

Abstract
We investigated the capacity of dietary (-)-epicatechin (EC) to mitigate insulin resistance through the modulation of redox-regulated mechanisms in a rat model of metabolic syndrome (MetS). Adolescent rats were fed a regular chow diet without or with high fructose (HFr) (10% (w/v)) in drinking water for 8 weeks, and a group of HFr-fed rats was supplemented with EC in the diet. HFr-fed rats developed insulin resistance which was mitigated by EC supplementation. Accordingly, the activation of components of the insulin signaling cascade (insulin receptor (IR), IRS-1, Akt and ERK1/2) was impaired, while negative regulators (PKC, IKK, JNK and PTP1B) were upregulated in the liver and adipose tissue of HFr rats. These alterations were partially or totally prevented by EC supplementation. In addition, EC inhibited events which contribute to insulin resistance: HFr-associated increased expression and activity of NADPH oxidase, activation of redox-sensitive signals, expression of NF-κB-regulated pro-inflammatory cytokines and chemokines, and some sub-arms of endoplasmic reticulum stress signaling. Collectively, these findings indicate that EC supplementation can mitigate HFr-induced insulin resistance and are relevant to define interventions that can prevent/mitigate MetS-associated insulin resistance.
INTRODUCTION
The metabolic syndrome (MetS) is defined as a cluster of symptoms that include increased waist circumference, plasma tryglicerides (TG), and fasting glycemia, reduced high-density lipoprotein (HDL) cholesterol, and hypertension [1]. The increasing incidence of MetS, which currently affects 34% of the world population, is associated with the development of insulin resistance and type 2 diabetes (T2D), obesity, and cardiovascular disease, constituting a major public health concern worldwide [2, 3].
Diet can play a major role in the prevention of MetS and its associated pathologies. Flavonoids are naturally occurring plant compounds that have a multiplicity of biological effects. The ability of flavonoids to modulate cell signaling could contribute to the health benefits associated with the consumption of fruit and vegetables [4]. Significantly, epidemiological studies show that the consumption of fruits and vegetables in humans decreases the risk for MetS [5-8]. Among flavonoids, the flavan-3-ol (-)-epicatechin (EC) is one of the most abundant in human diets. EC is present in large concentrations in fruit and vegetables (e.g. cocoa, grapes, tea, berries) and derived foods [9]. EC has a basic chemical structure of two aromatic rings linked by an oxygenated heterocycle with a hydroxyl group in position 4 (Fig. 1A). Consumption or supplementation with EC or EC-containing foods in humans and experimental animals is associated with improvement of several MetS hallmarks, including: a- decreased blood pressure and improved vascular function [10-14]; b- improved insulin sensitivity [15-20]; c- decreased plasma cholesterol [11]; d- improved oxidative stress parameters [11], and e- decreased risk for cardiometabolic disorders [21]. Previously, we demonstrated that in differentiated 3T3-L1 adipocytes EC prevents tumor necrosis factor alpha (TNFα)-induced signals that perpetuate inflammation and contribute to insulin resistance [22]. Importantly, consumption of cocoa flavanols (a particularly pure source of EC and its derived procyanidins), is associated with improvements in parameters of insulin sensitivity in healthy human adults [17], glucose-intolerant hypertensive subjects [15], and overweight/obese individuals [18].
Figure 1. Effects of EC supplementation on metabolic parameters in HFr-fed rats.
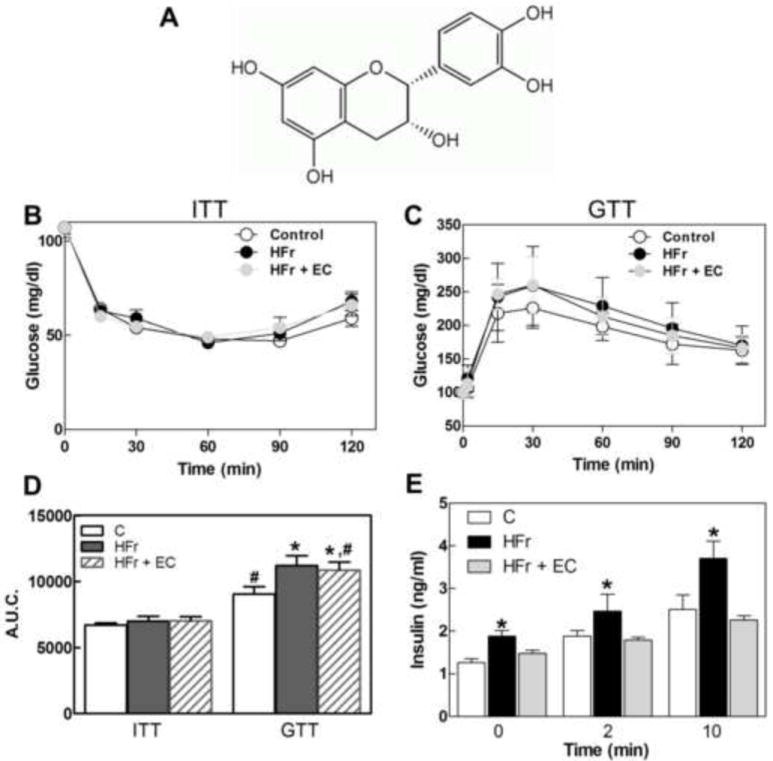
A- (-)-Epicatechin chemical structure, B- ITT, C-GTT, D- Area under the curve from ITT and GTT performed on week 7 on the respective treatments, and E-plasma insulin concentration during GTT in rats fed control diet and regular drinking water (empty circles and empty bars), a control diet and drinking water supplemented with 10% (w/v) fructose (black circles and black bars), or a diet supplemented with 20 mg EC/kg body weight and fructose-containing drinking water (grey circles and grey bars). Results are shown as means ± SE and are the average of 8 animals/group. D- Values having different symbols (*,#) are significantly different; E- *are significantly different from the other groups at the corresponding time points; (p < 0.05, one way ANOVA).
Increased production of cellular oxidants via the activation of NADPH oxidases (NOX) has been proposed as one contributing mechanism to the development of insulin resistance in MetS [23-25]. Increased oxidant production can activate redox-sensitive signals that: i) negatively regulate insulin signaling pathway (c-Jun N-terminal kinase (JNK), inhibitor of nuclear factor κB (IκB) kinase (IKK)), and ii) promote and sustain chronic inflammation and oxidative stress (NF-κB). EC has been previously shown to inhibit NOX activity [26], mechanism that could provide EC with the capacity of improve insulin sensitivity in MetS.
In this study we evaluated the capacity of dietary EC to influence insulin resistance in a rat model of MetS, induced by consumption of a high fructose (HFr) diet. The contribution of redox-dependent mechanisms to the capacity of EC to improve fructose-induced impairment of the metabolic phenotype were investigated. Dietary EC supplementation improved insulin sensitivity in HFr-fed rats. The beneficial effect of EC is due, at least in part, to decreased NOX activity and expression, mitigation of chronic redox signaling activation, inflammation, and endoplasmic reticulum (ER) stress. Our findings support the relevance of designing effective dietary or supplementation interventions for the prevention/amelioration of MetS-induced T2D.
MATERIALS AND METHODS
Materials
Cholesterol, HDL cholesterol, and triglyceride concentrations were determined using kits purchased from GTLab (Buenos Aires, Argentina). Antibodies for eIF2α, p-eIF2α (Ser51), ERK, p-ERK (Thr202/Tyr204), JNK, p-JNK (Thr183/Tyr185), NOX2, NOX4, p47phox, β-tubulin, MCP-1, TNFα, p-PERK (Thr980), PERK, PTP1B, sXBP1, cATF6, IRE1α, p-IR (Tyr1162/Tyr1163), and IR were from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibodies for p-PKCδ (Thr505), p65, p-p65, IKβα, p-IκBα (Ser32), IKKα, p-IKKα/β (Ser178/180), p-AKT (Ser473), AKT, and were obtained from Cell Signaling Technology (Danvers, MA). Antibody for p-IRE1α (Ser724) was purchased from Abcam (Cambridge, MA). Antibodies for p-IRS1 (Tyr608) and IRS1 were from Millipore Corp. (Billerica, MA). Catalogue numbers for all antibodies are included in supplemental table 1. PVDF membranes and protein standards were obtained from BIO-RAD (Hercules, CA). The ECL Western blotting system was from Thermo Fisher Scientific Inc. (Piscataway, NJ). Fructose was purchased from Saporiti Labs (Buenos Aires, Argentina). EC and all other reagents were from the highest quality available and were purchased from Sigma (St. Louis, MO).
