

Abstract
Autism spectrum disorder (ASD) and intellectual disability (ID) are neurodevelopmental disorders with large genetic components, but identification of pathogenic genes has proceeded slowly because hundreds of loci are involved. New exome sequencing technology has identified novel rare variants and has found that sporadic cases of ASD/ID are enriched for disruptive de novo mutations. Targeted large-scale resequencing studies have confirmed the significance of specific loci, including chromodomain helicase DNA binding protein 8 (CHD8), sodium channel, voltage-gated, type II, alpha subunit (SCN2A), dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), and catenin (cadherin-associated protein), beta 1, 88 kDa (CTNNB1, beta-catenin). We review recent studies and suggest that they have led to a convergence on three functional pathways: (i) chromatin remodeling; (ii) wnt signaling during development; and (iii) synaptic function. These pathways and genes significantly expand the neurobiological targets for study, and suggest a path for future genetic and functional studies.
Keywords: autism genetics, autism spectrum disorder, copy number variant, exome sequencing, intellectual disability, single nucleotide variant
Introduction
The identification of genes underlying ID and ASD has been most successful for syndromic Mendelian or monogenic disorders – for example, FMR1 (Fragile-X syndrome, [1]), MECP2 (Rett syndrome, [2]), or UBE3A (Angelman syndrome, [3]). Together, however, these syndromes are estimated to account for less than 10% of ASD/ID, suggesting the presence of additional genes and etiologies. Initial population-based studies failed to identify single genes of major effect and few major common risk variants have been replicated, despite the strong observed heritability of these diseases [4–7]. By contrast, targeted and genome-wide microarray studies revealed that large de novo copy number variants (CNVs) were significantly enriched among probands when compared to unaffected siblings and/or controls [8–14], a finding that echoed the earlier discovery of large chromosomal aberrations in ASD and ID. Both initial and subsequent higher-resolution studies estimate that 8% of sporadic ASD cases carried a de novo CNV, as compared with only 2% of unaffected siblings [11,12]. Furthermore, among children with general developmental delay (DD) and ID, rare large de novo CNVs are thought to account for up to 15% of disease burden [13]. Although individually rare, some of these CNVs were in fact recurrent mutations, mediated by locus-specific genomic instability [14], and many of these same recurrent CNVs observed initially in patients with ID [15] or ASD [16] have been identified in adults with epilepsy [17], bipolar disorder [18], or schizophrenia [19,20], suggesting overlap in the genetic etiology of these disorders.
The discovery of an aggregate burden of large de novo CNVs and the identification of recurrent events signaled a new paradigm for ASD and ID genetics. Although specific CNVs are individually rare, combined they account for a significant fraction of cases, indicating the presence of considerable locus heterogeneity of ASD and ID. The de novo nature of these CNVs, together with their absence in the general population, suggests they represent a class of highly deleterious and highly penetrant mutations. Their underlying genetic model does not explicitly fit a recessive model of disease because CNVs are primarily present as hemizygous deletions or duplications. These mutations alter the dosage of genes but do not completely abolish their presence. Collectively, these observations support a complex disease/rare variant model for ASD, in which a proportion of etiologic risk is conferred by very rare variants and de novo mutations.
The commoditization of next-generation or ‘massively parallel’ sequencing represents a turning point in human genetics and makes it possible to discover sequence-level variants across nearly all coding regions (‘the exome’) or the whole genome (Box 1). These methods were first applied to confirm point mutations underlying Mendelian disorders [21], and subsequent pilot studies demonstrated that family-based (trio) exome sequencing could discover pathogenic mutations in simplex ID [22] or ASD [23]. In the past year, this paradigm of de novo mutation discovery using exome sequencing of parent–child trios has been expanded to about 1000 ASD or ID families, resulting in the first detailed picture of how de novo coding mutations contribute to these disorders.
Box 1. Current sequencing technologies and their limitations.
