

Abstract
The type I interferon (IFN) response protects cells from viral infection by inducing hundreds of interferon-stimulated genes (ISGs), some of which encode direct antiviral effectors1–3. Recent screening studies have begun to catalogue ISGs with antiviral activity against several RNA and DNA viruses4–13. However, antiviral ISG specificity across multiple distinct classes of viruses remains largely unexplored. Here we used an ectopic expression assay to screen a library of more than 350 human ISGs for effects on 14 viruses representing 7 families and 11 genera. We show that 47 genes inhibit one or more viruses, and 25 genes enhance virus infectivity. Comparative analysis reveals that the screened ISGs target positive-sense single-stranded RNA viruses more effectively than negative-sense single-stranded RNA viruses. Gene clustering highlights the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS, also known as MB21D1) as a gene whose expression also broadly inhibits several RNA viruses. In vitro, lentiviral delivery of enzymatically active cGAS triggers a STING-dependent, IRF3-mediated antiviral program that functions independently of canonical IFN/STAT1 signalling. In vivo, genetic ablation of murine cGAS reveals its requirement in the antiviral response to two DNA viruses, and an unappreciated contribution to the innate control of an RNA virus. These studies uncover new paradigms for the preferential specificity of IFN-mediated antiviral pathways spanning several virus families.
To identify IFN-induced effectors targeting diverse viruses, we used our established flow cytometry-based ISG screening platform (Fig. 1a, see Methods)4. We screened over 350 common ISGs for inhibitory or enhancing effects on 14 viruses, including one double-stranded DNA (dsDNA) virus, six positive-sense single-stranded RNA (+ssRNA) viruses and seven negative-sense single-stranded RNA (−ssRNA) viruses (Extended Data Table 1 and Fig. 1 legend for abbreviations). Viruses were screened in HeLa cells, Huh7 hepatoma cells or human STAT1−/− fibroblasts14. Notably, infection of most +ssRNA viruses was inhibited by greater than 50% when any of multiple ISGs were expressed, whereas screens for vaccinia virus (VV), human metapneumovirus, respiratory syncytial virus, measles virus and Bunyamwera virus had few or no genes that inhibited virus infection by more than 50% (Fig. 1b and Supplementary Tables 1 and 2).
Figure 1. Flow cytometry-based screens for identifying inhibitory or enhancing ISGs against 14 viruses.

a, Schematic of the ectopic expression screen showing cells transduced with lentiviral vectors expressing an inhibitory ISG (IFITM3) and red fluorescent protein (RFP), a control (Fluc), or an enhancing ISG (MCOLN2). Cells were infected with influenza A virus expressing GFP, and GFP-positive cells were quantified by flow cytometry. b, Dot plots of virus infectivity in the presence of expressed ISGs. Data sets were normalized to the average of each screen, which is indicated by a yellow dotted line. The 50% inhibitory and 150% enhancing effects are denoted by red and green dotted lines, respectively. Bunyamwera virus, BUNV; Coxsackie B virus, CVB; equine arterivirus, EAV; influenza A virus, FLUAV; human metapneumovirus, HMPV; measles virus, MV; Newcastle disease virus, NDV; o’nyong-nyong virus, ONNV; human parainfluenza virus type 3, PIV3; poliovirus, PV; respiratory syncytial virus, RSV; Sindbis virus AR86, SINV-A; Sindbis virus Girdwood, SINV-G.
Extended Data Table 1.
Classification of viruses screened in this study
| Virus | Genome content | Family | Genus |
|---|---|---|---|
| vaccinia virus | dsDNA | Poxviridae | Orthopoxvirus |
| coxsackie B virus | (+)ssRNA | Picornaviridae | Enterovirus |
| poliovirus | (+)ssRNA | Picornaviridae | Enterovirus |
| equine arterivirus | (+)ssRNA | Arteriviridae | Arterivirus |
| Sindbis virus | (+)ssRNA | Togaviridae | Alphavirus |
| O’nyong nyong virus | (+)ssRNA | Togaviridae | Alphavirus |
| influenza A virus | (−)ssRNA† | Orthomyxoviridae | Influenzavirus A |
| human parainfluenza virus type 3 | (−)ssRNA | Paramyxoviridae | Respirovirus |
| Newcastle disease virus | (−)ssRNA | Paramyxoviridae | Avulavirus |
| human metapneumovirus | (−)ssRNA | Paramyxoviridae | Metapneumovirus |
| respiratory syncytial virus | (−)ssRNA | Paramyxoviridae | Pneumovirus |
| measles virus | (−)ssRNA | Paramyxoviridae | Morbillavirus |
| Bunyamwera virus | (−)ssRNA† | Bunyaviridae | Orthobunyavirus |
Two strains of SINV, AR86 and Girdwood, were included in the screens.
Segmented −ssRNA genomes; all other −ssRNA viruses have non-segmented genomes.
dsDNA, double-stranded DNA; +ssRNA, positive-sense single-stranded RNA; −ssRNA, negative-sense single-stranded RNA.
Confirmatory assays were performed on selected ISGs to verify the primary screening hits. A total of 159 assays representing 96 unique genes were performed (Fig. 2a, b). Of these, 125 assays representing 72 unique genes yielded results that were consistent with the primary screens. We identified 47 inhibitory and 25 enhancing ISGs. Of these, 25 inhibitory ISGs suppressed infectivity by more than 50%, and 4 enhancing ISGs increased infectivity by more than 150%. We also confirmed by plaque assay that ISGs with antiviral effects against green fluorescent protein (GFP)-tagged poliovirus could inhibit the parental non-GFP strain (Extended Data Fig. 1). Comparative analysis of the confirmatory assays on RNA viruses indicated that the ectopically expressed ISGs with the strongest inhibitory (<50%) and enhancing (>150%) effects were biased towards the +ssRNA viruses (Fig. 2c).
Figure 2. Confirmatory assays for selected inhibitory and enhancing ISGs.

a, b, Independent confirmatory assays against DNA and +ssRNA viruses (a) and –ssRNA viruses (b) were carried out using new lentivirus stocks. Data were normalized to a Fluc control, highlighted in a yellow box and by dotted line. Data are presented as box and whisker plots; whiskers extend to show the highest and lowest values. Data represent six technical replicates to control for intra-assay variability. Statistical significance was determined by one-way analysis of variance (ANOVA) (*P <0.05, **P <0.01, ***P <0.001; NS, not significant). The 50% inhibitory and 150% enhancing effects are denoted by red and green dotted lines, respectively. c, Comparative analysis showing the frequency with which confirmed inhibitory or enhancing ISGs targeted +ssRNA or –ssRNA viruses.
Extended Data Figure 1. Antiviral effects of ISGs on virus production of a non-GFP poliovirus.