Animals and animal care
All procedures were in agreement with standards for the care of laboratory animals as outlined in the NIH Guide for the Care and Use of Laboratory Animals. All procedures were performed according to institutional guidelines for animal experimentation and were approved by the Technical and Science Secretary at the National University of Cuyo, School of Medicine; and by the Animal Resource Services of the University of California, Davis, which is accredited by the American Association for the Accreditation of Laboratory Animal Care. Rats were housed under conditions of controlled temperature (21 - 25°C) and humidity with a 12 h light/dark cycle. Thirty-day-old male Wistar rats, weighing 100-130 g, were randomly divided in 3 groups (10 rats per group) that were fed a standard rat chow (Gepsa-Feeds, Buenos Aires, Argentina) (Table 2, supplemental material) and water ad libitum (control group; C), chow diet and fructose (10% w/v)-supplemented water (high fructose group; HFr), or chow diet supplemented with 20 mg EC/Kg body weight and the fructose-supplemented water (HFr + EC). The amount of EC used for dietary supplementation was based on levels that are feasible to reach through dietary supplementation in humans, on our preliminary studies using the same amount of the EC isomer, catechin, and on studies by other groups in models of hypertension and streptozotocin-induced type 1 diabetes [27, 28]. Food and water intake were recorded twice per week. The average daily food intake is shown in Table 1. The concentration of EC in the diet was adjusted weekly to account for changes in body weight and food intake. After 8 weeks on the dietary treatments, and after overnight fast rats were weighed and 5 animals per group were intraperitoneally injected with insulin (10 mU/g B.W. human insulin (HumulinR; Eli Lilly)) or saline, and euthanized after 10 min. Blood was collected from the abdominal aorta into heparinized tubes, and plasma obtained after centrifugation at 1,000 × g for 15 min at 4°C. Epididymal and mesenteric adipose tissue, and liver were collected and weighed. Tissues were flashfrozen in liquid nitrogen and then stored at -80°C for further analysis.
Table 1.
Metabolic parameters from rats fed for 8 weeks without (control) or with 10% (w/v) fructose in the water, in the absence (HFr) or presence (HFr + EC) of 20 mg EC/kg BW. Values are shown as means ± SE (n = 10). Values having different superscripts are significantly different (P < 0.05, one way ANOVA)
| Parameter | Groups | ||
|---|---|---|---|
| Control | HFr | HFr + EC | |
| Food intake (g/d) | 24.9 ± 1.5a | 19.9 ± 0.8b | 18.1 ± 1.3b |
| Water intake (ml/d) | 45 ± 2a | 53 ± 4a,b | 57 ± 4b |
| Fructose intake (g/d) | - | 5.3 ± 0.7 | 5.7 ± 0.5 |
| BW (g) | 344 ± 36 | 313 ± 34 | 325 ± 40 |
| Epididymal fat (mg)/BW | 10.5 ± 2.4 | 9.2 ± 2.7 | 12.2 ± 3.3 |
| Mesenteric fat (mg)/BW | 2.3 ± 0.7 | 2.3 ± 0.8 | 2.7 ± 1.0 |
| Liver (mg)/BW | 26.5 ± 2.1 | 26.2 ± 2.1 | 27.1 ± 2.6 |
| Plasma Chol (mg/dl) | 46 ± 2 | 51 ± 3 | 46 ± 5 |
| Plasma HDL (mg/dl) | 19.5 ± 1.3a | 15.7 ± 1.5a | 24.6 ± 1.4b |
| Plasma LDL (mg/dl) | 26 ± 2a | 35 ± 3b | 20 ± 3a |
| Plasma TG (mg/dl) | 46.8 ± 9.1a | 96.8 ± 23.1b | 79.9 ± 26.8c |
| Liver TG (mg/g) | 71 ± 9a | 90 ± 17b | 80 ± 22a |
| Liver Chol (mg/g) | 169 ± 21 | 176 ± 32 | 167 ± 22 |
| Fasting glucose (mg/dl) | 88.2 ± 5.9 | 84.7 ± 11.7 | 86.3 ± 5.8 |
| Fed glucose (mg/dl) | 124.7 ± 13.8 | 116.4 ± 4.9 | 124.0 ± 14.0 |
| Insulin (nmol/l) | 1.26 ± 0.09a | 1.96 ± 0.26b | 1.48 ± 0.17a |
(-)-Epicatechin mitigates high fructose-induced insulin resistance.
(-)-Epicatechin mitigates metabolic syndrome phenotypes.
(-)-Epicatechin improves insulin sensitivity through redox-dependent mechanisms.
Metabolic measurements
Glucose was measured in blood collected from the tail using a glucometer (Accu-Chek Performa, Roche, Argentina). Total and HDL cholesterol, TG (GTLab, Buenos Aires) and insulin (Coat-A-Count, Siemens, CA) concentrations were determined using commercial kits following manufacturer’s guidelines. Hepatic lipids were extracted with chloroform/methanol as described previously [29, 30] and assayed for TG and cholesterol content as described above. Fed glucose measurements were taken in tail bleeds using a glucometer (Home Aide Diagnostics) between 7-9 am and, where indicated, from rats fasted for 12 h. For insulin tolerance tests (ITT), rats were fasted for 4 h and injected intraperitoneally with 1 mU/g body weight human insulin. Blood glucose values were measured before and at 15, 30, 60, 90 and 120 min post-injection. For glucose tolerance tests (GTT), overnight fasted rats were injected with D-glucose (2g/kg B.W.), and blood glucose was measured before and at 15, 30, 60, 90 and 120 min post-injection. ITT and GTT tests done after 4 weeks on the respective diets were similar among the groups.
Western blot analysis
Tissues were homogenized in radio-immunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% w/v sodium dodecylsulfate, 1% w/v Triton X-100, 1% sodium deoxycholate, 5 mM EDTA, 1 mM NaF, 1 mM sodium orthovanadate and protease inhibitors). Homogenates were centrifuged at 15,000 × g for 30 min, the supernatant collected, and protein concentration measured [31]. Aliquots of total cell lysates containing 25-40 μg protein were denatured with Laemmli buffer, separated by reducing 10-12.5% polyacrylamide gel electrophoresis, and electroblotted to PVDF membranes. Membranes were blotted for 2 h in 5% (w/v) bovine serum albumin and subsequently incubated in the presence of the corresponding primary antibodies (1:1,000 dilution for all the antibodies except TNFα, 1:500) overnight at 4°C. After incubation for 90 min at room temperature in the presence of the secondary antibody (HRP conjugated) (1:10,000 dilution) the conjugates were visualized using enhanced chemiluminescence (Amersham Biosciences). Pixel intensities of immunoreactive bands were quantified using FluorChem Q Imaging software (Alpha Innotech).
