

Abstract
The liver is endowed with the ability to regenerate hepatocytes in response to injury. When this regeneration ability is impaired during liver injury, oval cells, which are considered to be postnatal hepatic progenitors, proliferate and differentiate into hepatocytes. Here we have demonstrated that 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) activates the nuclear receptor constitutive active/androstane receptor (CAR), resulting in proliferation of oval cells in mouse liver. Activation of CAR by DDC was shown by hepatic nuclear CAR accumulation and cytochrome P450 (CYP)2B10 mRNA induction after feeding a 0.1% DDC-containing diet to Car +/+ mice. After being fed the DDC diet, Car +/+, but not Car−/− mice, developed severe liver injury and an A6 antibody-stained ductular reaction in an area around the portal tract. Oval cell proliferation was confirmed by laser capture microdissection and real-time PCR; mRNAs for the two oval cell markers epithelial cell adhesion molecule and TROP2 were specifically induced in the periportal region of DDC diet-fed Car +/+, but not Car−/− mice. Although rates of both hepatocyte growth and death were initially enhanced only in DDC diet-fed Car +/+ mice, growth was attenuated when oval cells proliferated, whereas death continued unabated. DDC-induced liver injury, which differs from other CAR activators such as phenobarbital, occurred in the periportal region where cells developed hypertrophy, accumulated porphyrin crystals and inflammation developed, all in association with the proliferation of oval cells. Thus, CAR provides an excellent experimental model for further investigations into its roles in liver regeneration, as well as the development of diseases such as hepatocellular carcinoma.
Keywords: CAR, hepatic progenitor, laser capture microdissection, nuclear receptor, oval cell proliferation
The liver is endowed with the ability to regenerate hepatocytes in response to various types of injuries. It has also been suggested that when this regeneration ability is impaired, the postnatal hepatic progenitor oval cells proliferate.1 Oval cells are believed to differentiate into hepatocytes that restore liver functions, whereas they are also considered to be liver cancer stem cells.2 Chemical exposure is an injury that causes oval cell proliferation in rodents, and this has been utilized to investigate the molecular mechanism by which therapeutic treatment often results in liver injury, leading to proliferation of hepatic progenitor cells.3 Feeding a 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)-containing diet is one of the most efficient rodent models for chemical-induced oval cell proliferation; DDC is a porphyrinogenic chemical that causes oval cells in the liver to proliferate, but the molecular mechanism by which DDC elicits oval cell proliferation remains unknown at the present time. Here we utilized the DDC model to investigate the role of the nuclear xenobiotic receptor constitutive active/androstane receptor (CAR) in the proliferation of hepatic oval cells.
CAR (NR1I3), a member of the nuclear steroid/thyroid hormone receptor superfamily, was first characterized as a phenobarbital (PB)-activated factor that induces transcription of the cytochrome P450 2B (CYP2B) gene in livers, thus regulating hepatic drug metabolism.4 Subsequent studies showed that CAR also regulates other hepatic genes such as phosphoenoylpyruvate carboxylkinase, glucose 6-phosphatase and carnitine palmitoyltransferase, thus widening its role into hepatic energy homeostasis and energy-mediated diseases such as diabetes.5–7 Exemplified by the fact that CAR promotes the development of hepatocellular carcinoma in mice after chronic treatment with PB, drug activation of CAR can also become a risk factor that promotes the development of various drug-induced diseases including non-alcoholic steatohepatitis.8 Utilizing partial hepatectomized Car−/− mouse, we previously demonstrated that liver regeneration is more efficient in the presence of CAR.9 However, it has not been determined if CAR is involved in oval cell proliferation.
Here we have used Car−/− mice in C3H/HeNCrlBR, as well as C57BL/6 strains, Pxr−/− mice and their corresponding wild-type mice, feeding them with 0.1% DDC-containing diets for 2, 7 and 14 days. With the exception of the Car−/− mice, all DDC-fed mice were found to develop severe liver injury (ie, hepatomegaly and cholestasis), a ductular reaction and oval cell proliferation. We then confirmed that DDC activates CAR, and investigated oval cell proliferation in the livers of mice fed with DDC diets. Proliferating cell nuclear antigen (PCNA) and TUNEL assays were employed to examine cell growth and death, whereas laser capture microdissection (LCM) combined with real-time PCR were performed to identify the genes that were induced in the livers after feeding the DDC diets. An anti-A6 antibody was utilized to show a ductular reaction, as well as oval cell proliferation. Expression levels of the two oval cell markers epithelial cell adhesion molecule (EpCAM) and TROP2 mRNAs were measured to determine proliferation of oval cells. We also identified CAR-regulated genes that were differentially expressed in the hepatic centrilobular and/or periportal regions of DDC-fed Car +/+ and Car−/− mice, which may have roles in liver injury and oval cell proliferation.