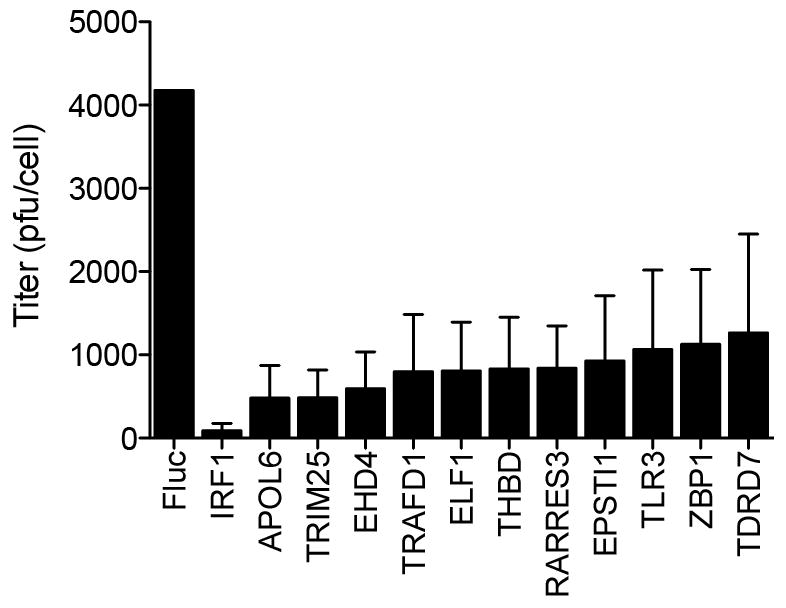
HeLa cells were transfected with plasmids encoding ISGs and 48 h later infected with P1M (10 m.o.i.) for 16 h. Lysates were collected and viral titres determined by plaque assay on HeLa cell monolayers, as described in Methods. Plaque assays were performed in duplicate. Data represent the average of three independent experiments. Error bars represent s.d.
We next performed a hierarchical clustering analysis of the primary screening data to group viruses and ISGs with one another (Fig. 3a). We considered only viruses screened in STAT1−/− fibroblasts, including three +ssRNA viruses from previous studies: two flaviviruses, West Nile virus (WNV) and yellow fever virus, and one alphavirus, chikungunya virus4. The clustering data revealed a division of viruses into two major groups representing either +ssRNA and –ssRNA viruses. Within these groups, several related viruses, including the flaviviruses, alpha-viruses and paramyxoviruses, clustered together. We repeated the analysis in the absence of 1, 2 or 6 of the more potent ISGs and obtained similar results, indicating that the clustering was not skewed by a selection of dominant genes (Extended Data Fig. 2a–e). In a second clustering analysis, viruses were grouped based on the presence of ISG names in a list of the top 30 genes from the primary screens (Supplementary Tables 1 and 2). This analysis revealed a similar division of +ssRNA and –ssRNA viruses (Extended Data Fig. 2f). A co-occurrence analysis of the top 20 antiviral genes from 7 +ssRNA and 5 –ssRNA virus screens further supported the hierarchical clustering studies (Extended Data Fig. 3). These data suggest that subsets of ISGs may target similar viruses and raise the possibility that therapeutics targeting specific antiviral host molecules may be broad spectrum across related viruses.
Figure 3. cGAS activates an IRF3-driven antiviral program independently of canonical IFN/STAT1 signalling.

a, Hierarchical clustering analysis of 22 ISGs and 13 viruses screened in STAT1−/− fibroblasts. CHIKV, chikungunya virus; YFV, yellow fever virus. b, OAS2 gene expression in STAT1−/− fibroblasts transduced with lentiviruses expressing IRF7, IRF1, cGAS and Fluc. Data represent one of two experiments performed in duplicate. c, Microarray analysis of STAT1−/− fibroblasts transduced with lentiviruses expressing Fluc or cGAS. Data show a subset of genes (green) induced 2.5-fold with P <0.05, n = 3 (using Benjamini–Hochberg false discovery rate correction). Data represent one of two independent experiments. d, Top, western blot of STING expression in STAT1−/− fibroblasts and Huh7 cells. Bottom, infectivity of Venezuelan equine encephalitis virus (VEEV) in STAT1−/− fibroblasts and Huh7 cells transduced with lentivirus expressing Fluc or cGAS. Virus infectivity was normalized to Fluc control. e, OAS2 mRNA expression (top) and western blots (bottom) of cGAS, phosphorylated IRF3 (p-IRF3) and control actin in STAT1−/−fibroblasts and Huh7 cells transduced with lentiviruses expressing Fluc or cGAS. f, Antiviral gene expression in STAT1−/− fibroblasts that were depleted of STING by siRNA (Extended Data Fig. 5a) before transduction with lentiviruses expressing Fluc or cGAS. NSC, non-silencing siRNA control. g, Infectivity of VEEV in STAT1−/− fibroblasts transduced with Fluc control, wild type (WT) or point mutant (E225A, D227A) cGAS, or IRF1. Virus infectivity was normalized to Fluc control. h, OAS2 mRNA induction in STAT1−/−fibroblasts stably expressing cGAS or an empty cassette. RNA samples were processed at the indicated cell passage number. In d–g, data represent the means of two or three independent experiments performed in triplicate. In h, data represent one of two independent experiments with similar results. Error bars represent s.d. Statistical significance was determined by t-test or one-way ANOVA (*P <0.05, **P <0.01, ***P <0.001; NS, not significant).
Extended Data Figure 2. Hierarchical clustering.

a–e, Analyses were performed as described in Methods. In each cluster, one or more ISGs were removed from the analysis in Fig. 3a to determine whether virus clustering is driven by a subset of one or more dominant genes. Blue and green bars underscore +ssRNA and −ssRNA viruses, respectively. f, The top 30 ISG inhibitors from the primary screens were compiled as a gene list and transformed to a binarized vector for clustering using MATLAB Statistics Toolbox (see Methods). A dendrogram was generated from the clustering analysis to show how viruses relate to each other with respect to the ISGs that target them.
Extended Data Figure 3. Co-occurrence of top 20 antiviral ISGs from primary screens.

ISGs were assigned a frequency on the basis of the number of times the gene appeared in a list of the 20 most inhibitory genes from 7 +ssRNA or 5 −ssRNA virus screens. A frequency of 1 indicates an occurrence of 100% across all gene lists. Co-occurrence is reflected by adjacent red and blue bars.
To distinguish direct ISG effectors from transcriptional regulators, we tested inhibitory ISGs for interferon-stimulated response element (ISRE)-dependent transcription. Of the 68 genes tested, only IRF1, IRF2, TLR3 and MYD88 directly activated an ISRE-driven reporter plasmid (Extended Data Fig. 4a). We also tested whether 4 ISGs with virus enhancing activity could impair IFN-mediated ISRE activation. In contrast to SOCS1, a known negative regulator of IFN signalling, none of the ISGs had an effect (Extended Data Fig. 4b). These data indicate that most ISG ‘hits’ do not affect ISRE-dependent gene transcription. They may have direct effector mechanisms or may regulate other pathways, as suggested by Gene Ontology analysis. (Supplementary Table 3).
Extended Data Figure 4. Effects of ISGs on ISRE-dependent transcription.

a, 293 cells were transduced with lentiviruses expressing ISGs, followed by transfection with an ISRE reporter plasmid expressing Fluc. Cells were assayed for Fluc activity 24 h after transfection. b, 293 cells were co-transfected with ISG-expressing plasmids and an ISRE reporter plasmid. The cells were then treated overnight with 1,000 U ml−1 interferon-α (IFN-α), followed by Fluc activity assay. Data represent the average of three independent experiments performed in triplicate. Error bars represent s.d. Statistical significance was determined by one-way ANOVA or t-test. *P <0.05, ***P <0.001.
The clustering analysis grouped the antiviral transcription factor IRF1 and cyclic GMP-AMP synthase cGAS (Fig. 3a). We first identified the gene encoding cGAS (formerly C6orf150) as antiviral in previous screens4, and our current studies confirmed this with additional viruses. Because both cGAS and IRF1 are broadly antiviral, we proposed that cGAS, like IRF1, might upregulate antiviral gene transcription. We found that STAT1−/− fibroblasts transduced with lentiviruses expressing cGAS and IRF1, but not IRF7 or firefly luciferase (Fluc), had increased messenger RNA levels of the ISG OAS2 (Fig. 3b). We extended these findings with microarray analysis and showed that lentiviral-mediated expression of cGAS induced 60 genes by at least twofold compared to Fluc control. (Fig. 3c and Extended Data Table 2). Many of these genes are ISGs, and more than half of them overlap with IRF1-induced transcripts in the same cellular background4. These results indicate that in STAT1−/−fibroblasts, lentiviral-mediated expression of cGAS induces an antiviral program independently of canonical IFN signalling.