NADPH oxidase activity and protein carbonyls
The lucigenin-derived chemiluminescence assay was used to determine NAD(P)H oxidase activity in liver membrane fractions. Liver tissue (100 mg) was homogenized in 1 ml of ice-cold Krebs buffer (20 mM HEPES pH 7.4, containing 119 mM NaCl, 4.7 mM KCl, 1 mM MgSO4, 0.4 mM NaH2PO4, 5 mM NaHCO3, 1.25 mM CaCl2, and protease inhibitors), and then centrifuged at 800 × g, at 4°C for 10 min. The supernatant was collected and then centrifuged at 100,000 × g for 1 h at 4°C. The supernatant (cytosolic fraction) was discarded and the pellet (membrane fraction) was resuspended in Krebs buffer and protein concentration measured [31]. To determine NOX activity, aliquots containing 10 μg of protein were added to Krebs buffer containing NADPH (0.5 mM) and lucigenin (5 μM). Light emission was measured for 10 min at 30 s intervals using a VICTOR 1420, Multi-label Counter (WALLAC Oy, Turku, Finland). The area under the curve was calculated and results referred to controls values.
Protein carbonyl content was evaluated using the OxyBlot Protein Oxidation Detection Kit Millipore Corp. (Billerica, MA) following the manufacturer’s protocol. Briefly, protein carbonyl groups in total tissue homogenates were initially derivatized by reaction with 2,4-dinitrophenylhydrazine (DNP). Western blots were performed as previously described and DNP-proteins detected with a specific HRP-antibody.
RNA isolation and real-time PCR
RNA was extracted from liver and adipose tissue using TRIzol reagent (Invitrogen). cDNA was generated using high-capacity cDNA Reverse Transcriptase (Applied Biosystems). Expression of NOX2 and NOX4 was assessed by reverse transcription PCR (iCycler, BioRad) and normalized to TATA-Box binding protein (TBP). For RT-PCR, Absolute blue qPCR premix (Fisher Scientific) was mixed with each primer. The following primers were used: NOX2 primers: 5’-CCAGTGAAGATGTGTTCAGCT-3’ (Forward), 5’-GCACAGCCAGTAGAAGTAGAT-3’ (Reverse); NOX4 primers: 5’-AGTCAAACAGATGGGATA-3’ (Forward), 5’-TGTCCCATATGAGTTGTT-3’ (Reverse); TBP primers: 5’-CAGCCTTCCACCTTATGCTC-3’ (Forward), 5’-TGCTGCTGTCTTTGTTGCTC-3’ (Reverse). The threshold cycle (Ct) was determined and the relative gene expression was calculated as follows: fold change=2-Δ(ΔCt), where ΔCt=Cttarget gene-CtTBP (cycle difference) and Δ(ΔCt)=CtHfr/HFr+Ec-CtC.
Statistical analysis
Data were analyzed by one-way analysis of variance (ANOVA) using Statview 5.0 (SAS Institute Inc., Cary, NC). Fisher least significance difference test was used to examine differences between group means. A P value < 0.05 was considered statistically significant. Data are shown as mean ± SEM.
Results
EC treatment improves metabolic parameters in HFr-fed rats
To investigate whether EC treatment regulates metabolic status in vivo, thirty-day-old rats were fed high fructose (HFr) and HFr with EC-supplemented diets, as detailed in Methods. Adolescent rats were studied since adult humans have a particularly high intake of fructose [32] which renders them a segment of the population particularly susceptible to the adverse effects of excess dietary fructose. Furthermore, high fructose consumption in adolescents is associated with cardiometabolic risk markers and visceral adiposity, both components of MetS [33].
After 8 weeks on the corresponding diets, metabolic parameters were assessed (Table 1). The average daily food intakes of HFr and HFr-EC rats were significantly lower than in controls; the daily water intake was significantly higher in the HFr-EC rats compared with control and HFr groups. Thus, rats provided fructose in the drinking water had a daily fructose intake of 5.3 and 5.7 g/rat for the HFr and HFr +EC groups, respectively. After 8 weeks on the respective diets, body weight, epidydimal and mesenteric adipose tissue and liver weights were comparable among groups (Table 1). High fructose consumption caused dyslipidemia, as evidenced by elevated plasma TG and LDL-cholesterol in HFr-fed rats compared with controls. EC supplementation partially or completely prevented HFr-induced plasma TG and LDL increase, respectively, and increased HDL cholesterol levels over control values (Table 1). Fasting and fed glucose levels were comparable among groups. Of note, HFr-fed rats required a higher concentration of insulin to maintain glucose levels compared with controls, suggestive of insulin resistance. In addition, EC treatment decreased the amount of insulin required to maintain comparable glucose concentration (Table 1). To assess insulin sensitivity, rats were subjected to ITT as detailed in Methods (Fig. 1B). Comparable response was observed for ITT values and the area under the curve (AUC) was similar among groups (Fig. 1D). On the other hand, HFr-treated rats exhibited decreased ability to clear glucose from the circulation compared with controls during an intraperitoneal GTT (Fig. 1C). AUC for the GTT was significantly (p < 0.05) higher in the HFr group compared with controls (Fig. 1D). In the EC-supplemented rats the GTT AUC was comparable to HFr values. It is worth noting that plasma insulin concentrations at 0, 2 and 10 min during the GTT were higher in HFr rats compared with controls while HFr-EC treated rats exhibited insulin concentrations that were comparable to controls (Fig. 1E). Together, these data indicate that EC consumption improves the systemic metabolic status of rats with chronic consumption of HFr.
EC treatment enhances insulin signaling in the liver and adipose tissue
To investigate the molecular basis for altered glucose tolerance in HFr-fed rats, we determined alterations in insulin signaling. Fasted control (C), HFr and HFr + EC rats were injected with saline or insulin (as detailed in Methods), then basal and insulin-stimulated signaling was determined in the liver and epididymal adipose tissue (Fig. 2). In the adipose tissue, insulin-induced insulin receptor (IR) tyrosyl phosphorylation at Tyr1162/Tyr1163 was attenuated in HFr rats compared with controls, while EC supplementation enhanced insulin-induced IR phosphorylation and restored it to control levels (Fig. 2A). Similarly, insulin-induced IR substrate 1 (IRS1) tyrosyl phosphorylation at Tyr608 (one of the PI3K binding sites) was attenuated in HFr rats compared with controls but was elevated upon EC supplementation (Fig. 2A). Consistent with IR and IRS1 tyrosyl phosphorylation, HFr-attenuated insulin-stimulated extracellular signal-regulated kinases 1/2 (ERK) phosphorylation (Thr202/Tyr204) and was elevated upon EC supplementation. Moreover, insulin-stimulated Akt Ser473 phosphorylation was decreased in HFr rats compared with controls but was elevated upon EC supplementation. Further, basal ERK and Akt phosphorylation was higher in HFr rats supplemented with EC than in adipose tissue from control and HFr animals. Comparable effects of HFr feeding and EC supplementation on insulin signaling were observed in the liver (Fig. 2B). Together, these data demonstrate that the HFr-induced attenuation of insulin signaling in liver and adipose tissue is mitigated by EC supplementation.
Figure 2. EC supplementation enhances insulin signaling in epididymal adipose and liver tissues in HFr-fed rats.

After 8 weeks on the corresponding diets, rats were fasted overnight then injected with saline or insulin (10 mU/g body weight) and then sacrificed after 10 minutes. Phosphorylation of IR, IRS1, ERK1/2, and Akt are shown for A- epididymal adipose tissue and B-liver. Bands were quantified and results for the HFr and HFr + EC were referred to control group values (C). Results are expressed as the ratio of phosphorylated/total protein level. Results are shown as mean ± SEM of 5 animals/treatment. *, # are significantly different between them and from the insulin untreated groups, &are significantly different from the insulin untreated control and HFr groups, and ** are significantly different from all other groups (p<0.05, one way ANOVA test).
Serine phosphorylation of IRS1 by the kinases JNK, IKK, or protein kinase C (PKC), and IR tyrosine dephosphorylation by protein-tyrosine phosphatase 1B (PTP1B), lead to downregulation of the insulin signaling cascade. High fructose consumption caused the activation of kinases IKKα/β, JNK and PKC, as evaluated by phosphorylation levels, and increased PTP1B expression. Phosphorylation of IKKα/β (Ser 178/180), JNK (Thr183/Tyr185) and PKCδ (Thr505), and PTP1B protein expression were high in both the adipose tissue (Fig. 3A) and liver (Fig. 3B) of the HFr rats. EC supplementation prevented HFr-induced increased IKKα/β, JNK and PKC phosphorylation and PTP1B expression in both tissues.
