

Abstract
Computational and theoretical methods are advancing protein design as a means to create and investigate proteins. Such efforts further our capacity to control, design and understand biomolecular structure, sequence and function. Herein, the focus is on some recent applications that involve using theoretical and computational methods to guide the design of protein sequence ensembles, new enzymes, proteins with novel cofactors, and membrane proteins.
Overview
Nature’s proteins fold to yield a variety of functionalities, including self-assembly, enzymatic catalysis and highly selective molecular recognition. The structure and function of proteins can potentially be specified through the careful selection of sequence. During the course of evolution, Nature varies the physical and chemical properties of a protein through mutation and variation of sequence to identify proteins having what are often precisely tuned functionalities. In the laboratory, well-folded proteins may also potentially be created through the careful choice of sequences. To do so, however, can be subtle and nontrivial. Proteins can contain tens to thousands of amino acid residues. Even for a single sequence, many conformations are possible by varying backbone and side-chain degrees of freedom, resulting in a large configurational space; this is true even for protein design, where the backbone tertiary structure is often largely predetermined. The interactions that stabilize proteins and guide the molecule to its folded state are largely noncovalent: van der Waals, hydrophobic, electrostatic, and hydrogen-bonding interactions. Given the subtlety of these interactions, reliable quantitative estimates of stability with respect to unfolding can be difficult to obtain computationally. In addition, the large numbers of candidate sequences can lead to a further combinatorial complexity in protein design: a small protein of only 100 amino acids has more than 10130 possible sequences using just the 20 naturally occurring amino acids. Nonetheless, theoretical methods have been proposed to surmount many of these difficulties and make accessible the design and study of nonnatural protein sequences and protein assemblies. Most methods take as input a target structure, which can be natural or created via computational modeling. An energy based objective function is used to quantify the compatibility between sequences and the target structure so as to identify either individual sequences or the properties of the ensemble of sequences, where the focus in each case is on sequences consistent with the targeted structure and functional properties. Optimization-based methods for identifying low-energy sequences include dead-end elimination, Monte Carlo simulated annealing, genetic algorithms, and optimization theory approaches [1-3]. On the other hand, probabilistic methods characterize the ensemble of sequences and use thermodynamic self-consistent field concepts or Monte Carlo sequence sampling to estimate the site-specific probabilities of the amino acids at variable residues [4,5]. In addition to the design of novel nonbiological protein-based molecular systems, computational protein design may be used to better understand protein stability and folding, to facilitate the study of natural proteins, and to enhance or tune the functions of natural proteins.
Herein we present some recent applications of computational methods for the design and engineering of proteins, where the designed proteins have been experimentally realized. Often a known protein is re-engineered so as to augment stability and functionality. The engineering of new functionality into proteins has seen recent advances, particularly with regard to catalytic activity. New protein structures (and sequences) have also been computationally designed, which may incorporate nonbiological components. Membrane proteins are a rich and important new frontier with regard to the design and redesign of proteins.
Exploring molecular diversity and combinatorial experiments
The specificities of protein-protein interactions have been explored using computational protein design. An approach for computationally designing specificity into coiled coil protein-interactions has been developed and used to identify partners for basic-region leucine zipper (bZIP) transcription factors [6*]. With the aid of experimental studies employing protein microarrays, many of the designed peptides, including those binding important oncoproteins, were selective for their intended targets. The results suggest that naturally occurring bZIPs have only sparsely sampled the range of possible interactions. Computational design methods may potentially be used to explore the interplay between stability and specificity in these transcription factors. Using similar microarrays, the effective “toolkit” for coiled-coils has been expanded by measuring pairwise interactions of 48 synthetic coiled coils and 7 human bZIP coiled coils using peptide microarrays [7]. Twenty-seven pairs of interacting peptides were identified that preferentially associate. Of special interest are heterospecific peptide pairs that participate in mutually orthogonal interactions, i.e., pairs that associate in a monogamous, selective fashion. Such “interactome” information can be used to create highly specific protein-protein intereactions and networks of interacting pairs, with potential appliations in synthetic biology.