Whole-genome sequencing (WGS) provides the ‘most complete’ view of genomic variation and can detect SNVs, indels, and CNVs irrespective of frequency in a genome-wide fashion (although the power to detect events across the genome is not uniform based on local genomic composition). High costs and difficulty in the interpretation of nongenic variants are bottlenecks in wide-scale application of WGS.
Exome sequencing combines next-generation sequencing technologies with the targeted capture and amplification of exons (approximately 40 Mbp of the genome) in order to reduce the total amount of sequencing needed for the accurate determination of mutations in (or near) exons. However, the efficacy of the capture depends on the percentage GC nucleotide composition of the targeted sequence (also known as ‘GC capture bias’). For exons with especially high GC content, exome sequencing can fail to produce enough coverage for accurate variant detection and calling. As shown in Figure I, several of the most GC-rich exons (blue bars) in the SHANK3 gene (right) have inadequate coverage (black lines indicate mean coverage of a single family), preventing accurate assessment of variants in those exons. By contrast, the lower GC content (green histogram) of the CHD8 gene (left) results in far higher and more uniform coverage of exons.
Molecular inversion probe (MIP) sequencing is a cost-effective targeted assay that uses custom oligonucleotides to efficiently capture target sequence. Unlike exome sequencing, however, MIPs use flanking primer arms and polymerase extension to capture the desired DNA target, thus reducing GC capture and other biases. In addition, MIP assays can be performed as highly multiplexed reactions, allowing for sequencing of targeted in genes in up to 192 individuals per sequencing reaction.
Genome-wide association studies (GWAS) are a widely used assay that leverage SNP markers (detected via hybridization to oligonucleotide arrays) to tag genomic regions and associate them with disease based on a comparison of cases and controls. Historically, GWAS studies have been limited to assaying common genetic variation and cannot detect novel or rare SNVs.
In this review, we synthesize the results of recent large-scale exome sequencing studies of ASD and ID [24–29] and summarize their implications for human neurodevelopmental genetics. There are three themes. (i) Exome sequencing of ASD/ID families has revealed a significant excess of de novo mutations in probands when compared to unaffected siblings and has identified novel candidate genes contributing to the neurological deficits. We note that the strongest effects are observed for de novo loss-of-function (or truncating) mutations (see Glossary), which prematurely truncate the protein due to frameshift and nonsense mutations. (ii) Both CNV and exome sequencing data suggest that no single gene will account for more than 1% of autism cases; rather, rare mutations in hundreds of genes may contribute to ASD or ID. (iii) Analyses of network connectivity further implicate potentially important neurodevelopmental and synaptic pathways in ASD and ID. Collectively, these studies represent a significant step forward for neurodevelopmental disorders providing a springboard for understanding their neurobiological underpinnings. We aim to focus on the molecular convergence revealed by these studies; for readers interested in other aspects of this topic, we suggest excellent reviews on ASD neurobiology [30], de novo mutation [31,32], and exome sequencing [33]. We emphasize that although this review is focused on the insights gained by considering a de novo/rare variant model of ASD and ID genetics, other genetic etiologies are implicated in ASD as well (for reviews, see [34,35]) and no single etiology is likely to be fully independent of other etiologies or of environmental factors {see [36] for a review; see also the CHARGE (CHildhood Autism Risks from Genetics and Environment) Study [37]}.
An increase in de novo loss-of-function mutations
Both de novo CNVs and single nucleotide variants (SNVs) can have, in principle, similarly disruptive effects on genes. Crucially, however, the detection of de novo SNVs yields gene-level specificity, thus allowing individual pathogenic genes and neurobiological pathways to be identified. Moreover, a small subset of the de novo mutations (~4% for unaffected and ~9% of affected [26,27]) are disruptive (e.g., frameshift, premature stop codon, splice-donor defect) with respect to the protein’s biological function. Recurrent mutations of this type for a specific gene can strengthen the probability that the de novo mutation relates to phenotype. Because de novo protein-encoding SNVs are collectively more common mutation events (~1/generation) than large de novo CNVs (~0.02/generation), there is the exciting possibility that this type of mutation may explain a larger faction of the genetic etiology of ASD.