Extended Data Table 2.
List of genes induced twofold or more in STAT1−/− fibroblasts transduced with lentivirus expressing cGAS compared to Fluc control
| Gene | FC | p value | Accession |
|---|---|---|---|
| PARP3 | 2.0 | 0.040 | NM_001003931.1 |
| LAMP3 | 2.0 | 0.024 | NM_014398.2 |
| CEACAM1 | 2.0 | 0.015 | NM_001024912.1 |
| GBP1 | 2.0 | 0.013 | NM_002053.1 |
| C6orf192 | 2.1 | 0.011 | NM_052831.2 |
| PODXL | 2.1 | 0.024 | NM_001018111.1 |
| VNN2 | 2.1 | 0.043 | NM_004665.2 |
| C1R | 2.1 | 0.040 | NM_001733.4 |
| CDCP1 | 2.1 | 0.005 | NM_022842.3 |
| TDRD7 | 2.1 | 0.023 | NM_014290.1 |
| SAMD9 | 2.1 | 0.025 | NM_017654.2 |
| PARP12 | 2.1 | 0.014 | NM_022750.2 |
| CASZ1 | 2.1 | 0.015 | NM_017766.3 |
| APOBEC3G | 2.2 | 0.024 | NM_021822.1 |
| IFIT3 | 2.2 | 0.046 | NM_001031683.1 |
| ABI3BP | 2.2 | 0.028 | NM_015429.2 |
| LOC728855 | 2.2 | 0.041 | NR_024510.1 |
| LGALS3BP | 2.2 | 0.036 | NM_005567.2 |
| IFI44L | 2.2 | 0.023 | NM_006820.1 |
| ZC3HAV1 | 2.2 | 0.018 | NM_020119.3 |
| DHX58 | 2.2 | 0.039 | NM_024119.2 |
| OAS2 | 2.3 | 0.023 | NM_001032731.1 |
| ACSS1 | 2.3 | 0.025 | NM_032501.2 |
| HLA-DRA | 2.3 | 0.017 | NM_019111.3 |
| CADPS2 | 2.3 | 0.009 | NM_017954.9 |
| USP18 | 2.4 | 0.028 | NM_017414.3 |
| TMEM62 | 2.4 | 0.022 | NM_024956.3 |
| BTN3A2 | 2.4 | 0.018 | NM_007047.3 |
| LRRC17 | 2.4 | 0.023 | NM_001031692.1 |
| EPSTI1 | 2.4 | 0.037 | NM_033255.2 |
| PARP14 | 2.5 | 0.028 | NM_017554.1 |
| HLA-F | 2.5 | 0.007 | NM_018950.1 |
| IFIT3 | 2.5 | 0.032 | NM_001549.2 |
| APOBEC3G | 2.5 | 0.018 | NM_021822.1 |
| GIMAP2 | 2.5 | 0.018 | NM_015660.2 |
| GBP4 | 2.6 | 0.036 | NM_052941.3 |
| DDX60 | 2.6 | 0.023 | NM_017631.4 |
| ITGA2 | 2.6 | 0.028 | NM_002203.3 |
| UBE2L6 | 2.6 | 0.029 | NM_004223.3 |
| CASP1 | 2.6 | 0.034 | NM_033294.2 |
| HERC6 | 2.6 | 0.047 | NM_017912.3 |
| UBE2L6 | 2.6 | 0.011 | NM_004223.3 |
| ZMYND15 | 2.7 | 0.025 | NM_032265.1 |
| CXCL16 | 2.7 | 0.011 | NM_022059.1 |
| PMAIP1 | 2.8 | 0.013 | NM_021127.1 |
| IFIH1 | 2.8 | 0.027 | NM_022168.2 |
| HLA-H | 2.8 | 0.015 | NR_001434.1 |
| ZC3HAV1 | 2.9 | 0.037 | NM_024625.3 |
| OAS2 | 2.9 | 0.029 | NM_016817.2 |
| LRRC17 | 2.9 | 0.023 | NM_005824.1 |
| IFI35 | 3.0 | 0.015 | NM_005533.2 |
| MX1 | 3.1 | 0.012 | NM_002462.2 |
| SLC15A3 | 3.1 | 0.007 | NM_016582.1 |
| PSMB8 | 3.2 | 0.013 | NM_148919.3 |
| ISG20 | 3.2 | 0.002 | NM_002201.4 |
| PSMB8 | 3.3 | 0.007 | NM_148919.3 |
| IFIT3 | 3.3 | 0.007 | NM_001031683.1 |
| PSMB8 | 3.5 | 0.002 | NM_004159.4 |
| PLCG2 | 3.5 | 0.007 | NM_002661.2 |
| IL8 | 3.5 | 0.013 | NM_000584.2 |
| CASP1 | 3.7 | 0.012 | NM_033294.2 |
| TNFRSF1B | 3.7 | 0.029 | NM_001066.2 |
| HERC5 | 3.7 | 0.007 | NM_016323.2 |
| ARL9 | 3.8 | 0.013 | NM_206919.1 |
| PSMB9 | 3.9 | 0.006 | NM_002800.4 |
| RARRES3 | 3.9 | 0.012 | NM_004585.3 |
| OASL | 4.4 | 0.007 | NM_198213.1 |
| HLA-B | 5.0 | 0.005 | NM_005514.5 |
| IFI44 | 5.2 | 0.007 | NM_006417.3 |
| cGAS | 9.4 | 0.007 | NM_138441.2 |
FC, fold change.
During the course of these studies, murine cGAS was shown to be a cytosolic DNA-sensing enzyme that catalyses the production of cyclic GMP-AMP (cGAMP), a second-messenger activator of IFN antiviral responses15,16. The induction of IFN by cGAS appears to require a DNA, but not RNA, substrate to trigger a STING/IRF3 activation pathway. Nonetheless, we observed that cGAS induced an antiviral program that targeted several RNA viruses in STAT1−/− fibroblasts (Figs 2 and 3c), which are compromised in canonical IFN/STAT signalling14. We therefore proposed that lentiviral-driven cGAS expression triggers antiviral gene expression by direct STING/IRF3 activation. We confirmed STING expression in STAT1−/− fibroblasts (Fig. 3d), and showed that cells transduced with lentivirus expressing cGAS had a strong induction of phosphorylated IRF3 and OAS2 mRNA compared to control cells (Fig. 3e). OAS2 induction by cGAS was abrogated when STING expression was silenced with short interfering RNA (siRNA; Fig. 3f and Extended Data Fig. 5a), confirming a requirement for STING in the pathway. Consistent with this, IRF3 phosphorylation, OAS2 mRNA induction and viral inhibition were not observed when lentiviruses expressing cGAS were used to transduce Huh7 cells, which lack detectable levels of STING (Fig. 3d, e). These data indicate that, in STAT1−/− fibroblasts, lentiviral-driven cGAS expression activates IRF3 through STING and establishes a transcriptional program that inhibits infection of several RNA viruses.
Extended Data Figure 5. cGAS mechanistic studies.