Figure 3. Effects of EC supplementation on epididymal adipose and liver tissue insulin signaling in HFr-fed rats: inhibitory signaling.
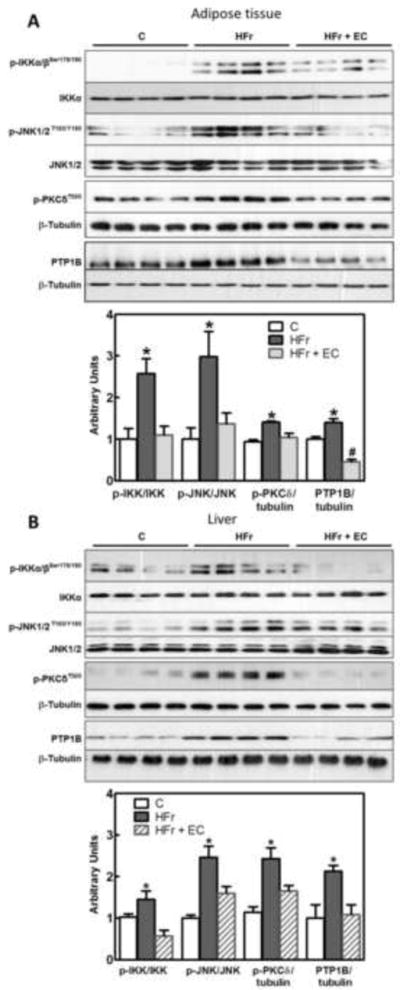
A,B- Phosphorylation of IKKα/β (Ser178/180, JNK (Thr183, Tyr185) and PKCδ (Thr505), and PTP-1B protein levels in epididymal adipose tissue (A) and liver (B) after 8 weeks on the corresponding diets. Bands were quantified and results for the HFr (HF) and HFr + EC (EC) were referred to control group values (C). Results are expressed referred to either total protein or β-tubulin levels. Results are shown as mean ± SEM of 8 animals/treatment. *, # are significantly different from all other groups (p<0.05, one way ANOVA test);
EC treatment mitigates HFr-induced increased NOX expression, oxidative stress, NF-κB activation and inflammation
Increased production of superoxide anion via NADPH oxidase (NOX) activation can trigger JNK, IKK/ NF-κB activation and contribute to the overall MetS pro-inflammatory scenario. Thus, the expression of NOX subunits NOX2 (gp91 phox), NOX4, and p47 was assessed. In HFr rats, protein levels of NOX2, NOX4, p47phox in liver and adipose tissue were significantly higher than in controls (Fig. 4A, B). EC supplementation prevented the effects of HFr feeding on NOX subunits except for p47phox (Fig. 4A, B). Further, evaluation of NOX2 and NOX4 mRNA demonstarted increased expression of NOX2 and NOX4, which was prevented by EC supplementation in both liver and adipose tissues (Fig. 4C). In the liver, NOX activity was 2.1-fold higher in the HFr group compared with controls, and EC supplementation prevented this increase (Fig. 4B). Together, these data suggest an increased capacity of liver and adipose tissues to generate superoxide anion in MetS, which is mitigated by EC supplementation. To evaluate if NOX upregulation was associated with tissue oxidative stress we measured tissue protein carbonyl levels. Protein carbonyls result from direct protein oxidation and from protein derivatization by by-products of lipid oxidation [34]. Indeed, higher levels of protein carbonylation were detected in liver and adipose tissue from rats fed HFr diet compared with controls, which were prevented by EC supplementation (Fig. 4D).
Figure 4. Effects of EC supplementation on epididymal adipose and liver tissue NOX upregulation.
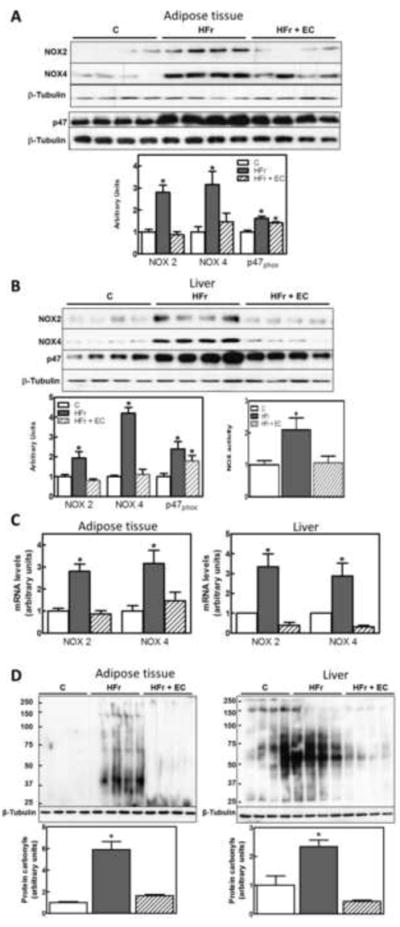
A,B: Protein levels of NOX subunits (NOX2, NOX4, p47) were measured by Western blot in A- adipose tissue and B- liver, and bands were quantified. B- NOX activity was measured in liver as described in methods. Results for the HFr (HF) and HFr + EC (EC) were referred to control group values (C). C- NOX2 and NOX4 mRNA were measured by quantitative real-time PCR, normalized against TATA-Box binding protein (TBP) and were referred to control group values (C). D- Protein carbonyls were measured as described in methods. Results are shown as mean ± SEM of 4-8 animals/treatment. *Significantly different from other groups (p<0.05, one way ANOVA test).
Inflammation is a major contributor to insulin resistance. Oxidative stress and activation of the redox-sensitive transcription factor NF-κB are at the center of a self-feeding cycle that perpetuates inflammation. Consumption of fructose for 8 weeks led to the activation of the NF-κB pathway, as evidenced by increased IKKα/β (Ser178/180) (Fig. 3A,B) phosphorylation, IκBα phosphorylation (Ser32) and degradation of the NF-κB inhibitory peptide (IκBα) in adipose tissue and liver (Fig. 5A). Other NF-κB activation step, p65 phosphorylation (Ser536) was also significantly higher in liver from the HFr rats than in controls. The activation of these steps in the NF-κB signaling pathway was not observed in the HFr rats supplemented with EC.
Figure 5. Effects of EC supplementation on epididymal adipose and liver tissue activation of the pro-inflammatory NF-κB signaling pathway.
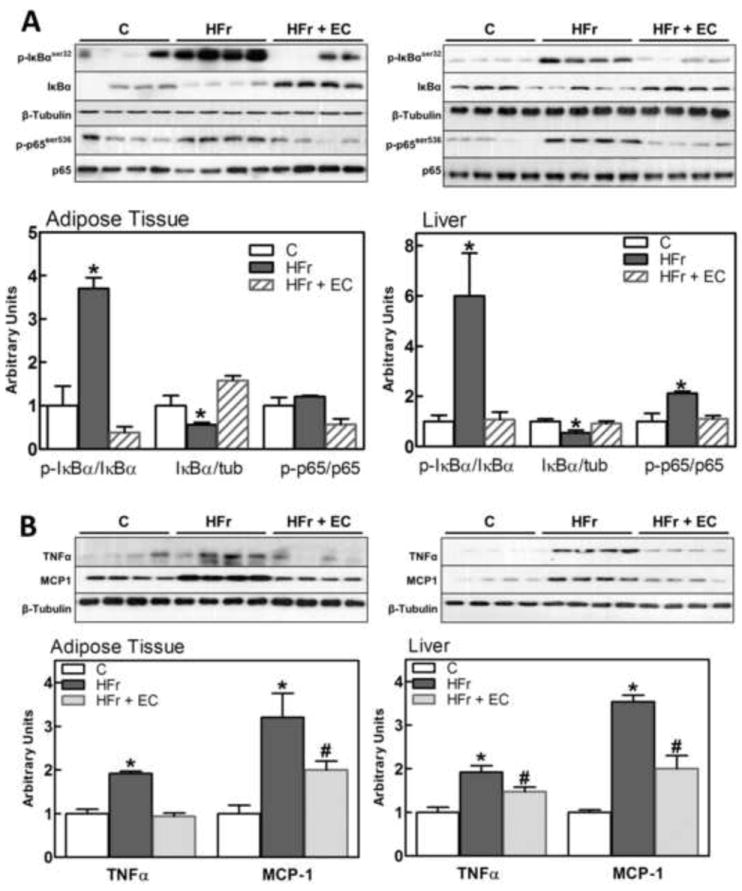
Different steps in the NF-κB pathway were evaluated in rat epididymal adipose tissue and liver after 8 weeks on the corresponding diets, measuring: A- phosphorylation (Ser32) and total levels of IκBα, and phosphorylation of p65 (Ser536); B- TNFα and MCP-1 (NF-κB target genes) protein levels. Bands were quantified and results for the HFr (HF) and HFr + EC (EC) were referred to control group values (C). Results are shown as mean ± SEM of 5 animals/treatment. *,# are significantly different from the untreated controls, and are significantly different among them. (p<0.05, one way ANOVA test).