In all, six recent exome sequencing studies of trios (mother, father, and affected child) and quads (also includes an unaffected sibling) of sporadic ASD [24–27] or sporadic ID [28,29], together comprising 1078 families (Table 1) have been performed. Three of the ASD studies included unaffected siblings in order to compare mutation rates between affected probands and siblings. Although these studies found a slightly elevated rate of mutation in probands versus their unaffected siblings (1.02 vs. 0.79 mutations per offspring), the type of mutation was critical: probands had two- to threefold more disruptive de novo mutations in comparison to their siblings, or to a random model of mutation [26,27]. Overall, among the 593 ASD quads, there were 80 such mutations in probands with ASD, but only 36 in siblings [odds ratio (OR) = 2.41, P < 1 × 10−4, Fisher’s exact test; Table 1]. The reported enrichment of missense mutations in probands has been less robust, with study estimates for enrichment between 1- and 1.34-fold, but analysis of all quads does show weak statistical enrichment (OR = 1.29, P = 0.03). It is likely that some missense mutations are pathogenic whereas others are benign, a distinction that is likely to be dependent on the context of the mutation and affected proteins themselves.
Table 1.
Six recent family-based exome studies of ASD and ID
| Study details | N | Synonymousb | Missenseb | Nonsense, splice-site, indelsb | ||
|---|---|---|---|---|---|---|
| Iossifov et al. (2012) [26] | ASDa | Quads | 343 | 79:69 | 207:207 | 59:28 |
| Sanders et al. (2012) [27] | ASDa | Quads | 200 | 29:39 | 110:82 | 15:5c |
| O’Roak et al. (2012) [24] | ASDa | Quads | 50 | 14:16 | 40:31 | 6:3 |
| Trios | 159 | 54 | 115 | 29 | ||
| Neale et al. (2012) [25] | ASD | Trios | 175 | 50 | 101 | 18 |
| de Ligt et al. (2012) [28] | ID | Trios | 100 | 16 | 48 | 14 |
| Rauch et al. (2012) [29] | ID | Trios | 51 | 11 | 56 | 20 |
| Total OR (ASD) (95% CI) |
1078 | 122:124 0.98 (0.73–1.31:1) |
357:320 1.29 (1.01–1.63) |
80:36 2.41 (1.58–3.75) |
Trios/quads from the Simons Simplex Collection (SSC).
Counts refer to the number of mutations in probands or, when separated by a colon, to probands and siblings (e.g., probands:siblings).
Not including indels.
Overall, these studies suggest that protein-truncating de novo SNVs contribute to the risk of ASD for about 10–15% of probands [26,27], although this fraction is almost certainly a conservative estimate, because an unknown fraction of de novo events are still missed using current sequencing methods and bioinformatics tools (see Figure I in Box 1). It is important to note that the six current exome studies focused primarily on a de novo mutation genetic model for the development of disease. Recent results highlight the effect of transmitted CNVs [38,39], as well as a renewed emphasis on the effect of common variation in ASD [40] based on the study of data generated from the same samples. Among the ID studies, it is also presently difficult to estimate the impact, due to the smaller number of sequenced ID families and the lack of information from family-based controls (i.e., unaffected siblings). Indeed, the quad-based studies for ASD reviewed here have been instrumental in uncovering the effect of de novo variation in probands, especially in comparison to their siblings, which serve as an internal control. By contrast, trio-based and case-control study designs are less informative with respect to observed mutation rates and are susceptible to stratification effects [41]. Finally, the pattern and impact of very low frequency variants in normal controls is not well understood, because most efforts have focused on patients with disease [although some projects such as the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project have begun to address this question].
Figure I.