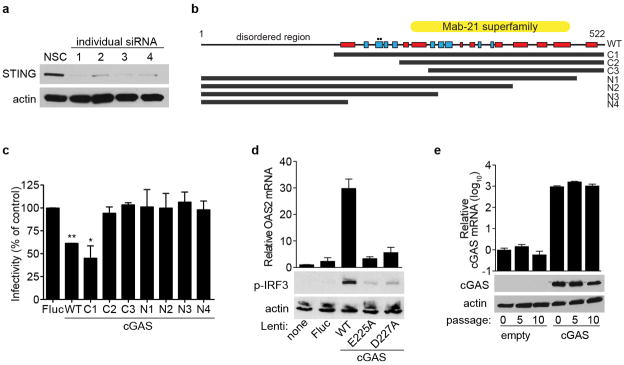
a, STAT1−/− fibroblasts were transfected with individual siRNAs targeting STING. Cell lysates were processed 48 h after transfection for western blot with anti-STING- or anti-actin-specific antibodies. From these results, siRNA no. 1 was chosen for additional studies. b, Schematic of cGAS protein sequence and truncation mutants. Red box, α-helix; blue box, β-sheet. Circles denote catalytic residues E225A and D227A. c, STAT1−/− fibroblasts were transduced with lentivirus expressing control or cGAS (wild type and truncation mutants). Cells were infected 48 h after transduction with VEEV-GFP and infectivity was monitored by FACS. Data represent the mean of two independent experiments. Error bars represent s.d. Statistical significance was determined by one-way ANOVA. *P <0.05, **P <0.01. d, STAT1−/− fibroblasts were transduced with lentivirus expressing control or cGAS (wild type and mutants). Cells were collected 48 h after transduction and total RNA was analysed for OAS2 mRNA induction relative to RPS11 (top), or protein lysates were analysed for phospho-IRF3 and actin expression by western blot (bottom). e, STAT1−/− fibroblasts were transduced with a puromycin-selectable lentivirus expressing cGAS and placed under selection. At various passages, cells were collected and total RNA was analysed for OAS2 mRNA induction relative to RPS11 (top), or protein lysates were analysed for cGAS and actin expression by western blot (bottom). Western blot and cGAS mRNA data represent one of two independent experiments, each showing similar results. OAS2 mRNA data are presented as the average of two independent experiments, each performed in triplicate. Error bars represent s.d.
We next probed the mechanism of cGAS activation by performing genetic analyses to identify functional domains and residues (Extended Data Fig. 5b). Deletion analyses of cGAS localized the antiviral activity to the carboxy-terminal domain, with the first 164 amino acids being dispensable (Extended Data Fig. 5c). Active site mutants (E225A, D227A) showed no antiviral activity (Fig. 3g) and were impaired in IRF3 phosphorylation and OAS2 mRNA induction (Extended Data Fig. 5d). These results are in agreement with recent studies showing the requirement for these residues in the synthesis of cGAMP15,17–19. Our data indicate that the antiviral effect of cGAS requires an active enzyme, and by extension, an activating substrate. We proposed that the lentivirus itself provides the trigger. Accordingly, we predicted that once cells stabilize from transient lentiviral infection, cGAS expression from the provirus would be less activating as the cells were passaged. Indeed, over at least 10 passages, we observed a progressive decrease in OAS2 levels in cGAS-expressing and control cells (Fig. 3h), despite continuous and high levels of cGAS mRNA and protein in cGAS-expressing cells (Extended Data Fig. 5e). These data suggest that transient delivery of lentivirus may trigger the formation of a DNA-based substrate that reacts with cGAS to activate IRF3. A recent report supports this hypothesis by showing that cGAS can sense reverse-transcribed retroviral DNA20. However, given the selectivity of this effect against several +ssRNA viruses, we cannot rule out other mechanisms of cGAS activation.
We next determined whether these in vitro studies predict physiologically relevant functions of antiviral molecules. We generated mice with a targeted deletion of cGas exon 2, which contains the active site (Extended Data Fig. 6a, b). Knockout mice bred in normal Mendelian ratios and showed no overt growth or developmental defects. Gene expression analysis from the spleen (Fig. 4a), lungs, and bone marrow-derived macrophages (BMMO) (Extended Data Fig. 6c) of wild-type and knockout mice confirmed reduced cGas mRNA (Fig. 4a). As cGAS is activated in vitro by DNA15,17,19, we challenged mice with two DNA viruses, murine gammaherpesvirus 68 (MHV68) and VV. Viral titres of MHV68 were 2.0-fold higher in the spleen and 3.5-fold higher in the lungs of cGas−/− mice compared to wild-type mice (Fig. 4b, c). VV had a notable mortality phenotype, with all cGas−/− mice succumbing to infection, whereas 70% of wild-type mice recovered (Fig. 4d). We next infected BMMO from wild-type and cGas−/− mice with MHV68 or VV and observed increased titres of both viruses in cGas−/− cells (Fig. 4e, f). cGas−/− BMMO were also refractory to the >200-fold induction of Ifnb (also known as Infb1) mRNA observed in MHV68-infected wild-type BMMO (Fig. 4g). These data provide direct genetic evidence that cGAS is required for innate control of DNA viruses in mice. A recently published study used cGas-deficient ‘gene-trap’ mice21, which served as our starting point before excision of cGas exon 2 by sequential crossings to FlpE-deleter and Cre-expressing mice (Extended Data 6a). This study demonstrated that cGas-deficient gene-trap mice were also more vulnerable to infection by a DNA virus, herpes simplex virus 1. Thus, two variants of mice lacking cGAS establish a role for this sensor in the antiviral immunity to DNA viruses.
Extended Data Figure 6. Gene-targeting strategy to create cGas knockout mice.

a, Mice expressing a cGas exon 2 gene-trap cassette were crossed to FlpE-expressing mice to generate conditional knockouts. These mice were then crossed to Cre-expressing mice to generate the knockout allele with a deletion of exon 2, which contains the cGAS catalytic sites. Mice were backcrossed to remove Cre, and cGas+/− mice were intercrossed to derive cGas−/− mice. b, PCR products from genomic DNA of cGas+/+, cGas+/− and cGas−/− mice using primers outlined in a. c, qRT–PCR of relative cGas expression in lungs (left) or BMMO (right) from wild-type B6 and cGas−/− mice. Data from lung represent means of three mice per group. Data from BMMO were derived from two independent experiments performed in triplicate. Error bars represent s.e.m. Statistical significance was determined by t-test. *P <0.05, ***P <0.001.
Figure 4. Requirement for cGAS in controlling viral infection in vivo.

a, Quantitative PCR with reverse transcription (qRT–PCR) of relative cGas expression in spleens of wild-type (B6) and cGas−/− mice. n = 3 mice per group. Error bars represent s.e.m. Statistical significance was determined by t-test. b, c, Viral titres in spleen (b) and lungs (c) of wild-type and cGas−/− mice after infection with 106 plaque-forming units (p.f.u.) of MHV68, n = 15 mice per group. Statistical significance was determined by Mann–Whitney test. d, Survival curves of wild-type and cGas−/− mice infected with 8000 p.f.u. of VV, n = 10 (wild type), n = 9 (cGas−/−). Statistical significance was determined by log rank test. e, f, i, Viral titres from BMMO after infection with 10 multiplicity of infection (m.o.i.) MHV68 (e), 0.1 m.o.i. VV (f) or 0.1 m.o.i. WNV (i). Data represent three (e, f) or four (i) independent experiments performed in triplicate. Error bars represent s.e.m. Statistical significance was determined by t-test. g, qRT–PCR of relative Ifnb expression in BMMO infected with MHV68 for 6 h. Data represent means of three experiments performed in triplicate. Error bars represent s.d. Statistical significance was determined by t-test. h, Survival curves of wild-type and cGas−/− mice infected with 100 p.f.u. of WNV, n = 10 (wild type), n = 9 (cGas−/−). j, qRT–PCR of baseline gene expression (Ifnb, Ifit1, Ifit2, Oas1a, actin) in wild-type and cGas−/− BMMO. Data represent means of two experiments performed in triplicate. Error bars represent s.d. Statistical significance was determined by t-test. In all panels, *P <0.05, **P <0.01, ***P <0.001; NS, not significant.
