DY. Elucidating rice cell metabolism under flooding and drought stresses using flux-based modeling and analysis. Plant Physiology. 2013;162:2140–2150. doi: 10.1104/pp.113.220178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasanthi-Kudahettige R, Magneschi L, Loreti E, Gonzali S, Licausi F, Novi G, Beretta O, Vitulli F, Alpi A, Perata P. Transcript profiling of the anoxic rice coleoptile. Plant Physiology. 2007;144:218–231. doi: 10.1104/pp.106.093997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis NE, Nagarajan H, Palsson BO. Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nature Reviews Microbiology. 2012;10:291–305. doi: 10.1038/nrmicro2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licausi F, van Dongen JT, Giuntoli B, Novi G, Santaniello A, Geigenberger P, Perata P. HRE1 and HRE2, two hypoxia-inducible ethylene response factors, affect anaerobic responses in Arabidopsis thaliana. The Plant Journal. 2010;62:302–315. doi: 10.1111/j.1365-313X.2010.04149.x. [DOI] [PubMed] [Google Scholar]
- Licausi F, Kosmacz M, Weits DA, Giuntoli B, Giorgi FM, Voesenek LA, Perata P, van Dongen JT. Oxygen sensing in plants is mediated by an N-end rule pathway for protein destabilization. Nature. 2011;479:419–422. doi: 10.1038/nature10536. [DOI] [PubMed] [Google Scholar]
- Loreti E, Alpi A, Perata P. Alpha-amylase expression under anoxia in rice seedlings: an update. Russian Journal of Plant Physiology. 2003a;50:737–742. [Google Scholar]
- Loreti E, Yamaguchi J, Alpi A, Perata P. Gibberellins are not required for rice germination under anoxia. Plant and Soil. 2003b;253:137–143. [Google Scholar]
- Loreti E, Poggi A, Novi G, Alpi A, Perata P. A genome-wide analysis of the effects of sucrose on gene expression in Arabidopsis seedlings under anoxia. Plant Physiology. 2005;137:1130–1138. doi: 10.1104/pp.104.057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magneschi L, Perata P. Rice germination and seedling growth in the absence of oxygen. Annals of Botany. 2009;103:181–196. doi: 10.1093/aob/mcn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis OV, Kloos DU, Land S, Lewicki-Potapov B, Michael H, Munch R, Reuter I, Rotert S, Saxel H, Scheer M, Thiele S, Wingender E. TRANSFAC: transcriptional regulation, from patterns to profiles. Nucleic Acids Research. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Gehring C, MacPherson CR, Kaur M, Maqungo M, Reuben S, Muyanga S, Shih MD, Wei FJ, Wanchana S, Mauleon R, Radovanovic A, Bruskiewich R, Tanaka T, Mohanty B, Itoh T, Wing R, Gojobori T, Sasaki T, Swarup S, Hsing Y, Bajic VB. The promoter signatures in rice LEA genes can be used to build a co-expressing LEA gene network. Rice. 2008;1:177–187. [Google Scholar]
- Miro B, Entila F, Ismail AM. Role of aldehyde dehydrogenase in submergence tolerance during anaerobic germination in rice. 11th ISPA Conference 2013; Los Banos. 2013. [Google Scholar]
- Mo ML, Palsson BO, Herrgard MJ. Connecting extracellular metabolomic measurements to intracellular flux states in yeast. BMC Systems Biology. 2009;3:37. doi: 10.1186/1752-0509-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocquot B, Prat C, Mouches C, Pradet A. Effect of anoxia on energy charge and protein synthesis in rice embryo. Plant Physiology. 1981;68:636–640. doi: 10.1104/pp.68.3.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty B, Wilson PM, Aprees T. Effects of anoxia on growth and carbohydrate-metabolism in suspension-cultures of soybean and rice. Phytochemistry. 1993;34:75–82. [Google Scholar]
- Mohanty B, Krishnan SP, Swarup S, Bajic VB. Detection and preliminary analysis of motifs in promoters of anaerobically induced genes of different plant species. Annals of Botany. 2005;96:669–681. doi: 10.1093/aob/mci219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty B, Herath V, Wijaya E, Yeo HC, de Los Reyes BG, Lee DY. Patterns of cis-element enrichment reveal potential regulatory modules involved in the transcriptional regulation of anoxia response of japonica rice. Gene. 2012;511:235–242. doi: 10.1016/j.gene.2012.09.048. [DOI] [PubMed] [Google Scholar]