MATERIALS AND METHODS
Materials
DDC was purchased from Sigma Chemicals (St Louis, MO). Control diets (Basal Diet 5755) and control diets supplemented with 0.1% DDC were both purchased from TestDiet (Richmond, IN). All other chemicals were obtained from commercial sources at the highest grade of purity available. Wild-type pcDNA3.1/mCAR that contained the full-length mouse CART176V mutant and (NR1)5-tk-luciferase reporter were reported previously.10
Animals and Treatment
All protocols and procedures were approved by the National Institutes of Health Animal Care and Use Committee and were in accordance with National Institutes of Health guidelines. Car +/+ and Car −/− mice on a C3H/HeNCrlBR (hereafter just called C3He) background were generated and characterized previously.11 Car +/+, Car −/−, Pxr +/+ and Pxr −/− mice on a mixed genetic background of 129/Sv and C57BL/6 were changed to C57BL/6 background by repeated backcrossing to C57BL/6J mice (Charles River Laboratories, Wilmington, MA) until microsatellite analysis showed that mice contained 98% of the C57BL/6 markers. All animals were housed in a temperature-controlled environment, with 12-h light/dark cycles with access to standard chow and water ad libitum. Age-matched groups of 10–12-week-old male mice were used for all the experiments. Three to six mice were used for each treatment group. Mice from each genotype were randomly divided into experimental groups and fed either a control diet or the control diet supplemented with 0.1% DDC. During the experimental period, the individual body weights and food intake were recorded twice a week. No significant differences were found in food and water consumptions among different groups of mice. 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene (TCPO-BOP) in PBS was ip injected once a week at a dose of 3 mg/kg body weight after 6 and 13 days of feeding with PBS (100 μl/ 25 g body weight) injected in control. Male mice were then killed and their sera and livers were collected after 2, 7 or 14 days of feeding the DDC diets. Each experiment was performed independently at least twice. Serum levels of alanine aminotransferase, alkaline phosphatase, total bilirubin, direct bilirubin, triglyceride, total cholesterol and total bile acids were measured using reagents and controls from Diagnostic Chemicals and the Cobas Mira plus CC analyzer (Roche Diagnostics).
Histology
Liver tissue specimens were fixed in 10% formalin, processed, embedded in paraffin, sectioned and stained with hematoxylin-eosin (H&E) or hematoxylin. Immunohistochemical analysis for A6, an oval cell specific marker, PCNA and CYP2B10 were performed by the avidin–biotin–peroxidase complex method (Vectastain ABC kit, Vector Laboratories, Burlingame, CA), using rat anti-A6 antibody (given by Dr VM Factor, NCI), a rabbit polyclonal anti-PCNA antibody (Santa Cruz Biotechnology, Santa Cruz, CA) and a rabbit polyclonal anti-CYP2B10 antibody,12 respectively. DAB substrates for peroxidase were used to visualize the antibody binding. At a magnification of ×200, total and PCNA-positive hepatocytes were counted in 12 periportal or centrilobular areas for each group in a blinded fashion. Apoptosis was directly detected using an ApopTag peroxidase in situ apoptosis detection kit (Millipore, Temecula, CA) according to the manufacturer’s instructions. Sections were deparaffinized and incubated with proteinase K and rinsed in terminal deoxynucleotidyl transferase reaction buffer for 60 min at 37 °C. Biotinylated nuclei were detected with avidin peroxidase and H2O2 containing DAB. At a magnification of × 200, the number of total and TUNEL positive hepatocytes were measured in nine areas for each group in a blinded fashion.
Laser Capture Microdissection
LCM was carried out on frozen liver tissue sections as described.13 Briefly, the sections were stained with 1% Cresyl Violet acetate, and periportal or centrilobular areas (Zone1 or 3) were captured by a MMI CellCut, laser capture microscope (The Molecular Machines & Industries, Haslett, MI). Forty to 60 regions (0.03–0.04 mm2 each) were collected, and within the portal cuts, both portal triad and hepatocytes were included (Supplementary Figure 1). Individual areas from four serial sections were dissected from the slides and captured in the caps of microcentrifuge tubes for RNA extractions. LCM was verified by light microscopic analysis. RNAs were extracted from the LCM samples using the Arcturus PicoPure Kit (MDS Analytical Technology, Sunnyvale, CA) and quantified by spectrophotometer.
Real-Time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen), the synthesis of first strand cDNA was performed using high capacity cDNA archive kit (Applied Biosystems, Foster City, CA) and real-time PCR analysis was performed using the 7900HT fast real-time PCR system (Applied Biosystems). Gene-specific primers were designed using Prime Express software (Applied Biosystems) or purchased as predesigned TaqMan gene expression assays containing a gene specific probe and primer mixture (Applied Biosystems). The assay identification number of predesigned TaqMan gene expression assays (gene, assay ID number) and sequences of the primers (mRNA, forward primer/reverse primer, 5′–3′) used in this study are described in Supplementary Table 1. The TaqMan rodent glyceraldehyde-3-phosphate dehydrogenase control reagent (Applied Biosystems) was used as an internal control. Real-time PCR data were obtained using TaqMan Universal PCR Master Mix or SYBR Green Master Mix (Applied Biosystems).