Computational protein design can also guide the design of libraries of protein sequences. Different methods of generating combinatorial libraries of green fluorescent protein variants have been compared [8], and the percentage of fluorescent, functional proteins was largest when a structure-based computational method was used to design the library. A greater diversity of color was observed in designed libraries while fluorescence was maintained. With structure-based computational methods, function is more likely preserved even as the extent of mutations increases among the libraries. Similar structurally guided design methods have been used to generate focused combinatorial libraries of mCherry (red fluorescent protein) mutants [9]. The design sought to introduce interactions likely to cause red-shifts without destabilizing the protein, largely via destabilization of the electronic ground state and stabilization of the excited state of the natural chromophore. After screening only 5000 clones, three mutants were identified having emission wavelengths greater than 630 nm. Crystal structures of two of these mutants are consistent with the models and the interactions expected to produce the red shift.
Computational protein design in the context of combinatorial libraries has been used to design a protein-protein interface [10]. A variant of the ubiquitin-ligase E6AP has been designed to bind to a nonnatural partner, the NEDD8-conjugating enzyme Ubc12. Three E6AP libraries were constructed: a naive library in which all 20 amino acids were allowed at the 13 positions on the target-binding surface of E6AP; a library that varied the same 13 residues but disallowed mutations computationally identified to destabilize E6AP; and a directed library that used docking and sequence optimization. Both of the directed libraries showed more than 30-fold enrichment over the naive library after the first round of screening and yielded tight binders (KD < 100 nM) after four rounds of selection, whereas multiple rounds of selection with the naive library failed to identify binders with KD < 50 μM.
Computational, structure-based methods have been used to characterize the sequence space of peptides recognized by PDZ domains [11]. From analysis of phage display data, 70-80% of the most frequently observed amino acids by phage display are predicted within the top five ranked amino acids, and the computational results recover changes in specificity upon mutation.
New functionalities: catalysis and sensing
Enzymatic activity is a long-standing goal in protein design. Recently proteins have been computationally designed that catalyze reactions for which there is no known natural enzyme. A suitable protein scaffold is selected by identifying those consistent with positioning side chains to bind the substrate and stabilize transition states or high energy intermediates. As in nature, often the common triose phosphate isomerase (TIM) barrel fold is identified. Proteins have been so designed to bind and stabilize high energy intermediates involved in retro-aldol reactions [12*] and in Kemp eliminations [13*]. In each case, large fractions (32/72 and 8/59) of the experimentally studied designed proteins possessed enzymatic activity. For the Kemp elimination catalyst, in vitro evolution methods identified proteins having a 200-fold increase in kcat/Km compared the initial computational design. Both designed enzymes have impressive rate enhancements of several orders of magnitude compared to the uncatalyzed reactions, although their efficiencies (kcat/Km ~ 1-103 M-1 s-1) do not yet rival natural enzymes [12,13]. With the retroaldolase, substrate binding interactions and a shift in the the pKa of the catalytic lysine account for much of the enzyme’s rate acceleration [14]. The approach has been applied to the design of biomolecular Diels-Alder enzymes, two of which have the targeted catalytic activity and stereo specificity [15].
Computational protein design has been used to re-engineer loops near active sites of enzymes and alter specificity [16]. A loop in human guanine deaminase has been redesigned so that interactions are made with a new substrate ammelide. Compared to the wild type, the redesigned enzyme is 100-fold more active on ammelide and 104-fold less active on guanine (kcat/Km =0.15 M-1 s-1). The structure of the designed protein was confirmed by X-ray crystallographic analysis and is in close agreement with the computational model.
A perimplasmic binding protein that has been computationally re-engineered to bind trinitrotoluene (TNT) has been incorporated into a signaling pathway in plants, where presence of the ligand leads to a degreening response in the leaves of a detector plant [17]. The work provides an example of how engineered plants may be used as environmental sensors.