Our in vitro studies linked cGAS antiviral function to RNA virus inhibition through IRF3, and initial evidence suggests that lentivirus is the trigger. However, some RNA viruses were not targeted by this lentivirus/cGAS/IRF3 axis (Fig. 2a, b), prompting us to explore whether endogenous cGAS modulates RNA virus infection. Notably, cGas−/− mice were more vulnerable to lethal WNV infection compared to wild-type mice (Fig. 4h). We did not detect an increase in viral burden in brains of cGas−/− mice (Extended Data Fig. 7), although extensive time courses and tissue profiling were not performed. However, when we infected wild-type and cGas−/− BMMO with WNV, we detected a modest yet significant fourfold increase in viral titres in cGas−/− cells (Fig. 4i). We assessed the kinetics of WNV-mediated activation of BMMO by monitoring mRNA induction of Ifnb, several ISGs (Ifit1, Ifit2, Oas1a), chemokines (Ccl5 and Cxcl10) and cytokines (Tnfa, Il6, Il1b). WNV induced most of these genes to similar levels in both wild-type and cGas−/− cells (data not shown). However, basal mRNA levels of Ifnb, all ISGs, and chemokines were significantly reduced in uninfected cGas−/− BMMO (Fig. 4g, j and Extended Data Fig. 8a). Activation of cGas−/− BMMO by agonists of RIG-I-like receptors and RNA-activated Toll-like receptors was also modestly impaired (Extended Data Fig. 8b). Together, these studies implicate a role for cGAS in controlling an RNA virus and in regulating basal immune responses. cGAS may, therefore, set the antiviral tone of the cell. We propose that, in the absence of cGAS, basal mRNA levels of some antiviral genes are reduced, making cells more vulnerable to some RNA viruses. As WNV RNA is predominantly controlled by RIG-I-like-receptor-mediated signalling through IRF3 (refs 22, 23), cGAS may be triggered by endogenous ligands to confer antiviral effects against RNA viruses. Alternatively, cGAS or protein complexes containing cGAS may have a more flexible functionality with respect to nucleic acid triggering than previously anticipated17,19, such that some viral RNA species can trigger cGAMP production and downstream antiviral responses.
Extended Data Figure 7. Viral burden in mice infected with WNV.
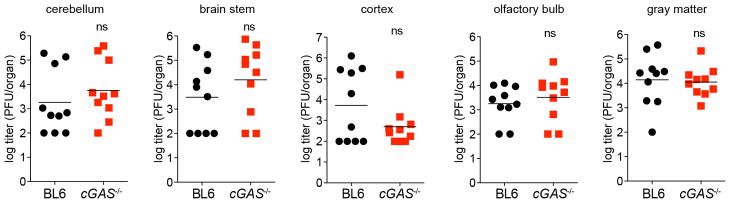
Wild-type or cGas−/− mice were infected with WNV and viral titres in several regions of the brain were determined by plaque assay. n = 10 mice per group. Statistical significance was determined by t-test.
Extended Data Figure 8. Role for cGAS in BMMO activation.
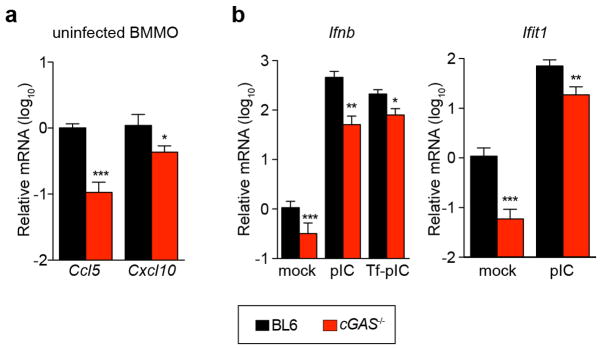
a, BMMO from wild-type and cGas−/− mice were analysed for baseline expression of chemokines Ccl5 and Cxcl10 by RT–PCR. b, BMMO from wild-type and cGas−/− mice were treated with polyIC (pIC) or transfected with polyIC (Tf-pIC) and Ifnb and Ifit1 levels were determined by qRT–PCR. In both panels, gene expression levels are relative to the housekeeping gene RPS29 and normalized to mock-treated wild-type cells. Data represent two experiments performed in triplicate. Error bars represent s.d. Statistical significance was determined by t-test. *P <0.05, **P <0.01, ***P <0.001.
The studies presented here validate the utility of the ISG screening platform to identify critical molecules in innate immunity and lay a foundation for further studies on mechanisms of novel antiviral molecules. Our in vivo data indicate that cGAS is pivotal in protecting the host from both DNA and RNA viruses, underscoring an unappreciated role for this key antiviral molecule in the innate immune response.
METHODS
Viruses and cells
Huh7, HeLa and 293T cells were maintained in DMEM (Invitrogen) with 10% FCS and 0.1 mM non-essential amino acids. NIH-3T12 cells were grown in DMEM supplemented with 5% FCS, 100 U penicillin per ml, 100 μg streptomycin per ml and 2 mM L-glutamine. STAT1−/− fibroblasts (an SV40 large T antigen immortalized human skin fibroblast line) were grown in RPMI (Invitrogen) with 10% FCS. The construction, characterization and generation of viral stocks for the following viruses have been previously described: CVB-GFP (derived from infectious clone pMKS1-GFP)24, PV-GFP (strain P1M, derived from infectious clone pPVM-2A144-GFP)25, EAV-GFP (derived from infectious clone pEAV211-GFP2aT)26, SINV-A-GFP and SINV-G-GFP (derived from infectious clones pS300-GFP and pG100-GFP)27, ONNV-GFP (derived from infectious clone pONNV.GFP)28, VEEV-GFP (derived from pTC83-GFP infectious clone)4, FLUAV-GFP (based on strain PR8)29, PIV3-GFP (based on strain JS)30, NDV-GFP (based on strain Hitchner B1)31, HMPV-GFP32 (based on isolate CAN97-83), RSV-GFP (based on strain A2)32, MV-GFP (MVvac2-GFP, based on vaccine strain, Edmonston lineage measles virus)33 and BUNV-GFP34 (based on rBUN-del7GFP). VV-GFP was propagated in BSC-40 cells. Viral stocks were prepared by three freeze–thaw cycles, followed by centrifugation at 1,000g to remove cellular debris. VV Western Reserve was obtained from the ATCC, propagated in Vero cells and purified by ultracentrifugation through a 36% sucrose cushion. MHV68 clone WUMS was obtained from the ATCC and propagated in NIH-3T12 cells. The WNV strain was isolated and passaged as described previously35.
Plasmids and molecular cloning
The production of the lentiviral-based ISG expression library has been described in detail4. To characterize human cGAS (MB21D1/C6orf150), we used the pENTR.C6orf150 (Genecopoeia, NCBI accession AK097148) plasmid as a starting point for all modifications. Standard PCR was used to generate mutants of cGAS that were progressively deleted of amino acids from the N or C termini. Overlap extension PCR was used to generate point mutants (E225A, D227A) in the wild-type protein. All mutant cGAS sequences were moved into the lentivirus by Gateway cloning, using pENTR.C6orf150 plasmids and pTRIP.CMV.IVSB.IRES.TagRFP-DEST in an LR reaction (Invitrogen) as previously described4. Primer sequences for mutagenesis are available upon request.