Western Blot
The nuclear extracts were prepared as previously described14 and resolved on a SDS-10% polyacrylamide gel, transferred to an Immobilon-P membrane (Millipore, Bedford, MA), and incubated overnight at 4 °C with anti-CAR antibody (1:5000 dilutions; Perseus Proteomics, Tokyo, Japan). Subsequently, they were incubated with HRP-conjugated anti-mouse IgG (1:5000 dilution; Santa Cruz Biotechnology) and protein bands were visualized by ECL Plus Western blotting detection (Amersham Biosciences).
Data Analysis
All experimental data are shown as means±s.d., with the significant differences determined by a one-way factorial analysis of variance for each group. The levels of significance for all statistical analyses were set at P<0.05 or P<0.01.
RESULTS
DDC Activates CAR in the Liver
Upon activation by therapeutic drugs such as PB, CAR accumulates in the nucleus, directly binding to the Cyp2b10 gene and inducing its transcription.4,15 Feeding a 0.1% DDC-containing diet (hereafter called DDC diet) for 2 days resulted in the nuclear accumulation of CAR in Car+/+/C3He, but not the Car−/−/C3He mice (Figure 1a). Using LCM, centrilobular and periportal regions of the livers were separately collected, from which RNAs were prepared for real-time PCR (Supplementary Figure 1). CAR mRNA was present in both the centrilobular and periportal regions, with levels in the centrilobular region 5 to 10-fold higher than in the periportal region. Levels of CAR mRNA were not affected by DDC treatment (Figure 1b). The hepatic levels of eight other nuclear receptors were also examined and were found not to have altered expression by feeding DDC (Supplementary Table 2). Induction of CYP2B10 mRNA was observed in both centrilobular and periportal hepatocytes in the livers of DDC diet-fed Car +/+/C3He, but not the Car−/−/C3He mice (Figure 1c). In addition to CYP2B10 mRNA, the six other cytochrome P450 mRNAs were neither induced nor repressed: CYP1A1, CYP2A5, CYP2C29, CYP2E1, CYP3A11 and CYP4A10 mRNAs (Supplementary Table 2). These results indicate that DDC activates CAR, inducing CYP2B10 in both centrilobular and periportal hepatocytes. However, DDC appears to be one of many therapeutics such as PB that indirectly activate CAR, since like PB, DDC did not activate a CAR-responsive reporter gene in cell-based transient transfection assays (data not shown).
Figure 1.
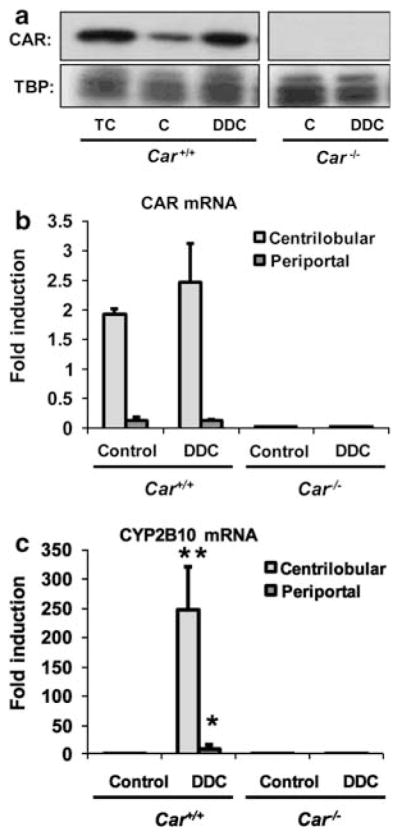
Constitutive active/androstane receptor (CAR) activation by 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC). (a) Western blotting analysis of nuclear CAR. Nuclear extracts were prepared from the liver of 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP)-treated (TC), control (C) and DDC diet-fed Car +/+ mice, and 20 μg of protein was subjected to western blot analysis, using an anti-CAR antibody as described in the Materials and Methods section. Results of real-time PCR assays for CAR mRNA (b) and cytochrome P450 (CYP)2B10 mRNA (c) are also shown. Total RNA was isolated from microdissected periportal lesions and centrilobular regions using the Arcturus PicoPure kit (MDS Analytical Technology). Real-time PCR was performed as indicated in Materials and Methods section. TBP was used as a loading control for nuclear proteins. The bar diagram shows the fold induction (mean±s.d.) relative to the gene expression in whole liver lysates from control diet -fed Car +/+ mice. * and ** indicate a significant difference at P<0.05 and P<0.01, respectively, in comparison with control diet.