Cofactors and de novo designed proteins
With the addition of cofactors, proteins can acquire functionally related properties not accessible with the amino acids alone. Such cofactors range from metal ions to large macromolecules, e.g., porphyrin based systems. Recently, protein structures have been designed from scratch to accommodate such cofactors. In this regard, four-helix bundle motifs are particularly versatile as the structure can be geometrically parameterized and varied using a few geometric variables that dictate the global structure [18]. Such methods have been used to create di-iron and di-manganese proteins, which are convenient systems for controlling and investigating binding and catalytic properties exhibited by these metalloproteins [19-22]. A 114-residue helical protein that presents such a di-nuclear binding site has been computationally designed, and was found to be highly stable, particularly with the metals present, despite having a large number of ionizable groups within its interior [23]. Recently, the structure of the Zn-version of the protein has been elucidated using a synthesis of NMR spectral data and quantum chemical and modeling calculations, where the calculations are especially useful in refining the structure near the dimetal site. This experimentally determined structure is in excellent agreement with the template structure used in the computational design of the protein [24*].
Computational de novo design can be used to arrive at novel protein assemblies containing nonbiological cofactors. Such systems have potential applications to biomaterials, electrooptics, and as tools to explore charge and excitation transfer in proteins. Incorporating nonbiological cofactors into proteins can potentially yield biomolecular systems with properties that are not accessible using only natural amino acids and biological cofactors. For large, complex cofactors there may be no appropriate scaffolds among known natural protein structures. Novel structures and proteins must be created. Computational design has been applied to arrive at a protein framework that encapsulates a synthetic cofactor [25], yielding a computationally designed protein that selectively binds two equivalents of a non-biological cofactor, a diphenyl-derivatized iron porphyrin (DPP-Fe). The protein structures and sequences are created so as to be consistent with coordination of the porphyrins metal ion by His and provide a complementary interior binding cavity. This approach has been extended to create modular metalloporphyrin peptide assemblies of varying lengths [26*]; the two-metalloporphyrin array has been elongated into a four-metalloporphyrin array consistent with a coiled-coil repeat. Experimental investigations of these elongated proteins are consistent with the targeted cofactor-binding stoichiometry and specificity. Recently, a related protein has been computationally designed that selectively binds a nonbiological photoactive zinc porphyrin (DPP-Zn), which assembles as an A2B2 heterotetramer [27]. These efforts exemplify how computational design can be used to create linear assemblies of ordered electronically and optically active cofactors. As opposed to oligomeric symmetric structures, single-chain helical proteins can provide interiors that better complement asymmetric cofactors. Such proteins may be over-expressed and noncovalently bind synthetic non-natural porphyrin cofactors. Experiments involving such a computationally designed protein are consistent with a high specificity of binding to the targeted Fe-DPP cofactor and suggest the protein is well-structured [28]. Designed amphipathic helical bundle proteins that bind related cofactors can be directed to self-order at aqueous interfaces, conferring vectorial orientation to the proteins and their cofactors [29,30].
Membrane proteins
Proteins associated with lipid bilayers are integral to the structure and function of biological membranes. Designing novel membrane proteins has applications toward better understanding membranes, membrane proteins, membrane-associated biomolecular processes, and controlling membrane integrity and transport. Since they are essential to many cellular processes, membrane proteins are among the most common drug targets. Computational protein design has been applied to refine models of G-protein coupled receptors. The effects of mutations on the oligomerization state are used to confirm atomistic models and better understand these important receptors [31].
Transmembrane proteins in particular span the bilayer and have large numbers of exterior hydrophobic residues that complement the bilayer’s hydrophobic interior. Consequently, these proteins are typically aggregation prone and difficult both to obtain and to purify. Most structural and many biophysical studies of membrane proteins involve their dispersal in aqueous media. This can be accomplished using detergents, lipids, auxiliary proteins, and reconstitution in membrane-like environments (bilayers, disks, micelles, vesicles and oriented bilayers). Such approaches can be used to obtain diffraction-quality crystals or dispersed protein suitable for NMR studies. Obtaining such solubilizing conditions for biophysical and structural studies, however, remains subtle and labor intensive.