Lentivirus production and transduction assays
Lentiviral stocks were generated in 293T cells by co-transfection of with plasmids expressing (1) the TRIP.CMV. IVSb.ISG.ires.TagRFP lentivirus; (2) HIV gag-pol; and (3) the vesicular stomatitis virus glycoprotein (VSV-G) in a ratio of 1:0.8:0.2. For puromycin-selectable lentiviruses, we used the SCRPSY lentiviral backbone, which has been described previously11. Supernatants were collected at 48 h and 72 h, pooled, cleared by centrifugation at 1,000g and stored at −80 °C. For transduction assays, Huh7, HeLa or STAT1−/− fibroblasts were seeded into 24-well plates at a density of 7 × 104 cells per well and transduced with lentiviral pseudoparticles by spinoculation at 1,000g for 45 min at 37 °C in medium containing 3% FBS, 20 mM HEPES and 4 μg ml−1 polybrene. For confirmatory experiments (Fig. 2), new lentiviral stocks were generated for selected ISGs that had Z-scores less than −1.5 or greater than 2.0 in the initial screens. The less stringent cutoff of Z < −1.5 was chosen to include more ISGs from –ssRNA screens. Confirmatory experiments were performed under the same infection conditions as described above. The data from the confirmatory assays was stratified according to the frequency and relative magnitude with which ISGs affected RNA virus infectivity, using progressive 50% cutoffs to delineate strong versus modest effectors.
Virus infections
Before ISG screens, all GFP reporter viruses were optimized for infection in their respective target cells. Dose response and time course assays were carried out to determine the optimal volume of virus needed to infect 25–50% (approximately 0.5 m.o.i.) of the cell population during the first round of replication, before onset of viral spread. ISG screens and confirmatory assays were carried out under these optimized conditions, and cells were infected with each virus for the following time periods: VV-GFP (8 h), CVB-GFP (6 h), PV-GFP (8 h), EAV-GFP (19 h), SINV-GFP (10 h), VEEV-GFP (6 h), ONNV-GFP (17 h), FLUAV-GFP (8 h), PIV3-GFP (24 h), NDV-GFP (8 h), HMPV-GFP (18 h), RSV-GFP (23 h), MV (24 h), BUNV (11 h). For FLUAV and HMPV, trypsin was not added to the infected cells, thereby preventing release of virions from the cell surface and blocking viral spread.
Bioinformatics (clustering, co-occurrence and Gene Ontology)
To cluster ISGs with respect to the viruses they inhibit, 22 ISGs that inhibited at least one virus by more than 50% in confirmatory assays were selected. Replication data from the primary screens for each of these 22 genes was compiled. A web-based tool, http://www.hiv.lanl.gov/content/sequence/HEATMAP/heatmap.html, which uses heatmap.2 of the gplots package of the R statistical computing and graphics software environment, was used to generate a heatmap based on the similarity of ISG effects on virus infectivity. In brief, a data set in which ISGs were set as columns and viruses set as rows, with replication values corresponding to each position, was uploaded to the server. The algorithm used the ‘average’ cluster method and ‘Euclidean’ distance method. The heatmap was created with nine colours representing the quantitative range of virus infectivity, and dendrograms were generated to show the hierarchical relationships of both ISGs and viruses.
In a second clustering analysis, selected viruses were clustered hierarchically according to the appearance of the top 30 ISG names in the lists generated from the primary screening data (see Supplementary Tables 1 and 2). Each gene list was transformed to an n-dimensional binarized vector, where n represents the number of unique gene names in the cumulative gene list for all selected viruses (n = 176 for the selected 12 viruses). Thus, each position of this vector corresponds to a unique gene from this cumulative list. A value of 1 or 0 at each position of this vector indicates the presence or absence, respectively, of the ISG in the current list. The binarized vectors for selected viruses were clustered using the linkage function of the MATLAB Statistics Toolbox with the weighted average distance (WPGMA) (‘weighted’) selected as the algorithm for computing distance between clusters and one minus the sample correlation between points (‘correlation’) as the distance metric. The dendrogram was constructed using dendrogram function of the MATLAB Statistics Toolbox. The dendrogram represents U-shaped lines connecting viruses in the hierarchical tree according to the clustering of binarized vectors (see above) of selected viruses. The height of each U represents the distance between the two viruses being connected.
The top 20 inhibitory ISGs from 12 screens were analysed for co-occurrence. A gene appearing in all lists was assigned a frequency of 1, or a fractional percentage of 1 if the gene appeared in fewer lists. The data were stratified by +ssRNA and −ssRNA viruses.
Gene ontology (GO) analysis was performed on the top 30 inhibitory genes from 12 screens, followed by statistical enrichment analysis using the Enrichment widget of STRING, as described36. In this analysis, GO terms associated with a known pathway were assigned to each protein in the list. The P values were determined by a hypergeometric test, corrected using the Benjamini–Hochberg false discovery rate procedure, and ranked based on enrichment value. We also included ‘electronic inferred annotations’ in the P values calculation to increase statistical significance. GO terms with P <0.05 were compiled in Supplementary Table 3. Full GO analyses for each virus are available upon request.
siRNA-mediated gene silencing
STING was depleted in STAT1−/− fibroblasts by siRNA-mediated gene silencing. Four individual siRNAs (Qiagen) at 20 nM were tested for knockdown using HiPerFect Transfection Reagent (Qiagen) according to the manufacturer’s reverse transfection protocol. Cells were collected 48 h after transfection and STING protein levels were monitored by western blot, as described below. One of the four siRNAs reduced STING expression to nearly undetectable protein levels, and was chosen for subsequent functional assays.
RNA and protein detection from cell cultures
For gene expression studies, total RNA from STAT1−/− fibroblasts transduced with lentiviral vectors was isolated 48 h after transduction using an RNeasy Mini Kit (Qiagen). 50 ng total RNA was analysed by qRT–PCR using QuantiFast SYBR Green RT–PCR kit with commercially available QuantiTect Primers specific for OAS2 and RPS11 housekeeping control (Qiagen) according to the manufacturer’s instructions. Reactions were run on a Roche 480 Light Cycler or ABI 7500 Fast Real Time PCR System, and gene expression was calculated using the ΔΔCT method. In separate experiments, total RNA was processed for microarray analysis using BeadArray technology (Illumina) as described previously4. For protein expression studies, cells were lysed in radio-immunoprecipitation assay buffer containing Complete Protease Inhibitor Cocktail (Roche) and PhosSTOP Phosphatase Inhibitor Cocktail (Roche). Protein concentration of cell lysates was determined by Bradford assay (Pierce). Lysates were separated on 4–20% SDS–PAGE gradient gels (BioRad), blotted to nitrocellulose membrane (Amersham) and processed by western blotting. Blots were blocked overnight in 5% milk in 1 × TBS (50 mM Tris-Cl, pH 7.5, 150 mM NaCl.) with 0.05% Tween-20 (TBS-T), followed by incubation with primary and secondary antibodies in 1% milk in TBS-T for 1 h and 30 min, respectively. Proteins were visualized by incubating blots with enhanced chemiluminescent substrate ECL (Pierce) and exposing blots to autoradiography film (Denville Scientific). Antibodies used in the study include: anti-STING (R&D Systems MAB7169), anti-phosho-IRF3 (Abcam ab76493), anti-MB21D1 (Sigma HPA031700), anti-actin (Abcam ab6276), goat anti-rabbit horseradish peroxidase (HRP) and goat anti-mouse HRP (Pierce).