CAR-Dependent Oval Cell Proliferation
Following feeding of a DDC diet for 14 days, there was hepatocellular hypertrophy in centrilobular and periportal areas, accompanied by proliferation of oval cells/biliary epithelial cells in the periportal areas in Car +/+/C3He mice (Figure 2a). Hypertrophy progressed before the ductular reaction, as early as 2 days after feeding DDC (Supplementary Figure 2). Neither hypertrophy nor ductular reaction developed in the livers of DDC diet-fed Car−/−/C3He mice. A6 is a biliary cell marker and is also known to be expressed in oval cells.16 Figure 2b shows the consecutive liver sections, which were stained by H&E and by an anti-A6 antibody. The biliary epithelial cells in the portal tracts were immunostained with an anti-A6 antibody in the livers of both the control diet-fed Car +/+/C3He and Car−/−/C3He mice. Feeding a DDC diet increased the staining intensity and expanded the area of the staining around a niche of the periportal regions in the livers of Car +/+/C3He, but not Car−/−/C3He mice. Furthermore, the A6-positive cells appeared to migrate into the surrounding region that largely consisted of hepatocytes. Expression of two members of the tumor-associated calcium signal transducer (TACSTD) family was examined, EpCAM (TACSTD1) has been used as an oval cell marker and TROP2 (TACSTD2) was recently characterized as a specific oval cell marker.17 Expression of these two mRNAs increased after DDC feeding; in only the Car +/+/C3He mice did these two mRNAs increase their expression levels and in only the periportal region where oval cells should have proliferated (Figure 2c). These results confirmed that DDC induced oval cell proliferation only when CAR was present.
Figure 2.
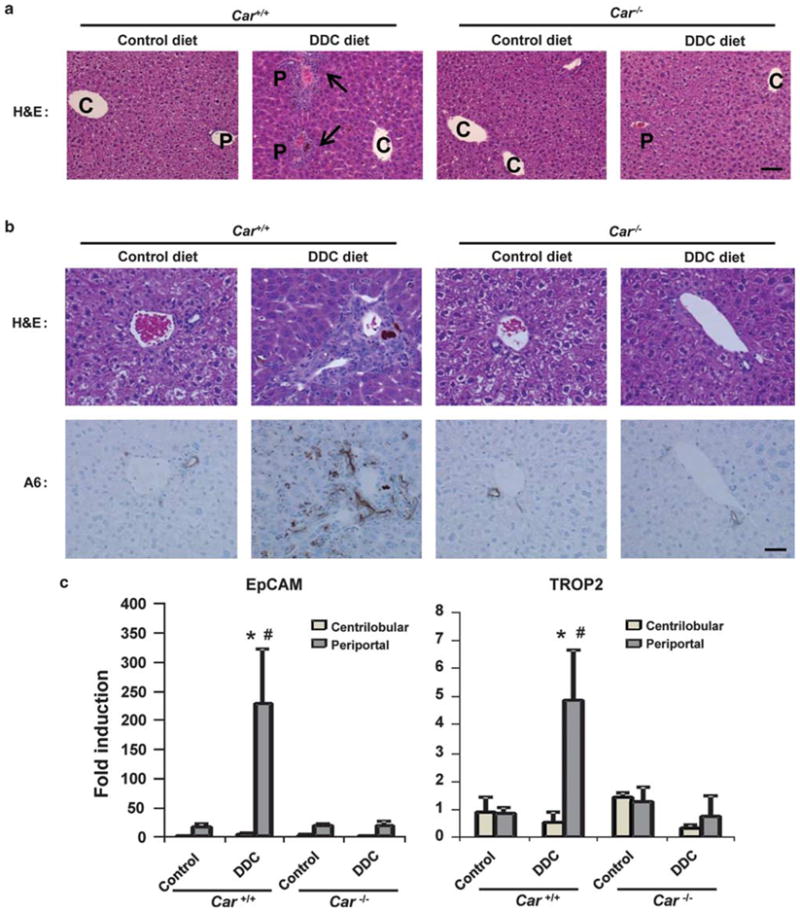
Constitutive active/androstane receptor (CAR)-dependent proliferation of oval cells. (a) Hematoxylin and eosin staining (H&E). The arrows indicate the ductular reactions and, C and P indicate central and portal veins, respectively. Scale of bar, 100 μm. (b) The upper and lower rows show H&E and anti-A6 antibody (A6) staining, respectively. Scale of bar, 200 μm. (c) Real-time PCR for the oval cell markers epithelial cell adhesion molecule (EpCAM) mRNA and TROP2 mRNA in total RNA isolated from microdissected centrilobular and periportal regions. The bars indicate the fold inductions (mean±s.d.) relative to their levels in total livers of the control diet-fed Car +/+ mice. * indicates a significant difference at P<0.05 in comparison with control diet and #indicates a significant difference at P<0.01 in comparison between Car +/+ and Car−/− mice.