An alternative is to redesign a membrane protein as a soluble variant, while seeking to retain the parent protein’s structure and some of its functionally related properties. Using computational design, water-soluble variants of transmembrane proteins have been designed, providing a proof of principle for creating variants that facilitate studies of their structures and function. Recently the solution structure of a computationally designed water-soluble variant of the bacterial potassium ion channel KcsA has been determined [32,33*]. The water-soluble protein contains 29 designed exterior mutations in each of the four 104-residue subunits. The structure is in good agreement with both the tertiary and quaternary structure of the membrane-soluble, wild-type structure. Not surprisingly, the water-soluble form exhibits larger structural fluctuations than the membrane form, but the residues in the selectivity filter fo the protein are highly sensitive to potassium ion concentration, consistent with their functional signifcance. These findings highlight the promise of developing water-soluble variants of membrane proteins—for biophysical studies and for therapeutic development—using computational redesign of sequence.
Transmembrane proteins can be computationally designed in order to control their activity and explore intra-membrane protein-protein interactions. Peptides have been computationally designed that target specific transmembrane helices to modulate the activity of integrins. The designed peptides discriminate between the transmembrane helices of two closely related integrins (αIIbβ3 and αvβ3), and the specificity is obtained using complementary peptide-helix van der Waals interactions [34*]. This approach has been applied to the design of an anti-RIIb peptide that targets isolated full-length integrin RIIb. The designed peptides take on transmembrane alpha-helical structures that do not disrupt the bilayer [35]. Computational design methods can be used to identify mutations that disrupt the interface between the β3 and the αIIβ and αv [36]. Destabilizing, stabilizing, and neutral mutations were computationally identified in the β3 region, yielding distinct sets of mutations that would modulate each interface. The predictions were then tested using cell-based fibrinogen and osteopontin binding assays. These results suggest that that the a stalk helical interface and its stability are involved in maintaining integrins in inactive conformations and illustrate how such design efforts can be used to understand integrin mutations.
A helical membrane protein PRIME has been computationally designed that positions two nonbiological iron diphenylporphyrins (FeDPP) so that they are poised to form a multicentered pathway for transmembrane electron transfer [37]. As with previous water soluble designs [26], four helices form bundle with D2 symmetry that binds two equivalents of the FeDPP cofator. Experimental characterization is consistent with the protein’s binding the cofactor with high affinity and specificity and with the expected stoichiometry and geometry.
Computational modeling has been used to probe intramembrane protein-protein interfaces, with an emphasis on the interactions that confer orientation and proximity [38]. A model membrane soluble protein, MS1, was used to explore the sensitivity of the dimer structure to sequence. Surprisingly, experimental studies reveal that the stability of MS1 dimer is greatest when Gly is at each “a” position of the heptad repeat (MS1-Gly), followed by Ala, Val, Ile in decreasing stability. MS1-Gly prefers to form antiparallel dimers, MS1-Ala forms a mixture of parallel and antiparallel dimers, while MS1-Val and MS1-Ile prefer parallel dimers. Calculations based on conformational searching and rotamer optimization were consistent with the measured stabilities and preferences for parallel/antiparallel orientations. These findings can be interpreted in terms of the van der Waals and electrostatic interactions present, which appear to dominate the stability and orientational preferences of the dimers.
Proteins have been designed to provide controllable integrity of a bilayer. A natural alpha-helical cell-lytic peptide, mastoparan X, has been re-engineered to bind metal cations. Binding of Zn(II) or Ni(II) stabilizes the peptide’s amphiphilic structure, leading to lysis of cells and vesicles [39].