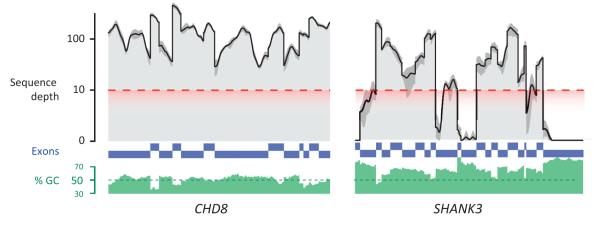
High genomic GC nucleotide content (green histogram) hinders whole-exome sequencing for some genes, such as SHANK3 (right). Individual coding exons are shown in blue with nongenic sequences removed. Black lines indicate mean sequence depth for a single ASD trio and dark gray intervals indicate maximum and minimum depth across the family. The red dashed line indicates the minimum threshold required for accurate variant detection.
Candidate genes yet few recurrent hits
Many strong neurobiological candidates have emerged from the genes disrupted by de novo mutations in these studies, including mutations in previously identified ASD/ID genes. Several mutations were identified in neurexin 1 (NRXN1) and neuroligin 1 (NLGN1); both are central components of the neurexin–neuroligin synaptic cell adhesion complex [42]. Numerous de novo mutations were identified in genes or loci linked to Mendelian disorders, many of which have features of ASD or ID. These loci and genes include MBD5 [mental retardation, autosomal dominant #1, Online Mendelian Inheritance in Man (OMIM) 156200], CHD7 (CHARGE syndrome, OMIM 214800), PTEN (Cowden syndrome, OMIM 158350), DYRK1A (in Down syndrome critical region, OMIM 190685), TSC2 (tuberous sclerosis, OMIM 613254), SETBP1 (Schinzel-Giedion syndrome, OMIM 269150), and RPS6KA3 (Coffin-Lowery syndrome, OMIM 303600). Finally, mutations in several genes mapped to critical deletion regions or association intervals initially discovered by large CNVs, including mutations in SYNRG (17q12 deletion syndrome), POLRMT (19p13.3 deletion), and CTTNBP2 – a potential candidate for the AUTS1 (7q31) deletion locus.
Recurrently mutated genes, however, were few. In summary, only three genes [CHD8, SCN2A, and synaptic Ras GTPase activating protein 1 (SYNGAP1)] had two independent truncating de novo mutations in any single study, and no gene had more than three mutations. Models designed to estimate the significance of recurrent de novo mutations based on gene size and context found nominal significance for CHD8, NTNG1 [24], and SCN2A [27], but most genes could not be conclusively implicated. Notably, however, a follow-up case-control study by Neale and colleagues of 935 cases found three additional truncating CHD8 mutations and one splice-site mutation in SCN2A, further strengthening the initial disease associations of these genes [25]. In addition, within a few weeks of these initial reports, a de novo translocation was discovered mapping to CHD8 [43].
Large-scale resequencing of candidate genes
Given the low rate of recurrence among genes with de novo mutations, estimates of overall locus heterogeneity for ASD have yielded between 300 and 1000 genes that could confer increased ASD risk when subjected to de novo mutation (Figure 1). Even if exome sequencing prices continue to fall, the cost to confirm the association for a significant fraction of these genes remains impractically high, especially if thousands or tens of thousands of samples are required, as has been suggested by CNV studies. Instead, targeted next-generation resequencing of candidate genes has proven to be instrumental in associating specific genes. In particular, de Ligt and colleagues resequenced five candidate genes in a confirmation series of 765 ID patients, identifying additional mutations in CTNNB1 and GATAD2B and markedly strengthening their association with ID. Similarly, we have successfully used a molecular inversion probe (MIP) assay to capture and sequence 44 candidate genes in 2446 ASD probands [44]. MIP resequencing generates a complete sequence across targeted regions, can be performed at high scale and low cost (under $1 per gene per sample), and delivers higher sensitivity for targeted loci than exome sequencing due to increased sequence coverage. Altogether, this assay yielded 27 new de novo mutations across 16 genes; of these mutations, 17 were disruptive SNVs, a fraction higher than expected by chance. The discovery of these mutations confirmed the association with ASD for CHD8 and DYRK1A and provided significant statistical support for four novel genes: GRIN2B (glutamate receptor, ionotropic, N-methyl d-aspartate 2B), TBR1 (T-box brain gene 1), PTEN (phosphatase and tensin homolog), and TBL1XR1 [transducin (beta)-like 1 X-linked receptor 1].