The cytokine TNFα and the chemokine monocyte chemotactic protein-1 (MCP-1) are NF-κB-regulated proteins that have central roles in MetS- and obesity-induced insulin resistance. TNFα and MCP-1 expression was significantly higher in adipose tissue and liver of HFr rats than in controls, while EC supplementation partially or totally prevented the HFr-associated upregulation (Fig. 5B).
EC supplementation attenuated HFr-induced ER stress in liver and adipose tissue
A growing body of evidence indicates that endoplasmic reticulum (ER) dysfunction is a contributor to metabolic diseases [35, 36]. When the folding capacity of the ER is exceeded, misfolded proteins accumulate and lead to ER stress [37]. Cells use adaptive mechanisms to mitigate ER stress and to restore homeostasis known as the unfolded protein response (UPR). This protective response consists of three major branches that are controlled by the ER transmembrane proteins PKR-like ER-regulated kinase (PERK), inositol requiring protein 1α (IRE1α), and activating transcription factor 6 (ATF6). We determined the effects of HFr-consumption and EC supplementation on UPR in liver and adipose tissue. After 8 w of HFr consumption the three branches of the UPR were markedly upregulated in adipose tissue (Fig. 6A) and liver (Fig. 6B), as assessed by PERK (Tyr980) and eIF2α (Ser51) phosphorylation, ATF6 cleavage IRE1α (Ser724) phosphorylation and the levels of X-box binding protein 1 spliced isoform (sXBP-1). Of note, EC supplementation of HFr rats attenuated specific subarms of the UPR. In adipose tissue, EC supplementation attenuated HFr-induced IRE1α (Fig. 6A). In addition, in the liver, EC supplementation partially or totally attenuated HFr-induced expression of the spliced XBP-1 isoform, IRE1α and PERK phosphorylation (Fig. 6B). Together, these data indicate that EC supplementation can attenuate sepcific sub-arms of ER stress induced by HFr feeding in liver and adipose tissue.
Figure 6. Effects of EC supplementation on parameters of ER stress in epididymal adipose tissue and liver.
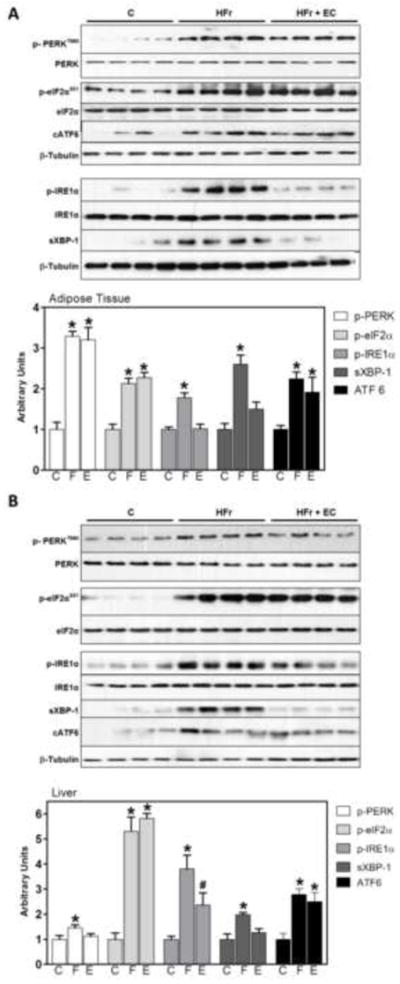
The three branches of the UPR response were evaluated by Western blot measuring PERK (Tyr980), eIF2α (Ser51), and IRE1α (Ser724) phosphorylation, sXBP-1 and cleaved ATF6 (cATF6) in A- adipose tissue and B- liver. Bands were quantified and results for the HFr (HF) and HFr + EC (EC) were referred to control group values (C). Phosphorylated/total ratios were calculated for PERK, eIFα and IRE1α; sXBP-1 and cATF6 were normalized to tubulin content. Results are shown as mean ± SEM of 5-8 animals/treatment. *Significantly different from other groups (p<0.05, one way ANOVA test).
Discussion
This work presents evidence that in vivo EC supplementation mitigates the impairment of metabolic parameters induced by high fructose feeding. This occurs in parallel with the inhibition by EC of events associated with altered tissue redox status and impaired insulin sensitivity: NOX upregulation, activation of redox-sensitive signals (JNK, IKK/NF-κB), inflammation, and ER stress. The capacity of EC to modulate NOX subunit expression and to directly inhibit NOX [26] is likely a central mechanism in the beneficial action of EC on MetS
The long-term consumption of fructose-rich diets by humans [38] and experimental animals causes the typical pathogenic features of MetS: insulin resistance, obesity, dyslipidemia, and hypertension. In the current study, metabolic parameters in HFr-fed rats indicated an early stage of insulin resistance which was mitigated by EC supplementation. Accordingly with the present results, human consumption of food/food extracts containing EC is associated with improvements of insulin sensitivity. In this regard, a systematic review and meta-analysis of randomized, controlled trials showed a relationship between the short-term consumption of EC-rich cocoa and improvement in insulin sensitivity (HOMA-IR) in humans [20]. Consumption of cocoa extracts was associated with improvements in parameters of insulin sensitivity in different populations, including healthy human adults [17], glucose-intolerant hypertensive subjects [15], and overweight/obese individuals [18]. Findings that EC supplementation improved plasma and lipid profiles in the HFr rats suggest that EC can also influence the altered lipid metabolism associated with HFr consumption.
In the liver and adipose tissue, HFr consumption causes inflammation, oxidative and ER stress which can lead to insulin resistance [39-41]. The in vivo response to insulin by liver and adipose tissues was impaired in the HFr rats as evidenced by decreased insulin-mediated phosphorylation of IR, IRS-1, Akt and ERK1/2. However, EC supplementation improved or restored liver and adipose tissue response to insulin. Several mechanisms can act in concert to induce insulin resistance as a consequence of HFr consumption. Fructose preferential uptake by the liver leads to an overproduction of glyceraldehyde-3-P, and acetyl CoA which fuels de novo lipogenesis, and activates (diacylglycerol) novel PKC. HFr-induced metabolic burden can also increase oxidant production with a consequent activation of redox-sensitive signals (JNK, IKK/NF-κB). PKC, JNK and IKK activation can impair insulin sensitivity. One of the proposed mechanisms is the serine/threonine phosphorylation of IRS-1 by these kinases which can exert a negative regulation of the insulin pathway [42, 43]. This is further supported by findings that genetic deficits of JNK or IKK mitigate obesity-induced insulin resistance [44, 45]. In addition, insulin signaling is attenuated through the dephosphorylation of IR/IRS-1 by PTP1B [46, 47]. We observed that the impaired response to insulin in liver and adipose tissue from HFr rats was associated with high levels of PKC, JNK and IKK phosphorylation, and increased PTP1B expression, which were prevented by EC supplementation. Furthermore, in HFr rats supplemented with EC, basal ERK and Akt phosphorylation were higher than controls in liver and adipose tissue. Of note, it was previously shown that EC can activate ERK and Akt in cortical neuronal cultures [48].