CAR-Dependent Liver Injury and Cholestasis
Feeding on the DDC diet decreased the body weights and increased the liver weights of Car +/+/C3He mice, but not the Car−/−/C3He mice (Supplementary Table 3). The increased serum levels of ALT, ALP, bilirubin and bile acids suggested that the Car +/+/C3He mice, but not Car−/−/C3He mice developed liver injury and cholestasis after feeding the DDC diet (Figure 3a). Consistent with these observations, the two bilirubin and bile acid transporters NTCP and OATP1 were repressed in only the DDC diet-fed Car +/+/C3He mice (Figure 3b). The other transporters, which were neither induced nor repressed, were BSEP, MRP2, MDR1, MDR2, OATP2 and OATP4 (Supplementary Table 2). With respect to porphyria, both Car +/+/C3He and Car−/−/C3He mice accumulated numerous small porphyrin crystals in the centrilobular hepatocytes after feeding DDC (Figures 4d and e). Only the Car +/+/C3He mice were found to accumulate these porphyrin crystals in the periportal regions, which eventually grew into multiple larger crystals (Figures 4d and e). Neither 5-aminolevulinic acid synthase 1 mRNA, nor ferrochelatase mRNA was induced in the DDC diet-fed mice (Supplementary Table 2).
Figure 3.

Constitutive active/androstane receptor (CAR)-dependent liver injury. (a) The serum levels of ALT, ALP, total bilirubin (T-Bil) and total bile acids (TBA) in mice fed with the DDC diet for14 days. (b) Real-time PCR for NTCP and OATP1mRNAs using total RNA isolated from microdissected centrilobular and periportal regions. The bars indicate the fold inductions (mean±s.d.) relative to their levels in total livers of the control diet-fed Car +/+ mice. * and #indicate a significant difference at P<0.01 in comparison with control diet and in comparison between Car +/+ and Car−/− mice, respectively.
Figure 4.

Constitutive active/androstane receptor (CAR)-dependent inflammation, cell growth and death. (a–c) Total RNA was isolated from microdissected centrilobular and periportal regions and real-time PCR was performed for TNFα, TGFβ and growth arrest and DNA damage-induced protein 45 family (GADD45)β mRNAs as described in the Materials and Methods section. The bars indicate the fold inductions (mean±s.d.) relative to their levels in total livers of the control diet-fed Car +/+ mice. * and #indicate a significant difference at P<0.01 in comparison with control diet and in comparison between Car +/+ and Car−/− mice, respectively. (d and e) Car +/+ (a, b, c and d) and Car−/− (e, f, g and h) mice were fed with control diet (a and e) or the 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diets for 2 (b and f), 7 (c and g) and 14 (d and h) days. (d) Proliferating cell nuclear antigen (PCNA) assays: Sections were prepared from the livers of these mice and subjected to immunohistochemistry using an anti-PCNA antibody. The numbers of total and PCNA-positive hepatocytes were counted in nine different areas of centrilobular and of periportal regions in PCNA-stained pictures, with a 200-fold magnification to generate means±s.d. estimates for the PCNA positives in the left figure. The arrows indicate porphyrin depositions. The picture of the left side in each pair is centrilobular region, whereas the right is periportal region. * and #indicate a significant difference at P<0.01 in comparison with control diet and in comparison with centrilobular regions, respectively. Scale of bar is 200 μm. (e) TUNEL assays: Liver sections were immunostained with an anti-dUTP antibody. The numbers of total and TUNEL-positive hepatocytes were counted in nine different areas in TUNEL-stained pictures with a 200-fold magnification to generate means±s.d. for the TUNEL positives in the left figure. The means±s.d. are shown. The arrows indicate porphyrin depositions. * and #indicate a significant difference at P<0.01 in comparison with control diet and in comparison with Car +/+ mice fed with the DDC diet for 2 days. Scale of bar is 200 μm.
CAR-Dependent Inflammation, Cell Growth and Death
Oval cell proliferation has been suggested to occur when the regeneration of hepatocytes is hampered in injured liver. The expression of the liver-regeneration-related cytokine and growth/death-related factors, TNFα and HGF, TGFα, TGFβ, growth arrest and DNA damage-induced protein 45 family (GADD45)α, GADD45β and GADD45γ, were examined in the livers.18–20 Of these, TNFα, TGFβ and GADD45β were found to have their CAR-dependent inductions after feeding the DDC diet (Supplementary Table 2). Both TNFα and TGFβ mRNAs, which are known to be expressed in non-hepatic cells, were expressed at a higher level at the late stage of the feeding. These increases were largely restricted to the periportal region, indicating that this region became inflammatory (Figures 4a and b). These late increases in TNFα and TGFβ expression in the periportal region may also have partially coincided with oval cell proliferation. The expression of GADD45β mRNA increased sharply by 20- and 5-fold in the centrilobular and periportal hepatocytes, respectively, after 2 days of feeding in Car +/+/C3He mice (Figure 4c). These levels were maintained throughout the feeding, with an additional increase observed in the periportal region after 14 days, which may be due to increased expression of GADD45β in non-hepatic cells. Given the role of GADD45β as an anti-apoptotic factor, these time-dependent increases in GADD45β in both centrilobular and periportal regions, matched well to those of the cell growth rates in the DDC diet-fed Car +/+mice (Figures 4d and e).