Outlook
Computational protein design continues to provide tools to create proteins having desired structures, properties, and functionalities. Mutagenesis is a central tool for addressing molecular diversity associated with proteins and is regularly used to investigate protein structure and function. Computationally guided protein design stands to make such mutation-based studies more informative and effective and can also provide novel protein variants that are much easier to manipulate while retaining structural and functional properties of interest. The design of proteins having the efficiency and specificity of nature’s proteins is likely to remain challenging, but the synergy of combining computational design and experimental studies will continue to advance our abilities to engineer novel proteins, control biological activity, and better understand nature’s proteins. Lastly, incorporating nonnatural functions and components, e.g., nonbiological amino acids and cofactors, will yield protein-based systems and materials having properties not available with nature’s proteins.
Figure 1.
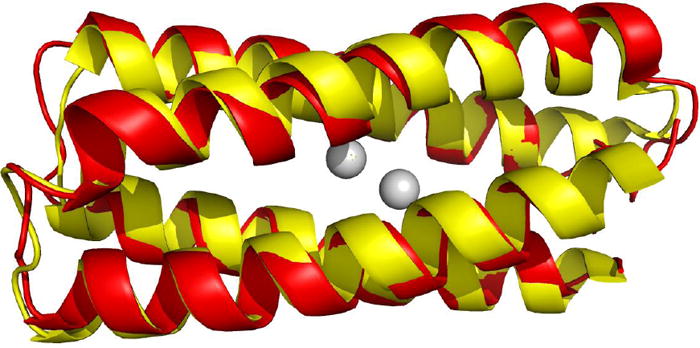
Superposition of the computational model of the single chain dinuclear metalloprotein DFsc (yellow) [23] and the experimentally determined structure obtained using NMR spectroscopy with QM/MM refinement (red) [24].
Figure 2.
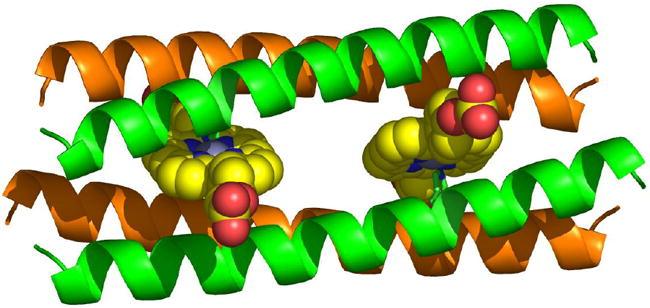
A2B2 protein designed to accommodate nonbiological cofactors (diphenyl Zn porphyrins) [27]. Protein comprises two distinct peptides (green, orange). Metal-coordinating His residue is rendered green.
Figure 3.
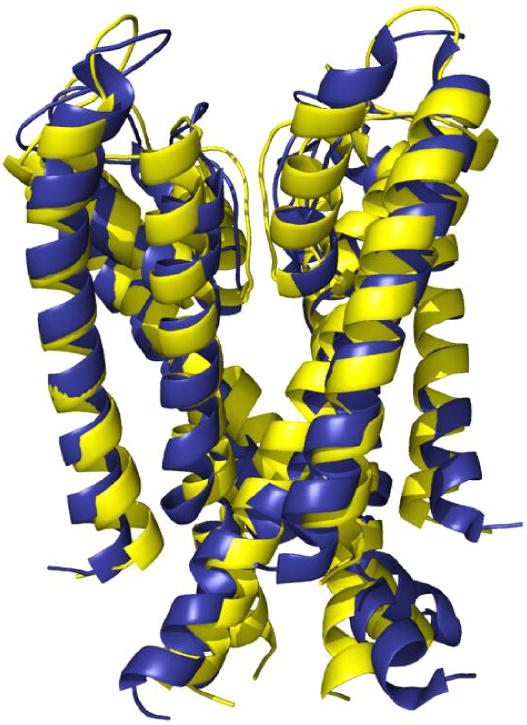
Superposition of model structure of water soluble variant of the transmembrane portion of the bacterial potassium ion channel KcsA (yellow) [32] and its experimentally determined structure (blue) (pdb accession code: 1K4E) [33].