Figure 1.
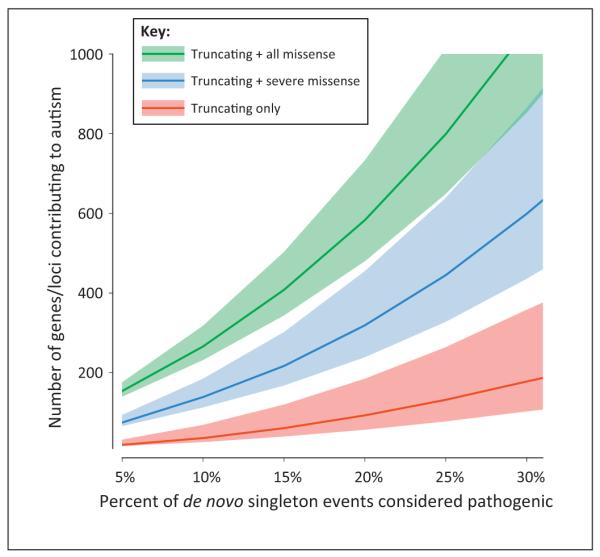
Estimating the number of autism spectrum disorder (ASD)/intellectual disability (ID) risk genes. We estimate the number of ASD and ID genes, using an adaptation of the ‘hidden species problem’ based on the ratio of genes with multiple de novo mutations to all genes with de novo mutations. For each estimate, all genes with recurrent de novo mutations are considered pathogenic, as well as a defined fraction of mutations in genes observed just once (because all de novo mutations are unlikely to be pathogenic). Including more of these singleton mutations as pathogenic, as well as including a broader range of mutation type, exponentially increases the number of ASD and ID risk loci. Thus, considering a disease model in which 15% of all truncating de novo mutations are sufficient and pathogenic, only approximately 50 genes are expected to be similarly sufficient in their pathogenicity; however, by including missense mutations, the number of loci rises dramatically [to over 400 loci when 15% of de novo single nucleotide variants (SNVs) are considered pathogenic]. Taken together, this model highlights the locus heterogeneity underlying the genetic etiology of ASD and ID and suggests that the etiology of a large proportion of ASD/ID cases may not be due to a single de novo mutation (truncating or missense); rather, these cases may be the result of a complex set of interactions between multiple mutations, including SNVs, indels, and copy number variants (CNVs). The shaded area indicates the 95% confidence interval (CI) around the estimate.
In summary, when considering only protein-disruptive mutations from six exome sequencing studies (four ASD and two ID) and including the resequencing of some of these candidate genes, a set of 11 genes (Table 2) show enrichment in cases with ASD/ID and account for approximately 2.2% of all cases. We have summarized the distribution of mutations, as well as the prevalence and coding location of mutations found in exome sequence from 6503 samples from the NHLBI Exome Sequencing Project (ESP) in Table 2. For several of these genes with recurrent de novo hits in ASD probands (CHD8, GRIN2B, DYRK1A), no truncating variants were observed in the ESP. Furthermore, while control mutations are sometimes found in genes in high frequency (e.g., frameshift in SYNGAP1 at 3.2% frequency in controls), these mutations are found exclusively near the carboxy terminus of the protein and outside of functional protein domains and are unlikely to affect protein function (Figure 2).
Table 2.