PCNA staining assays showed that cell growth transiently increased in the livers of DDC diet-fed Car +/+/C3He mice, but not Car−/−/C3He mice; the growth rates increased four-to five-fold in both centrilobular and periportal regions after 2 to 7 days of feeding the DDC diet, followed by a significant attenuation after 14 days (Figure 4d). No significant cell growth occurred in the DDC diet-fed Car−/−/C3He mice. In contrast to the transient increase in cell growth, TUNEL assays revealed that a gradual increase in cell death began 2 days after feeding the DDC diet, reaching a maximum at 7 days and was maintained thereafter in the Car +/+, but not Car−/− mice (Figure 4e). Because the rates of cell death were low, it was problematic to separately account for cell death in centrilobular and periportal hepatocytes. Thus, the increase in cell growth first preceded that of cell death, with the maximum rate of cell death occurring after 7 days of DDC feeding, implying that the regenerative ability of the livers could have been diminished at the time when oval cells proliferated after feeding the DDC diet.
Co-Treatment With CAR Ligand TCPOBOP and Oval Cell Proliferation
TCPOBOP is the most potent CAR-activating ligand in mice. Car +/+/C3He and Car−/−/C3He mice were either treated with TCPOBOP alone for 24 h, or fed the DDC diet for 14 days and co-treated with TCPOBOP for 24 h. Liver sections were prepared from these mice and subjected to immunohistochemical analysis using anti-A6 or anti-CYP2B10 antibodies (Supplementary Figure 3). First, H&E staining revealed that TCPOBOP caused hypertrophy in the centrilobular, but not the periportal hepatocytes. TCPOBOP neither induced a ductular reaction, nor affected the DDC-induced development of this reaction in the periportal regions (1st column of Figure 5); similar results were found with the anti-A6 antibody staining (2nd column of Figure 5). As can be seen in the 3rd column of Figure 5, immunostaining with an anti-CYP2B10 antibody show that TCPOBOP induced the expression of CYP2B10 protein predominantly in the centrilobular hepatocytes, whereas DDC induced it equally in both centrilobular and periportal hepatocytes. Non-hepatic cells within a ductular reaction were not stained by the anti-CYP2B10 antibody. Co-treatment with TCPOBOP intensified the DDC-induced staining pattern and further widened the area of the antibody-positive periportal hepatocytes to an area within the ductular reaction (3rd column of Figure 5). None of this induced staining was observed in the livers of Car−/− mice; H&E staining confirmed that Car−/− livers did not develop a ductular reaction (4th column of Figure 5), and neither anti-A6, nor anti-CYP2B10 antibodies stained any of the liver sections from Car−/−/C3He mice (5th and 6th columns of Figure 5).
Figure 5.

Immunohistochemical study for A6 and cytochrome P450 (CYP)2B10 in the livers after 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) co-treatment. Continuous series of the liver sections were stained by hematoxylin and eosin (H&E), an anti-A6 antibody or anti-CYP2B10 antibody. C and P indicate central and portal veins. The upper picture of each pair is centrilobular region, whereas the lower picture is periportal region. Scale of bar is 200 μm.
From these same livers, LCM was employed to separately collect the centrilobular and periportal regions for subsequent real-time PCR assays (Supplementary Figure 1). As observed in Figure 2, the DDC diet dramatically increased the mRNAs of oval cell markers TROP2 and EpCAM in the periportal regions; TCPOBOP treatment did not increase these mRNAs in the Car +/+/C3He mice (Figures 6a and b), which is consistent with the anti-A6 antibody staining shown in the 2nd column of Figure 5. In addition, co-treatment with TCPOBOP did not significantly affect the levels of these mRNAs in the livers of DDC diet-fed Car +/+/C3He mice, suggesting that it does not further facilitate oval cell proliferation. Similarly, TCPOBOP neither induced TGFβ and TNFα mRNAs, nor affected their levels in the liver of DDC diet-fed mice (Figures 6c and d). GADD45β mRNA was specifically induced by TCPOBOP in centrilobular hepatocytes, whereas DDC induced this mRNA in the centrilobular hepatocytes and to a greater extent, in the periportal region that was occupied by a ductular reaction (Figure 6e). Co-treatment with TCPOBOP and DDC resulted in high expression patterns of GADD45β mRNA in both centrilobular and periportal hepatocytes. The expression of CYP2B10 mRNA was induced by TCPOBOP in both the centrilobular and periportal regions of Car +/+/C3He mice (Figure 6f). Because LCM cut the periportal region that was largely occupied by non-hepatic cells in the DDC diet-fed Car +/+/C3He mice, CYP2B10 mRNA was not detected in this region. Consistent with the fact that TCPOBOP co-treatment extended the area of periportal hepatocytes closer to a niche within the ductular reaction, a significant expression of CYP2B10 mRNA was detected in the periportal fractions microdissected from the livers of DDC diet-fed mice (Figure 6c). None of these mRNAs were induced in Car−/−/C3He mice (Figure 6).