- Morris RT, O'Connor TR, Wyrick JJ. Osiris: an integrated promoter database for Oryza sativa L. Bioinformatics. 2008;24:2915–2917. doi: 10.1093/bioinformatics/btn537. [DOI] [PubMed] [Google Scholar]
- Nakazono M, Tsuji H, Li Y, Saisho D, Arimura S, Tsutsumi N, Hirai A. Expression of a gene encoding mitochondrial aldehyde dehydrogenase in rice increases under submerged conditions. Plant Physiology. 2000;124:587–598. doi: 10.1104/pp.124.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterlund T, Nookaew I, Bordel S, Nielsen J. Mapping condition-dependent regulation of metabolism in yeast through genome-scale modeling. BMC Systems Biology. 2013;7:36. doi: 10.1186/1752-0509-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey DM, Kim S-R. Identification and expression analysis of hypoxia stress inducible CCCH-type zinc finger protein genes in rice. Journal of Plant Biology. 2012;55:489–497. [Google Scholar]
- Plaxton WC. The organization and regulation of plant glycolysis. Annual Review of Plant Physiology and Plant Molecular Biology. 1996;47:185–214. doi: 10.1146/annurev.arplant.47.1.185. [DOI] [PubMed] [Google Scholar]
- Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, Kang J, Hyduke DR, Palsson BØ. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nature Protocols. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvarasu S, Ho YS, Chong WP, Wong NS, Yusufi FN, Lee YY, Yap MG, Lee DY. Combined in silico modeling and metabolomics analysis to characterize fed-batch CHO cell culture. Biotechnology and Bioengineering. 2012;109:1415–1429. doi: 10.1002/bit.24445. [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Yamaguchi-Shinozaki K. Gene networks involved in drought stress response and tolerance. Journal of Experimental Botany. 2007;58:221–227. doi: 10.1093/jxb/erl164. [DOI] [PubMed] [Google Scholar]
- Shlomi T, Cabili MN, Herrgard MJ, Palsson BO, Ruppin E. Network-based prediction of human tissue-specific metabolism. Nature Biotechnology. 2008;26:1003–1010. doi: 10.1038/nbt.1487. [DOI] [PubMed] [Google Scholar]
- Stamm P, Ravindran P, Mohanty B, Tan EL, Hao Yu H, Kumar PP. Insights into the molecular mechanism of RGL2-mediated inhibition of seed germination in Arabidopsis thaliana. BMC Plant Biology. 2012;12:179. doi: 10.1186/1471-2229-12-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiele I, Price ND, Vo TD, Palsson BO. Candidate metabolic network states in human mitochondria. Impact of diabetes, ischemia, and diet. The Journal of Biological Chemistry. 2005;280:11683–11695. doi: 10.1074/jbc.M409072200. [DOI] [PubMed] [Google Scholar]
- Wettenhall JM, Smyth GK. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics. 2004;20:3705–3706. doi: 10.1093/bioinformatics/bth449. [DOI] [PubMed] [Google Scholar]
- Xu K, Xu X, Fukao T, Canlas P, Maghirang-Rodriguez R, Heuer S, Ismail AM, Bailey-Serres J, Ronald PC, Mackill DJ. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature. 2006;442:705–708. doi: 10.1038/nature04920. [DOI] [PubMed] [Google Scholar]
- Yilmaz A, Mejia-Guerra MK, Kurz K, Liang X, Welch L, Grotewold E. AGRIS: the Arabidopsis gene regulatory information server, an update. Nucleic Acids Research. 2011;39:D1118–D1122. doi: 10.1093/nar/gkq1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun KY, Park MR, Mohanty B, Herath V, Xu F, Mauleon R, Wijaya E, Bajic VB, Bruskiewich R, de los Reyes B. Transcriptional regulatory network triggered by oxidative signals configures the early response mechanisms of japonica rice to chilling stress. BMC Plant Biology. 2010;10:16. doi: 10.1186/1471-2229-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Yu S, Zuo K, Luo L, Tang K. Identification of gene modules associated with drought response in rice by network-based analysis. PLoS ONE. 2012;7:e33748. doi: 10.1371/journal.pone.0033748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.