ISRE reporter assays
To test ISGs for activation of ISRE-dependent transcription, 1.5 × 104 293T cells in individual wells of a 96-well plate were transduced with lentiviruses expressing ISGs. 16–18 h later, transduced cells were transfected with the pISG54.ISRE-Fluc plasmid using X-treme Gene 9 Transfection reagent (Roche) following standard protocols. Cells were collected 24 h after transfection using 1 × cell culture lysis buffer (Promega) and Fluc activity was monitored using the Luciferase Assay System (Promega). To test for ISG-mediated suppression of IFN activity, a similar protocol was used, except that ISG and ISRE-Fluc plasmids were co-transfected before treatment with recombinant IFN-α (PBL Interferon Source).
Infection of mice
Mb21d1tm1a mice were obtained from the EUCOMM consortium and bred to FLPe-expressing mice (B6-Tg(CAG-FLPe)36, provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan37) to generate conditional knockout mice by removing the targeting cassette. Conditional knockout mice were bred to Cre-expressing mice (B6.C-Tg(CMV-cre)1Cgn/J, Jackson Laboratories) to generate mice with a deletion of exon 2, which includes the catalytic residues E211 and D213. Mice were backcrossed to C57BL/6J (B6) mice (Jackson Laboratories) to remove Cre allele. All mice were bred and maintained in a specific-pathogen-free barrier facility at Washington University in St Louis, Missouri, in accordance with federal and institutional guidelines. To determine MHV68 titres in tissues, mice were infected between 8 and 9 weeks of age with 106 p.f.u. of MHV68 by intraperitoneal injection in 0.3 ml PBS. Upon euthanization, organs were placed in 1 ml of complete DMEM and frozen at −80 °C. To determine susceptibility to VV Western Reserve, 8–9-week-old mice were anesthetized with ketamine/xylazine before intranasal inoculation with 8,000 p.f.u. in 50 μl MEM. Mice were also infected with 100 p.f.u. of WNV (strain New York 1999) in 50 μl via a subcutaneous route. Tissues were collected at day 8 after infection and analysed for viral burden by plaque assay on Vero cells. For these studies, 10 or 15 mice were used per experiment in order to achieve reliable statistics. All mice were monitored daily for weight loss and lethality, and mice that became moribund were euthanized.
Bone-marrow-derived macrophage infections
Primary bone-marrow-derived macrophages were prepared as described previously38. Cells were allowed to differentiate for 7 days, and then adherent cells were scraped and seeded in tissue culture-treated plates. For MHV68 experiments, cells were infected with MHV68 at a m.o.i. of 10 for 1 h with occasional rocking at 37 °C and 5% CO2. For viral growth curves, cells were washed three times with medium and incubated in DMEM supplemented with 10% FBS and 2 mM L-glutamine for the indicated period of time at 37 °C and 5% CO2, before being frozen at −80°C. For gene expression analysis, the inoculum was replaced with complete DMEM and the cells incubated for 6 h before being lysed in TRIzol reagent for total RNA extraction. For VV experiments, macrophages were infected at a m.o.i. of 0.1 in DMEM without serum. One hour later, cells were washed once with PBS, and incubated in DMEM supplemented with 2% FBS. At indicted times post infection, cells and media were frozen/thawed twice and then supernatants were serially diluted and plaqued on monolayers of Vero cells. For WNV experiments, cells were infected in 12-well plates at a m.o.i. of 0.1 or 3. Virus was collected from supernatants at specific times and titrated by plaque assay on Vero cells. For activation experiments, cells were stimulated with 2 μg ml−1 imiquimod (Invivogen), 2 μg ml−1 polyI:C (Invivogen) or transfected with 1 μg polyI:C using TransIT-LT1 (Mirus) for the indicated amount of time. Total RNA was collected from BMMO to assess gene expression levels by qRT–PCR, as described below.
Plaque assays
To determine the effects of ISGs on poliovirus production, HeLa cells were transfected in a 24-well plated with 500 ng lentiviral plasmids encoding ISGs using FugeneHD (Roche). Forty-eight hours after transfection, cells were infected with P1M (10 m.o.i.) for 16 h. Lysates were collected and viral titres determined by plaque assay on HeLa cell monolayers. Infections were performed at 37 °C for 1 h with occasional rocking before cells were overlaid with medium containing DMEM, 0.2% NaHCO3, 5% bovine calf serum, 1% penicillin-streptomycin and 0.8% Noble agar (Sigma). After 48 h incubation, plaques were visualized with crystal violet. Plaque assays to determine MHV68 and WNV viral titres from BMMO or mouse organs were performed on NIH-3T12 and Vero cells, respectively. Organs were thawed and homogenized with sterile 1.0 mm zirconia/silica beads and a mini-beadbeater (BioSpec Products) before dilution and plating onto cells. Infection was performed at 37 °C for 1 h with occasional rocking before cells were overlaid with medium containing 2% methylcellulose. After a 1-week incubation, plaques were visualized with 3% neutral red solution.
Determination of mRNA transcript levels in mouse cells and organs
Spleens were homogenized and macrophages were lysed in TRIzol reagent (Invitrogen) and processed according to the manufacturer’s instructions to isolate total RNA. RNA samples were treated with DNase I (Ambion) before first-strand cDNA synthesis with ImProm-II (Promega) and oligo(dT)15. qPCR was performed on a StepOnePlus machine using Power SYBR Green master mix (Applied Biosystems) and primers specific for cGas (5′-ACGAGAGCCGTTTTATCTCGTACCC-3′ and 5′-TGTCCGGAAGATTCACAGCATGTTT-3′) or ribosomal protein S29 (RPS29; 5′-AGCAGCTCTACTGGAGTCACC-3′ and 5′-AGGTCGCTTAGTCCAACTTAATG-3′). TaqMan quantitative PCR was performed using primers and probes specific for Ifnb (5′-CTGGAGCAGCTGAATGGAAAG-3′, 5′-CTTCTCCGTCATCTCCATAGGG-3′ and probe 5′-/56-FAM/CAACCTCACCTACAGGGCGGACTTCAAG/36-TAMSp/-3′39, Ifit1 (5′-GAGCCAGAAAACCCTGAGTACA-3′, 5′-AGAAATAAAGTTGTCATCTAAATC-3′ and probe 5′-/56-FAM/ACTGGCTATGCAGTCGTAGCCTATCGCC/36-TAMSp/-3′), Ifit2 (5′-CTGAAGCTTGACGCGGTACA-3′, 5′-ACTTGGGTCTTTCTTTAAGGCTTCT-3′and probe 5′-/56-FAM/AAAACCAAGCAATGGCGCTGGTTG/36-TAMSp/-3′), Oas1a (5′-TGAGCGCCCCCCA TCT-3′, 5′-CATGACCCAGGACATCAAAGG-3′ and probe 5′-/56-FAM/AGGAGGTGGAGTTTGATGTGCTG/36-TAMSp/-3′), Il6 (5′-GCCAGAGTCCTTCAGAGAGATACA-3′ and 5′-CTTGGTCCTTAGCCACTCCTTC-3′), Tnfa (5′-GGGTGATCGGTCCCCAAAGG-3′ and 5′-CTCCACTTGGTGGTTTGCTACGA-3′), Il1b (5′-GCACACCCACCCTGCAG-3′ and 5′-AACCGTTTTTCCATCTTCTTCTT-3′), Ccl5 (5′-CAAGTGCTCCAATCTTGCAGTC-3′ and 5′-TTCTCTGGGTTGGCACACAC-3′), Cxcl10 (5′-AGTGCTGCCGTCATTTTCTG-3′, 5′-ATTCTCACTGGCCCGTCAT-3′ and probe 5′-/56-FAM/AGTCCCACTCAGACCCAGCAGG/36-TAMSp/-3′) with AmpliTaq Gold (Applied Biosystems). Transcript levels were analysed using the ΔΔCt method, with RPS29 as the reference gene. qPCR products were confirmed by melt curve and/or agarose gel electrophoresis.