Figure 6.

Constitutive active/androstane receptor (CAR)-dependent induction of oval cell markers, growth factors, cytokines and cytochrome P450 (CYP)2B10 mRNAs after 1,4 bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP) co-treatment. Car +/+ and Car−/− mice were fed with control or 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) diets for 14 days. Half of these mice were co-treated with TCPOBOP for 2 days (C + TC and DDC + TC), whereas the other half was then treated with DMSO for the last 2 days (control and DDC). Centrilobular and periportal regions were microdissected from the livers of these mice, from which total RNA was isolated and real-time PCR analysis was performed for (a) TROP2, (b) epithelial cell adhesion molecule (EpCAM), (c) TGFβ, (d) TNFα, (e) growth arrest and DNA damage-induced protein 45 family (GADD)45β and (f) CYP2B10 mRNAs as described in the Materials and Methods section. The bars indicate the fold inductions (means±s.d.) relative to their mRNA levels in total livers of the control diet-fed Car +/+ mice. * and #indicate a significant difference at P<0.01 in comparison with control diet and at P<0.05 in comparison with co-treatment with TCPOBOP.
DISCUSSION
Using Car +/+ and Car−/− mice, our present study has now shown that activation of CAR is required for DDC to induce proliferation of oval cells in mouse livers. DDC-induced expression of the oval cell markers A6, EpCAM and TROP2, was not observed in the Car−/− livers. It has been suggested that oval cells differentiate into hepatocytes, helping the injured liver to regenerate and injured livers to recover normal functions. At the same time, recent studies have provided evidence indicating that oval cells can also become tumor stem cells, initiating tumor development in the liver.2,21 The finding of an essential role of CAR in oval cell proliferation should help us to further investigate the processes through which oval cells either differentiate into hepatocytes or become tumor stem cells.
DDC was found to cause hepatic hypertrophy and porphyria, which may have led to proliferation of oval cells in the liver.17,21 In fact, we found that porphyria dramatically increased when EpCAM and TROP2 mRNAs were strongly induced. PB is well known to cause liver hypertrophy, but only in the centrilobular regions.22,23 Our present study has shown that TCPOBOP causes hypertrophy in only the centrilobular regions. On the other hand, DDC caused hepatic hypertrophy in the centrilobular, as well as in the periportal regions, where a ductular reaction occurs and oval cells proliferate. Development of porphyria was observed in the absence of CAR, but it was limited to the centrilobular regions. Only when CAR was present did the periportal regions develop a severe porphyria. In addition to hypertrophy and porphyria reaction, the livers became inflammatory after feeding the DDC diet, but not after TCPOBOP treatment. TCPOBOP is the most potent known CAR activator, but is a weak hepatotoxic chemical. The fact that TCPOBOP treatment alone did not enable liver to proliferate oval cells, therefore, suggests that CAR activation by itself is not enough to induce oval cell proliferation in Car +/+/C3He mice. However, the strong activation of CAR by co-treatment with TCPOBOP appeared to facilitate migration of oval cells in and away from portal region. This CAR-dependent inflammation occurred only in the periportal regions, the mechanism of which is an important question for future investigations. Thus, DDC is characterized by its ability to cause CAR-mediated hypertrophy, porphyria and inflammation in the periportal regions, which can be critical in enabling DDC to induce proliferation of oval cells. In conclusion, the CAR-regulated genes, which have been associated with oval cell proliferation, are now identified. Further characterization of these genes and their periportal-specific expressions will help us to decipher the CAR-mediated mechanism of oval cell proliferation.
Supplementary Material
Acknowledgments
We extend our sincerest appreciations towards Drs Gordon Flake, Jim Squires and Mark Hoenerhoff at NIEHS for their critical reading of this manuscript and comments. We thank Ms Patricia Stockton for excellent technical assistance of LCM. We also thank DNA sequence and histology cores at NIEHS for their excellent assistances. This study was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health: Z01ES71005-01.