Acknowledgments
The author gratefully acknowledge support from the US Department of Energy (DE-FG02-04ER46156), the National Institutes of Health (P01 GM55876, R01 HL085303), the University of Pennsylvania’s Nano/Bio Interface Center through the National Science Foundation (NSF) NSEC DMR-0425780. The author thanks the Laboratory for Research on the Structure of Matter through NSF MRSEC DMR05-20020 for infrastructural support. The figures were rendered using PyMol (DeLano Scientific LLC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Park S, Fu X, Wang W, Yang X, Saven JG. Computational protein design and discovery. Annual Reports on the Progress of Chemistry Section C. 2004;100:195–236. [Google Scholar]
- 2.Floudas CA, Fung HK, McAllister SR, Monnigmann M, Rajgaria R. Advances in protein structure prediction and de novo protein design: A review. Chemical Engineering Science. 2006;61:966–988. [Google Scholar]
- 3.Lehmann A, Lanci CL, Petty TJ, II, Kang SG, Saven JG. Protein Design: Tailoring Sequence, Structure and Folding Properties. In: Muñoz V, editor. Protein Folding, Misfolding and Aggregation: Classical Themes and Novel Approaches. Royal Society of Chemistry Biomolecular Sciences; 2008. pp. 188–213. [Google Scholar]
- 4.Park S, Kono H, Wang W, Boder ET, Saven JG. Progress in the development and application of computational methods for probabilistic protein design. Computers & Chemical Engineering. 2005;29:407–421. [Google Scholar]
- 5.Yang X, Saven JG. Computational methods for protein design and protein sequence variability: biased Monte Carlo and replica exchange. Chemical Physics Letters. 2005;401:205–210. [Google Scholar]
- 6*.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–U852. doi: 10.1038/nature07885.. The authors probe specificity of protein interactions via protein design. An elegant use of computational and micro-array data explores the effectiveness of design and the range of possible interactions between bZIP domain partners.
- 7.Reinke AW, Grant RA, Keating AE. A synthetic coiled-coil interactome provides heterospecific modules for molecular engineering. J Am Chem Soc. 2010;132:6025–6031. doi: 10.1021/ja907617a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Treynor TP, Vizcarra CL, Nedelcu D, Mayo SL. Computationally designed libraries of fluorescent proteins evaluated by preservation and diversity of function. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:48–53. doi: 10.1073/pnas.0609647103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chica RA, Moore MM, Allen BD, Mayo SL. Generation of longer emission wavelength red fluorescent proteins using computationally designed libraries. Proc Natl Acad Sci U S A. 2010;107:20257–20262. doi: 10.1073/pnas.1013910107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guntas G, Purbeck C, Kuhlman B. Engineering a protein-protein interface using a computationally designed library. Proc Natl Acad Sci U S A. 2010;107:19296–19301. doi: 10.1073/pnas.1006528107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith CA, Kortemme T. Structure-based prediction of the peptide sequence space recognized by natural and synthetic PDZ domains. J Mol Biol. 2010;402:460–474. doi: 10.1016/j.jmb.2010.07.032. [DOI] [PubMed] [Google Scholar]
- 12*.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692.. De novo designed enzymes that catalyze nonbiological reactions. The combined presentation of design, functional characterization and structure makes for a strong presentation. The authors mention that these systems do not yet have the efficiency fo natural proteins, suggesting that much remains to be done in the field of enzyme design.
- 13*.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–U194. doi: 10.1038/nature06879.. De novo designed enzymes that catalyze nonbiological reactions. The combined presentation of design, functional characterization and structure makes for a strong presentation. The authors mention that these systems do not yet have the efficiency fo natural proteins, suggesting that much remains to be done in the field of enzyme design.