Supplementary Material
Acknowledgments
We thank the following investigators for contributing viral molecular clones or viral stocks: R. Cattaneo (MV), P. Collins (PIV3, HMPV, RSV), I Frolov (VEEV), M. Heise (SINV), S. Higgs (ONNV), P. Traktman (VV), J. L. Whitton (CVB). We thank E. Jouanguy and J.-L. Casanova for STAT1−/− fibroblasts. We acknowledge the support of C. Zhao, X. Wang and W. Zhang at The Rockefeller University Genomics Resource Center. We thank E. Castillo, B. Flatley, A. Webson, E. Duan and A. McLees for laboratory support, and D. Kraemalmeyer for managing mouse colonies. This work was supported in part by National Institutes of Health grants AI091707to C.M.R., AI057158 to I. Lipkin (Northeast Biodefense Center, subcontract to C.M.R.), DK095031 to J.W.S., AI057160 to H.W.V., HHSN272200900041CU19 to M.S.D. and H.W.V., AI104972 to M.S.D., AI083025, AI095611 and CEIRS contract HHSN266200700010C to A.G-S., GM076547 and GM103511 to J.D.A. and AI057160 to W.M.Y., an investigator of the Howard Hughes Medical Institute. M.D.G. is supported by T32 AR007279. Additional funding was provided by the Greenberg Medical Research Institute, the Starr Foundation and the Ronald A. Shellow, M.D. Memorial Fund (C.M.R.).
Footnotes
Online Content Any additional Methods, Extended Data display items and Source Data are available in the online version of the paper; references unique to these sections appear only in the online paper.
Supplementary Information is available in the online version of the paper.
Author Contributions J.W.S., D.A.M., M.S.D., H.W.V. and C.M.R. designed the project. J.W.S.,D.A.M., N.I., M.D.G.,B.S.,R.D., J.L.E.,K.B.M. and R.B.R. performed the experimental work. J.W.S., V.L. and A.V.R. performed clustering analyses. J.W.S., D.A.M., M.S.D., H.W.V. and C.M.R. analysed the results and wrote the manuscript. B.M., A.A., J.D.A., R.M.E., A.G.-S., V.R., M.D.G., W.M.Y., E.J.S. and M.S.D. contributed reagents and technical expertise.
Author Information Microarray Data were submitted to NCBI Gene Expression Omnibus (GEO) with accession number GSE52241. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Veer MJ, et al. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- 3.Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1:519–525. doi: 10.1016/j.coviro.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schoggins JW, et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu SY, Sanchez DJ, Aliyari R, Lu S, Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc Natl Acad Sci USA. 2012;109:4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metz P, et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology. 2012;56:2082–2093. doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 7.Fusco DN, et al. A genetic screen identifies interferon-α effector genes required to suppress hepatitis C virus replication. Gastroenterology. 2013;144:1438–1449. doi: 10.1053/j.gastro.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao H, et al. A functional genomic screen reveals novel host genes that mediate interferon-alpha’s effects against hepatitis C virus. J Hepatol. 2012;56:326–333. doi: 10.1016/j.jhep.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Burke CW, Ryman KD, Klimstra WB. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J Virol. 2007;81:11246–11255. doi: 10.1128/JVI.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson SJ, et al. Inhibition of HIV-1 particle assembly by 2′,3′-cyclic-nucleotide 3′-phosphodiesterase. Cell Host Microbe. 2012;12:585–597. doi: 10.1016/j.chom.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoggins JW, et al. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc Natl Acad Sci USA. 2012;109:14610–14615. doi: 10.1073/pnas.1212379109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karki S, et al. Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PLoS ONE. 2012;7:e37398. doi: 10.1371/journal.pone.0037398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng X, et al. C7L family of poxvirus host range genes inhibits antiviral activities induced by type I interferons and interferon regulatory factor 1. J Virol. 2012;86:4538–4547. doi: 10.1128/JVI.06140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dupuis S, et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nature Genet. 2003;33:388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 15.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kranzusch PJ, Lee ASY, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013;3:1362–1368. doi: 10.1016/j.celrep.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao P, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Civril F, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–337. doi: 10.1038/nature12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao D, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li XD, et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suthar MS, et al. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog. 2010;6:e1000757. doi: 10.1371/journal.ppat.1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M., Jr The essential, non-redundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J Virol. 2013;87:11416–11425. doi: 10.1128/JVI.01488-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feuer R, Mena I, Pagarigan R, Slifka MK, Whitton JL. Cell cycle status affects coxsackievirus replication, persistence, and reactivation in vitro. J Virol. 2002;76:4430–4440. doi: 10.1128/JVI.76.9.4430-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teterina NL, Levenson EA, Ehrenfeld E. Viable polioviruses that encode 2A proteins with fluorescent protein tags. J Virol. 2010;84:1477–1488. doi: 10.1128/JVI.01578-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van den Born E, Posthuma CC, Knoops K, Snijder EJ. An infectious recombinant equine arteritis virus expressing green fluorescent protein from its replicase gene. J Gen Virol. 2007;88:1196–1205. doi: 10.1099/vir.0.82590-0. [DOI] [PubMed] [Google Scholar]
- 27.Simmons JD, Wollish AC, Heise MT. A determinant of Sindbis virus neurovirulence enables efficient disruption of Jak/STAT signaling. J Virol. 2010;84:11429–11439. doi: 10.1128/JVI.00577-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brault AC, et al. Infection patterns of o’nyong nyong virus in the malaria-transmitting mosquito, Anopheles gambiae. Insect Mol Biol. 2004;13:625–635. doi: 10.1111/j.0962-1075.2004.00521.x. [DOI] [PubMed] [Google Scholar]
- 29.Manicassamy B, et al. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci USA. 2010;107:11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, et al. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J Virol. 2005;79:1113–1124. doi: 10.1128/JVI.79.2.1113-1124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park MS, et al. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J Virol. 2003;77:1501–1511. doi: 10.1128/JVI.77.2.1501-1511.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biacchesi S, et al. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology. 2004;321:247–259. doi: 10.1016/j.virol.2003.12.020. [DOI] [PubMed] [Google Scholar]
- 33.del Valle JR, et al. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J Virol. 2007;81:10597–10605. doi: 10.1128/JVI.00923-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi X, van Mierlo JT, French A, Elliott RM. Visualizing the replication cycle of bunyamwera orthobunyavirus expressing fluorescent protein-tagged Gc glycoprotein. J Virol. 2010;84:8460–8469. doi: 10.1128/JVI.00902-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ebel GD, Carricaburu J, Young D, Bernard KA, Kramer LD. Genetic and phenotypic variation of West Nile virus in New York, 2000–2003. Am J Trop Med Hyg. 2004;71:493–500. [PubMed] [Google Scholar]
- 36.Franceschini A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanki H, Suzuki H, Itohara S. High-efficiency CAG-FLPe deleter mice in C57BL/6J background. Exp Anim. 2006;55:137–141. doi: 10.1538/expanim.55.137. [DOI] [PubMed] [Google Scholar]
- 38.Hwang S, et al. Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe. 2012;11:397–409. doi: 10.1016/j.chom.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daffis S, Samuel MA, Keller BC, Gale M, Jr, Diamond MS. Cell-specific IRF-3 responses protect against West Nile virus infection by interferon-dependent and -independent mechanisms. PLoS Pathog. 2007;3:e106. doi: 10.1371/journal.ppat.0030106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.