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Laboratory Investigation website (http://www.laboratoryinvestigation.org)
References
- 1.Fausto N, Campbell JS. The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev. 2003;120:117–130. doi: 10.1016/s0925-4773(02)00338-6. [DOI] [PubMed] [Google Scholar]
- 2.Mishra L, Banker T, Murray J, et al. Liver stem cells and hepatocellular carcinoma. Hepatology. 2009;49:318–329. doi: 10.1002/hep.22704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duncan AW, Dorrell C, Grompe M. Stem cells and liver regeneration. Gastroenterology. 2009;137:466–481. doi: 10.1053/j.gastro.2009.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zelko I, Negishi M. Phenobarbital-elicited activation of nuclear receptor CAR in induction of cytochrome P450 genes. Biochem Biophys Res Commun. 2000;277:1–6. doi: 10.1006/bbrc.2000.3557. [DOI] [PubMed] [Google Scholar]
- 5.Ueda A, Hamadeh HK, Webb HK, et al. Diverse roles of the nuclear orphan receptor CAR in regulating hepatic genes in response to phenobarbital. Mol Pharmacol. 2002;61:1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- 6.Kodama S, Koike C, Negishi M, et al. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24:7931–7940. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konno Y, Negishi M, Kodama S. The roles of nuclear receptors CAR and PXR in hepatic energy metabolism. Drug Metab Parmacokinet. 2008;23:8–13. doi: 10.2133/dmpk.23.8. [DOI] [PubMed] [Google Scholar]
- 8.Yamazaki Y, Kakizaki S, Horiguchi N, et al. The role of the nuclear receptor constitutive androstane receptor in the pathogenesis of non-alcoholic steatohepatitis. Gut. 2007;56:565–574. doi: 10.1136/gut.2006.093260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tien ES, Matsui K, Moore R, et al. The nuclear receptor constitutively active/androstane receptor regulates type 1 deiodinase and thyroid hormone activity in the regenerating mouse liver. J Pharmacol Exp Ther. 2007;320:307–313. doi: 10.1124/jpet.106.112706. [DOI] [PubMed] [Google Scholar]
- 10.Ueda A, Matsui K, Yamamoto Y, et al. Thr176 regulates the activity of the mouse nuclear receptor CAR and is conserved in the NR1I subfamily members PXR and VDR. Biochem J. 2005;388:623–630. doi: 10.1042/BJ20041572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto Y, Moore R, Goldsworthy TL, et al. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res. 2004;64:7197–7200. doi: 10.1158/0008-5472.CAN-04-1459. [DOI] [PubMed] [Google Scholar]
- 12.Honkakoski P, Kojo A, Lang MA. Regulation of the mouse liver cytochrome P450 2B subfamily by sex hormones and phenobarbital. Biochem J. 1992;285:979–983. doi: 10.1042/bj2850979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Troadec MB, Fautrel A, Drénou B, et al. Transcripts of ceruloplasmin but not hepcidin, both major iron metabolism genes, exhibit a decreasing pattern along the portocentral axis of mouse liver. Biochim Biophys Acta. 2008;1782:239–249. doi: 10.1016/j.bbadis.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 14.Sueyoshi T, Moore R, Sugatani J, et al. PPP1R16A, the membrane subunit of protein phosphatase 1beta, signals nuclear translocation of the nuclear receptor constitutive active/androstane receptor. Mol Pharmacol. 2008;73:1113–1121. doi: 10.1124/mol.107.042960. [DOI] [PubMed] [Google Scholar]
- 15.Kawamoto T, Sueyoshi T, Zelko I, et al. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol. 1999;19:6318–6322. doi: 10.1128/mcb.19.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Preisegger KH, Factor VM, Fuchsbichler A, et al. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Invest. 1999;79:103–109. [PubMed] [Google Scholar]
- 17.Okabe M, Tsukahara Y, Tanaka M, et al. Potential hepatic stem cells reside in EpCAM+ cells of normal and injured mouse liver. Development. 2009;136:1951–1960. doi: 10.1242/dev.031369. [DOI] [PubMed] [Google Scholar]
- 18.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–S53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen LN, Furuya MH, Wolfraim LA, et al. Transforming growth factor-beta differentially regulates oval cell and hepatocyte proliferation. Hepatology. 2007;45:31–41. doi: 10.1002/hep.21466. [DOI] [PubMed] [Google Scholar]
- 20.Papa S, Zazzeroni F, Fu YX, et al. Gadd45beta promotes hepatocyte survival during liver regeneration in mice by modulating JNK signaling. J Clin Invest. 2008;118:1911–1923. doi: 10.1172/JCI33913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang X, Foster M, Al-Dhalimy M, et al. The origin and liver repopulating capacity of murine oval cells. Proc Natl Acad Sci USA. 2003;100:11881–11888. doi: 10.1073/pnas.1734199100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carthew P, Edwards RE, Nolan BM. The quantitative distinction of hyperplasia from hypertrophy in hepatomegaly induced in the rat liver by phenobarbital. Toxicol Sci. 1998;44:46–51. doi: 10.1006/toxs.1998.2473. [DOI] [PubMed] [Google Scholar]
- 23.Cunninghame ME, Evans JG, Butler WH. An ultra-structural study of spontaneous and phenobarbitone-induced nodules in the mouse liver. Int J Exp Pathol. 1991;72:695–703. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.