- 14.Lassila JK, Baker D, Herschlag D. Origins of catalysis by computationally designed retroaldolase enzymes. Proc Natl Acad Sci U S A. 2010;107:4937–4942. doi: 10.1073/pnas.0913638107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St Clair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy PM, Bolduc JM, Gallaher JL, Stoddard BL, Baker D. Alteration of enzyme specificity by computational loop remodeling and design. Proc Natl Acad Sci U S A. 2009;106:9215–9220. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Antunes MS, Morey KJ, Smith JJ, Albrecht KD, Bowen TA, Zdunek JK, Troupe JF, Cuneo MJ, Webb CT, Hellinga HW, et al. Programmable ligand detection system in plants through a synthetic signal transduction pathway. PLoS One. 2011;6:e16292. doi: 10.1371/journal.pone.0016292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grigoryan G, Degrado WF. Probing designability via a generalized model of helical bundle geometry. J Mol Biol. 2011;405:1079–1100. doi: 10.1016/j.jmb.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maglio O, Nastri F, Calhoun JR, Lahr S, Wade H, Pavone V, DeGrado WF, Lombardi A. Artificial di-iron proteins: solution characterization of four helix bundles containing two distinct types of inter-helical loops. Journal of Biological Inorganic Chemistry. 2005;10:539–549. doi: 10.1007/s00775-005-0002-8. [DOI] [PubMed] [Google Scholar]
- 20.Calhoun JR, Nastri F, Maglio O, Pavone V, Lombardi A, DeGrado WF. Artificial diiron proteins: From structure to function. Biopolymers. 2005;80:264–278. doi: 10.1002/bip.20230. [DOI] [PubMed] [Google Scholar]
- 21.Wei PP, Skulan AJ, Wade H, DeGrado WF, Solomon EI. Spectroscopic and computational studies of the de novo designed protein DF2t: Correlation to the biferrous active site of ribonucleotide reductase and factors that affect O-2 reactivity. Journal of the American Chemical Society. 2005;127:16098–16106. doi: 10.1021/ja053661a. [DOI] [PubMed] [Google Scholar]
- 22.Wade H, Stayrook SE, DeGrado WF. The structure of a designed diiron(III) protein: Implications for cofactor stabilization and catalysis. Angewandte Chemie-International Edition. 2006;45:4951–4954. doi: 10.1002/anie.200600042. [DOI] [PubMed] [Google Scholar]
- 23.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J Mol Biol. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 24*.Calhoun JR, Liu W, Spiegel K, Dal Peraro M, Klein ML, Valentine KG, Wand AJ, DeGrado WF. Solution NMR structure of a designed metalloprotein and complementary molecular dynamics refinement. Structure. 2008;16:210–215. doi: 10.1016/j.str.2007.11.011.. This work results in an experimentally determined structure of a di-nuclear metalloprotein that is in agreement with that resulting from the computational design. In addition, the authors show how quantum-mechanics/molecular-mechanics (QM/MM) modeling can be used to obtained high quality models consistent with NMR spectra, particularly for cases where the quality of the molecular force field used in refinement may be poor or incomplete.
- 25.Cochran FV, Wu SP, Wang W, Nanda V, Saven JG, Therien MJ, DeGrado WF. Computational de novo design and characterization of a four-helix bundle protein that selectively binds a nonbiological cofactor. Journal of the American Chemical Society. 2005;127:1346–1347. doi: 10.1021/ja044129a. [DOI] [PubMed] [Google Scholar]
- 26*.McAllister KA, Zou HL, Cochran FV, Bender GM, Senes A, Fry HC, Nanda V, Keenan PA, Lear JD, Saven JG, et al. Using alpha-helical coiled-coils to design nanostructured metalloporphyrin arrays. Journal of the American Chemical Society. 2008;130:11921–11927. doi: 10.1021/ja800697g.. The authors expand upon the computational design of helix bundle proteins to obtain extended systems with well positioned redox-active nonbiological chromophores.