The activation of redox-sensitive signaling (NF-κB), and associated expression of pro-inflammatory genes constitute a self-feeding cycle that contributes to insulin resistance. NOX is the major enzymatic source of cellular superoxide anion. Through the production of small and transient amounts of superoxide, NOX co-activates membrane receptor-mediated signaling cascades, including those triggered by cytokines (e.g. TNFα), growth factors, and hormones (e.g. insulin) [49-51]. However, sustained NOX and/or uncontrolled activation (e.g. chronic inflammation) leads to oxidative stress, which is proposed to be a major contributor to MetS and T2D development [52, 53]. EC can modulate NOX activation through its capacity to directly inhibit enzyme activity [26, 54], or to regulate the expression of its subunits [55]. We observed that HFr consumption caused an increase in the activity and mRNA and protein expression of NOX subunits (NOX2, NOX4, and p47). EC supplementation prevented hepatic NOX activation, and NOX2 and NOX4 increased mRNA and protein expression, but not that of p47. The latter could be due to different mechanisms of transcriptional/translational regulation, which can vary among tissues and stimuli [55]. NOX upregulation in HFr-fed rats was associated with increased liver and adipose tissue oxidative damage (protein carbonyls) which was prevented by EC supplementation. Thus, EC-mediated NOX downregulation could mitigate MetS-associate oxidative stress, which can both decrease the damage to cellular components and improve insulin sensitivity.
HFr consumption leads to the activation of the redox-regulated NF-κB with an associated increased expression of the NF-κB target proteins PTP-1B, MCP-1, and TNFα. MCP-1 acts recruiting pro-inflammatory cells, while TNFα activates mitogen activated protein kinases (MAPKs) and transcription factors AP-1 and NF-κB [56-58], which can act re-feeding the pro-inflammatory cycle. EC supplementation mitigated HFr-mediated NF-κB activation, and PTP1B, TNFα and MCP-1 expression. In line with these observations, we recently observed that EC inhibits TNFα-mediated activation of NF-κB, AP-1 and the MAPKs in 3T3-L1 adipocytes [22]. EC-mediated modulation of NF-κB can occur through the previously described inhibition of NOX, and also through specific interactions of EC with the NF-κB proteins p50 and p65 preventing their binding to DNA κB sites [59, 60] (reviewed in [61]).
Increasing evidence supports a major role of ER stress in the development of insulin resistance and T2D in obesity and MetS. Several pathogenic phenotypes of MetS, including inflammatory cytokines [62], hyperglycemia [63, 64]; obesity [36], high cholesterol [65], high fatty acids [62, 66] trigger ER stress in different cells and tissues. Importantly, markers of increased ER stress are observed in human mononuclear cells from MetS patients [64], and in adipose tissue from obese and insulin resistant individuals [67], which improve after weight loss [68]. Notably, chemical chaperones that relieve ER stress in liver cells, increase systemic insulin sensitivity and reduce fatty liver disease in obese mice [69]. We currently observed that HFr feeding activated the three branches of the UPR both in liver and adipose tissue, while EC supplementation mitigated the activation of select subarms: IRE1α in adipose tissue; XBP-1, IRE1α, and PERK in the liver. Attenuation of ER stress, and in particular of the associated negative regulator of the insulin signaling cascade JNK, can in part explain the capacity of EC to improve insulin sensitivity. The flavan-3-ol epigallocatechin gallate (EGCG) inhibits the ATPase activity of the UPR protein GRP78 [70]. EC is chemically different from EGCG, but some of the observed effects could be ascribed to the capacity of select flavonoids to mimic ATP and interfere with ATP-dependent enzymes. However, GRP78 inhibition by EC should affect all UPR sub arms, while we currently observed a sub arm-specific effect. Thus, additional studies to investigate the mechanim(s) underlying regulation of ER stress signaling by EC are warranted.
In summary, EC capacity to modulate liver and adipose tissue superoxide production (NOX down regulation), the consequent activation of the redox sensitive pathways JNK and IKK/NF-κB, and the UPR would converge in the mitigation of inflammation. This would decrease a self-feeding cycle which would ultimately lead into MetS-induced insulin resistance and T2D. Given the high and increasing prevalence of MetS and the associated T2D, there is an urgent need to identify nutritional factors that can co-operate to prevent and/or treat these conditions. The current findings indicate that EC is a dietary component that may have mitigating actions on MetS and the associated insulin resistance.
Supplementary Material
Acknowledgments
Supported by the University of California, Davis; NIFA CA-D-XXX-7244-H to P.I.O., and research grants from JDRF (1-2009-337), NIH grants R56DK084317 and R01DK090492 to F.G.H, 1K99DK100736 to A.B., USA.PICT 2011-1957; Programa I+D 2010 Universidad Nacional de Cuyo, PIP 11220120100558, and Ubacyt 20020120100177, Argentina
Abbreviations
- ATF6
activating transcription factor 6
- B.W
body weight
- EC
(-)-epicatechin
- eIF2α
eukaryotic initiation factor 2 alpha
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinases
- GTT
glucose tolerance test
- HFr
high fructose
- IκB
inhibitor of nuclear factor κB
- IKK
IκB kinase
- IR
insulin receptor
- IRS1
insulin receptor substrate 1
- IRE1α
inositol requiring enzyme 1 alpha
- ITT
insulin tolerance test
- JNK
c-Jun N-terminal kinase
- MAPKs
mitogen activated protein kinases
- MCP-1
monocyte chemotactic protein-1
- MetS
metabolic syndrome
- NOX
NADPH oxidase
- PERK
PKR-like ER-regulated kinase
- PKC
protein kinase C
- PTP1B
protein tyrosine phosphatase 1B
- TG
triglycerides
- T2D
type 2 diabetes
- TNFα
tumor necrosis factor alpha
- UPR
unfolded protein response
- XBP1
X-box binding protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC., Jr Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 2.Ford ES, Giles WH, Dietz WH. Prevalence of the metabolic syndrome among US adults: findings from the third National Health and Nutrition Examination Survey. JAMA. 2002;287:356–359. doi: 10.1001/jama.287.3.356. [DOI] [PubMed] [Google Scholar]
- 3.Cameron AJ, Shaw JE, Zimmet PZ. The metabolic syndrome: prevalence in worldwide populations. Endocrinol Metab Clin North Am. 2004;33:351–375. doi: 10.1016/j.ecl.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Fraga CG, Oteiza PI. Dietary flavonoids: Role of (-)-epicatechin and related procyanidins in cell signaling. Free Radic Biol Med. 2011;51:813–823. doi: 10.1016/j.freeradbiomed.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 5.Hostmark AT. The Oslo Health Study: a Dietary Index estimating high intake of soft drinks and low intake of fruits and vegetables was positively associated with components of the metabolic syndrome. Appl Physiol Nutr Metab. 2010;35:816–825. doi: 10.1139/H10-080. [DOI] [PubMed] [Google Scholar]
- 6.Esmaillzadeh A, Kimiagar M, Mehrabi Y, Azadbakht L, Hu FB, Willett WC. Fruit and vegetable intakes, C-reactive protein, and the metabolic syndrome. Am J Clin Nutr. 2006;84:1489–1497. doi: 10.1093/ajcn/84.6.1489. [DOI] [PubMed] [Google Scholar]