- 27.Fry HC, Lehmann A, Saven JG, DeGrado WF, Therien MJ. Computational design and elaboration of a de novo heterotetrameric alpha-helical protein that selectively binds an emissive abiological (porphinato)zinc chromophore. J Am Chem Soc. 2010;132:3997–4005. doi: 10.1021/ja907407m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, et al. De novo design of a single-chain diphenylporphyrin metalloprotein. Journal of the American Chemical Society. 2007;129:10732–10740. doi: 10.1021/ja071199j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zou HL, Therien MJ, Blasie JK. Structure and dynamics of an extended conjugated NLO chromophore within an amphiphilic 4-helix bundle peptide by molecular dynamics simulation. Journal of Physical Chemistry B. 2008;112:1350–1357. doi: 10.1021/jp076643j. [DOI] [PubMed] [Google Scholar]
- 30.Zou HL, Strzalka J, Xu T, Tronin A, Blasie JK. Three-dimensional structure and dynamics of a de novo designed, amphiphilic, metallo-porphyrin-binding protein maquette at soft interfaces by molecular dynamics simulations. Journal of Physical Chemistry B. 2007;111:1823–1833. doi: 10.1021/jp0666378. [DOI] [PubMed] [Google Scholar]
- 31.Taylor MS, Fung HK, Rajgaria R, Filizola M, Weinstein H, Floudas CA. Mutations affecting the oligomerization interface of G-protein-coupled receptors revealed by a novel de novo protein design framework. Biophysical Journal. 2008;94:2470–2481. doi: 10.1529/biophysj.107.117622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slovic AM, Kono H, Lear JD, Saven JG, DeGrado WF. Computational design of water-soluble analogues of the potassium channel KcsA. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1828–1833. doi: 10.1073/pnas.0306417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Ma DJ, Tillman TS, Tang P, Meirovitch E, Eckenhoff R, Carnini A, Xu Y. NMR studies of a channel protein without membranes: Structure and dynamics of water-solubilized KcsA. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16537–16542. doi: 10.1073/pnas.0805501105.. NMR based studies of a computationally designed water soluble variant of a bacterial potassium ion channel. The experimentally derived structure is in agreement with the structure of the parent membrane bound form.
- 34*.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, DeGrado WF. Computational design of peptides that target transmembrane helices. Science. 2007;315:1817–1822. doi: 10.1126/science.1136782.. Computational protein design is used to obtain membrane proteins that selectively bind to integral membrane proteins, in this case integrins, and modulate their function.
- 35.Caputo GA, Litvinov RI, Li W, Bennett JS, DeGrado WF, Yin H. Computationally designed peptide inhibitors of protein-protein interactions in membranes. Biochemistry. 2008;47:8600–8606. doi: 10.1021/bi800687h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donald JE, Zhu H, Litvinov RI, DeGrado WF, Bennett JS. Identification of interacting hot spots in the beta3 integrin stalk using comprehensive interface design. J Biol Chem. 2010;285:38658–38665. doi: 10.1074/jbc.M110.170670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korendovych IV, Senes A, Kim YH, Lear JD, Fry HC, Therien MJ, Blasie JK, Walker FA, Degrado WF. De novo design and molecular assembly of a transmembrane diporphyrin-binding protein complex. J Am Chem Soc. 2010;132:15516–15518. doi: 10.1021/ja107487b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Kulp DW, Lear JD, DeGrado WF. Experimental and computational evaluation of forces directing the association of transmembrane helices. J Am Chem Soc. 2009;131:11341–11343. doi: 10.1021/ja904625b. [DOI] [PubMed] [Google Scholar]
- 39.Signarvic RS, Degrado WF. Metal-binding dependent disruption of membranes by designed helices. J Am Chem Soc. 2009;131:3377–3384. doi: 10.1021/ja809580b. [DOI] [PMC free article] [PubMed] [Google Scholar]