- 7.Kouki R, Schwab U, Hassinen M, Komulainen P, Heikkila H, Lakka TA, Rauramaa R. Food consumption, nutrient intake and the risk of having metabolic syndrome: the DR’s EXTRA Study. Eur J Clin Nutr. 2011;65:368–377. doi: 10.1038/ejcn.2010.262. [DOI] [PubMed] [Google Scholar]
- 8.Panagiotakos DB, Pitsavos C, Skoumas Y, Stefanadis C. The association between food patterns and the metabolic syndrome using principal components analysis: The ATTICA Study. J Am Diet Assoc. 2007;107:979–987. doi: 10.1016/j.jada.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Harnly JM, Doherty RF, Beecher GR, Holden JM, Haytowitz DB, Bhagwat S, Gebhardt S. Flavonoid content of U.S. fruits, vegetables, and nuts. J Agric Food Chem. 2006;54:9966–9977. doi: 10.1021/jf061478a. [DOI] [PubMed] [Google Scholar]
- 10.Galleano M, Oteiza PI, Fraga CG. Cocoa, chocolate, and cardiovascular disease. Journal of cardiovascular pharmacology. 2009;54:483–490. doi: 10.1097/FJC.0b013e3181b76787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fraga CG, Actis-Goretta L, Ottaviani JI, Carrasquedo F, Lotito SB, Lazarus S, Schmitz HH, Keen CL. Regular consumption of a flavanol-rich chocolate can improve oxidant stress in young soccer players. Clin Dev Immunol. 2005;12:11–17. doi: 10.1080/10446670410001722159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heiss C, Finis D, Kleinbongard P, Hoffmann A, Rassaf T, Kelm M, Sies H. Sustained increase in flow-mediated dilation after daily intake of high-flavanol cocoa drink over 1 week. J Cardiovasc Pharmacol. 2007;49:74–80. doi: 10.1097/FJC.0b013e31802d0001. [DOI] [PubMed] [Google Scholar]
- 13.Taubert D, Roesen R, Lehmann C, Jung N, Schomig E. Effects of low habitual cocoa intake on blood pressure and bioactive nitric oxide: a randomized controlled trial. JAMA. 2007;298:49–60. doi: 10.1001/jama.298.1.49. [DOI] [PubMed] [Google Scholar]
- 14.Ottaviani JI, Momma TY, Heiss C, Kwik-Uribe C, Schroeter H, Keen CL. The stereochemical configuration of flavanols influences the level and metabolism of flavanols in humans and their biological activity in vivo. Free Radic Biol Med. 2011;50:237–244. doi: 10.1016/j.freeradbiomed.2010.11.005. [DOI] [PubMed] [Google Scholar]
- 15.Grassi D, Desideri G, Necozione S, Lippi C, Casale R, Properzi G, Blumberg JB, Ferri C. Blood pressure is reduced and insulin sensitivity increased in glucose-intolerant, hypertensive subjects after 15 days of consuming high-polyphenol dark chocolate. The Journal of nutrition. 2008;138:1671–1676. doi: 10.1093/jn/138.9.1671. [DOI] [PubMed] [Google Scholar]
- 16.Curtis PJ, Sampson M, Potter J, Dhatariya K, Kroon PA, Cassidy A. Chronic ingestion of flavan-3-ols and isoflavones improves insulin sensitivity and lipoprotein status and attenuates estimated 10-year CVD risk in medicated postmenopausal women with type 2 diabetes: a 1-year, double-blind, randomized, controlled trial. Diabetes Care. 2012;35:226–232. doi: 10.2337/dc11-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grassi D, Lippi C, Necozione S, Desideri G, Ferri C. Short-term administration of dark chocolate is followed by a significant increase in insulin sensitivity and a decrease in blood pressure in healthy persons. Am J Clin Nutr. 2005;81:611–614. doi: 10.1093/ajcn/81.3.611. [DOI] [PubMed] [Google Scholar]
- 18.Davison K, Coates AM, Buckley JD, Howe PR. Effect of cocoa flavanols and exercise on cardiometabolic risk factors in overweight and obese subjects. Int J Obes (Lond) 2008;32:1289–1296. doi: 10.1038/ijo.2008.66. [DOI] [PubMed] [Google Scholar]
- 19.Montagut G, Blade C, Blay M, Fernandez-Larrea J, Pujadas G, Salvado MJ, Arola L, Pinent M, Ardevol A. Effects of a grapeseed procyanidin extract (GSPE) on insulin resistance. J Nutr Biochem. 2010;21:961–967. doi: 10.1016/j.jnutbio.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 20.Shrime MG, Bauer SR, McDonald AC, Chowdhury NH, Coltart CE, Ding EL. Flavonoid-rich cocoa consumption affects multiple cardiovascular risk factors in a meta-analysis of short-term studies. J Nutr. 2011;141:1982–1988. doi: 10.3945/jn.111.145482. [DOI] [PubMed] [Google Scholar]
- 21.Buitrago-Lopez A, Sanderson J, Johnson L, Warnakula S, Wood A, Di Angelantonio E, Franco OH. Chocolate consumption and cardiometabolic disorders: systematic review and meta-analysis. BMJ. 2011;343:d4488. doi: 10.1136/bmj.d4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vazquez-Prieto MA, Bettaieb A, Haj FG, Fraga CG, Oteiza PI. (-)-Epicatechin prevents TNFalpha-induced activation of signaling cascades involved in inflammation and insulin sensitivity in 3T3-L1 adipocytes. Arch Biochem Biophys. 2012 doi: 10.1016/j.abb.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elnakish MT, Hassanain HH, Janssen PM, Angelos MG, Khan M. Emerging role of oxidative stress in metabolic syndrome and cardiovascular diseases: important role of Rac/NADPH oxidase. J Pathol. 2013;231:290–300. doi: 10.1002/path.4255. [DOI] [PubMed] [Google Scholar]
- 24.Henriksen EJ, Diamond-Stanic MK, Marchionne EM. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radic Biol Med. 2011;51:993–999. doi: 10.1016/j.freeradbiomed.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pepping JK, Freeman LR, Gupta S, Keller JN, Bruce-Keller AJ. NOX2 deficiency attenuates markers of adiposopathy and brain injury induced by high-fat diet. Am J Physiol Endocrinol Metab. 2013;304:E392–404. doi: 10.1152/ajpendo.00398.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steffen Y, Gruber C, Schewe T, Sies H. Mono-O-methylated flavanols and other flavonoids as inhibitors of endothelial NADPH oxidase. Arch Biochem Biophys. 2008;469:209–219. doi: 10.1016/j.abb.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Gomez-Guzman M, Jimenez R, Sanchez M, Zarzuelo MJ, Galindo P, Quintela AM, Lopez-Sepulveda R, Romero M, Tamargo J, Vargas F, Perez-Vizcaino F, Duarte J. Epicatechin lowers blood pressure, restores endothelial function, and decreases oxidative stress and endothelin-1 and NADPH oxidase activity in DOCA-salt hypertension. Free Radic Biol Med. 2012;52:70–79. doi: 10.1016/j.freeradbiomed.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 28.Quine SD, Raghu PS. Effects of (-)-epicatechin, a flavonoid on lipid peroxidation and antioxidants in streptozotocin-induced diabetic liver, kidney and heart. Pharmacological reports : PR. 2005;57:610–615. [PubMed] [Google Scholar]
- 29.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 30.Salmon DM, Flatt JP. Effect of dietary fat content on the incidence of obesity among ad libitum fed mice. Int J Obes. 1985;9:443–449. [PubMed] [Google Scholar]
- 31.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 32.Vos MB, Kimmons JE, Gillespie C, Welsh J, Blanck HM. Dietary fructose consumption among US children and adults: the Third National Health and Nutrition Examination Survey. Medscape J Med. 2008;10:160. [PMC free article] [PubMed] [Google Scholar]
- 33.Pollock NK, Bundy V, Kanto W, Davis CL, Bernard PJ, Zhu H, Gutin B, Dong Y. Greater fructose consumption is associated with cardiometabolic risk markers and visceral adiposity in adolescents. The Journal of nutrition. 2012;142:251–257. doi: 10.3945/jn.111.150219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berlett BS, Stadtman ER. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 35.Hummasti S, Hotamisligil GS. Endoplasmic reticulum stress and inflammation in obesity and diabetes. Circ Res. 2010;107:579–591. doi: 10.1161/CIRCRESAHA.110.225698. [DOI] [PubMed] [Google Scholar]
- 36.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 37.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 38.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, Hatcher B, Cox CL, Dyachenko A, Zhang W, McGahan JP, Seibert A, Krauss RM, Chiu S, Schaefer EJ, Ai M, Otokozawa S, Nakajima K, Nakano T, Beysen C, Hellerstein MK, Berglund L, Havel PJ. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–1334. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Suppl 2):S9–S15. doi: 10.2337/db06-S002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, Goto T, Westerbacka J, Sovijarvi A, Halavaara J, Yki-Jarvinen H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. 2002;87:3023–3028. doi: 10.1210/jcem.87.7.8638. [DOI] [PubMed] [Google Scholar]
- 41.Kotronen A, Seppala-Lindroos A, Bergholm R, Yki-Jarvinen H. Tissue specificity of insulin resistance in humans: fat in the liver rather than muscle is associated with features of the metabolic syndrome. Diabetologia. 2008;51:130–138. doi: 10.1007/s00125-007-0867-x. [DOI] [PubMed] [Google Scholar]
- 42.Samuel VT, Liu ZX, Wang A, Beddow SA, Geisler JG, Kahn M, Zhang XM, Monia BP, Bhanot S, Shulman GI. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117:739–745. doi: 10.1172/JCI30400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei Y, Pagliassotti MJ. Hepatospecific effects of fructose on c-jun NH2-terminal kinase: implications for hepatic insulin resistance. Am J Physiol Endocrinol Metab. 2004;287:E926–933. doi: 10.1152/ajpendo.00185.2004. [DOI] [PubMed] [Google Scholar]
- 44.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 45.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 46.Haj FG, Zabolotny JM, Kim YB, Kahn BB, Neel BG. Liver-specific protein-tyrosine phosphatase 1B (PTP1B) re-expression alters glucose homeostasis of PTP1B-/-mice. J Biol Chem. 2005;280:15038–15046. doi: 10.1074/jbc.M413240200. [DOI] [PubMed] [Google Scholar]
- 47.Matsuo K, Bettaieb A, Nagata N, Matsuo I, Keilhack H, Haj FG. Regulation of brown fat adipogenesis by protein tyrosine phosphatase 1B. PLoS One. 2011;6:e16446. doi: 10.1371/journal.pone.0016446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schroeter H, Bahia P, Spencer JP, Sheppard O, Rattray M, Cadenas E, Rice-Evans C, Williams RJ. (-)Epicatechin stimulates ERK-dependent cyclic AMP response element activity and up-regulates GluR2 in cortical neurons. J Neurochem. 2007;101:1596–1606. doi: 10.1111/j.1471-4159.2006.04434.x. [DOI] [PubMed] [Google Scholar]
- 49.Yang B, Rizzo V. TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am J Physiol Heart Circ Physiol. 2007;292:H954–962. doi: 10.1152/ajpheart.00758.2006. [DOI] [PubMed] [Google Scholar]
- 50.Jung Y, Kim H, Min SH, Rhee SG, Jeong W. Dynein light chain LC8 negatively regulates NF-kappaB through the redox-dependent interaction with IkappaBalpha. J Biol Chem. 2008;283:23863–23871. doi: 10.1074/jbc.M803072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldstein BJ, Mahadev K, Wu X. Redox paradox: insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes. 2005;54:311–321. doi: 10.2337/diabetes.54.2.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guichard C, Moreau R, Pessayre D, Epperson TK, Krause KH. NOX family NADPH oxidases in liver and in pancreatic islets: a role in the metabolic syndrome and diabetes? Biochem Soc Trans. 2008;36:920–929. doi: 10.1042/BST0360920. [DOI] [PubMed] [Google Scholar]
- 53.Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 54.Steffen Y, Schewe T, Sies H. (-)-Epicatechin elevates nitric oxide in endothelial cells via inhibition of NADPH oxidase. Biochem Biophys Res Commun. 2007;359:828–833. doi: 10.1016/j.bbrc.2007.05.200. [DOI] [PubMed] [Google Scholar]
- 55.Litterio MC, Jaggers G, Sagdicoglu Celep G, Adamo AM, Costa MA, Oteiza PI, Fraga CG, Galleano M. Blood pressure-lowering effect of dietary (-)-epicatechin administration in L-NAME-treated rats is associated with restored nitric oxide levels. Free Radic Biol Med. 2012;53:1894–1902. doi: 10.1016/j.freeradbiomed.2012.08.585. [DOI] [PubMed] [Google Scholar]
- 56.Chuang CC, Martinez K, Xie G, Kennedy A, Bumrungpert A, Overman A, Jia W, McIntosh MK. Quercetin is equally or more effective than resveratrol in attenuating tumor necrosis factor-{alpha}-mediated inflammation and insulin resistance in primary human adipocytes. Am J Clin Nutr. 2010;92:1511–1521. doi: 10.3945/ajcn.2010.29807. [DOI] [PubMed] [Google Scholar]
- 57.Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes. 2002;51:1319–1336. doi: 10.2337/diabetes.51.5.1319. [DOI] [PubMed] [Google Scholar]
- 58.Gil A, Maria Aguilera C, Gil-Campos M, Canete R. Altered signalling and gene expression associated with the immune system and the inflammatory response in obesity. Br J Nutr. 2007;98(Suppl 1):S121–126. doi: 10.1017/S0007114507838050. [DOI] [PubMed] [Google Scholar]
- 59.Mackenzie GG, Carrasquedo F, Delfino JM, Keen CL, Fraga CG, Oteiza PI. Epicatechin, catechin, and dimeric procyanidins inhibit PMA-induced NF-kappaB activation at multiple steps in Jurkat T cells. FASEB J. 2004;18:167–169. doi: 10.1096/fj.03-0402fje. [DOI] [PubMed] [Google Scholar]
- 60.Mackenzie GG, Oteiza PI. Modulation of transcription factor NF-kappaB in Hodgkin’s lymphoma cell lines: effect of (-)-epicatechin. Free Radic Res. 2006;40:1086–1094. doi: 10.1080/10715760600788396. [DOI] [PubMed] [Google Scholar]
- 61.Fraga CG, Litterio MC, Prince PD, Calabro V, Piotrkowski B, Galleano M. Cocoa flavanols: effects on vascular nitric oxide and blood pressure. J Clin Biochem Nutr. 2011;48:63–67. doi: 10.3164/jcbn.11-010FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL. Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology. 2004;145:5087–5096. doi: 10.1210/en.2004-0478. [DOI] [PubMed] [Google Scholar]
- 63.Werstuck GH, Khan MI, Femia G, Kim AJ, Tedesco V, Trigatti B, Shi Y. Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes. 2006;55:93–101. [PubMed] [Google Scholar]
- 64.Sage AT, Holtby-Ottenhof S, Shi Y, Damjanovic S, Sharma AM, Werstuck GH. Metabolic syndrome and acute hyperglycemia are associated with endoplasmic reticulum stress in human mononuclear cells. Obesity (Silver Spring) 2012;20:748–755. doi: 10.1038/oby.2011.144. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, Davis RJ, Flavell R, Brenner DA, Tabas I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280:21763–21772. doi: 10.1074/jbc.M501759200. [DOI] [PubMed] [Google Scholar]
- 66.Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology. 2006;147:3398–3407. doi: 10.1210/en.2005-1494. [DOI] [PubMed] [Google Scholar]
- 67.Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, Cheung P, Merali S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57:2438–2444. doi: 10.2337/db08-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ermakova SP, Kang BS, Choi BY, Choi HS, Schuster TF, Ma WY, Bode AM, Dong Z. (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006;66:9260–9269. doi: 10.1158/0008-5472.CAN-06-1586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.