Recurrent disruptive mutations in ID and ASD
| Genea,b | ID cases | ASD cases | Summaryc,d,e | ESP samples | |
|---|---|---|---|---|---|
| Variants | Frequency | ||||
| CHD8 | – | 9/2446 | 2 (O), 7 (O*) [+3 (N*)] | 0 | 0/6503 |
| SCN2A | 3/151 | 2/593 | 1 (L), 2 (R), 2 (S) [+1 (N*)] | 1 | 7/6503 |
| SYNGAP1 | 3/151 | – | 1 (L), 2 (R) | 1 | 207/6503f |
| GRIN2B | – | 3/2446 | 1 (O), 2 (O*) | 0 | 0/6503 |
| DYRK1A | – | 3/2446 | 1 (I), 1 (O), 1 (O*) | 0 | 0/6503 |
| ZNF292 | 1/151 | 1/593 | 1 (L), 1 (N) | 1 | 2/6503 |
| POGZ | – | 2/593 | 1 (I), 1 (N) | 1 | 1/6503 |
| KATNAL2 | – | 2/593 | 1 (O), 1 (S) | 1 | 1/6503 |
| TBR1 | – | 2/2446 | 1 (O), 1 (O*) | 0 | 0/6503 |
| CTNNB1 | 1/151 | 1/2446 | 1 (L), 1 (O*), [+1 (L*)] | 0 | 0/6503 |
| SETBP1 | 1/151 | 1/593 | 1 (O), 1 (R) | 3 | 58/6503f |
| ADNP | – | 2/2446 | 1 (O), 1 (O*) | 1 | 1/6500 |
| LRP2 | 1/151 | 1/593 | 1 (I), 1 (L) | 6 | 53/6500 |
| ARID1B | – | 2/2446 | 1 (O), 1 (O*) | 5 | 314/6500 |
Genes with two or more de novo truncating mutations observed in studies of ASD or ID are listed.
Additional abbreviations: KATNAL2, katanin p60 subunit A-like 2; LRP2, low density lipoprotein receptor-related protein 2; POGZ, pogo transposable element with ZNF domain; ZNF292, zinc finger protein 292.
The summary indicates studies in which mutations were discovered.
I, Iossifov et al. [26]; L, de Ligt et al. [28]; N, Neal et al. [25], O and O*: O’Roak et al. [24,44]; R, Rauch et al. [29]; S, Sanders et al. [27].
Mutations found in secondary replication screens or case-control studies are indicated in [brackets] with starred (*) reference. Truncating events found in the Exome Sequencing Project (ESP) database and their population frequencies are shown.
The truncating variants found in the ESP database in SYNGAP1 and SETBP1 genes fell at the extreme 3′ end of the gene, suggesting that they do not adversely affect gene function
Figure 2.

The location of de novo truncating mutations in the top five autism spectrum disorder (ASD) and intellectual disability (ID) genes. Red markers indicate locations of de novo mutations in ASD and ID cases; green markers indicate locations of truncating mutations in the Exome Sequencing Project (ESP) database of over 6500 samples (see Table 2 for details). Mutation codes: S, stop-gain; Fs, frameshift; Ss, splice-site mutation; ΔAA, amino-acid loss (non-frameshifting). Blocks indicate annotated protein domains from UniProt. Domain names, top to bottom: CD, chromodomain; DEX, helicase ATP-binding; HELC, helicase C-terminal; TM, transmembrane domain; IQ, IQ domain; PDZ, PDZ-binding motif; LOC, bipartite nuclear localization signal; STK, serine/threonine protein kinase; PH, pleckstrin homology domain; C2, C2 domain; SH3, SRC homology 3 domain.
Novel candidates and their neurobiology
Many of the top genes from recent exome studies are novel candidates for ASD and ID, including the strongest overall association: CHD8, an ATP-dependent chromodomain helicase that directly regulates CTNNB1 [45] as well as the p53 pathway [46]. The CHD8 protein has known binding activity with another chromodomain helicase, CHD7 [47], which is the key protein in CHARGE (Coloboma of the eye, Heart defects, Atresia, Retardation of growth, Genital and/or urinary abnormalities, and Ear abnormalities and deafness) syndrome, a rare syndrome with high ASD comorbidity [48]. In addition to directly interacting, both are homologs of the Drosophila trithorax group protein kismet and are components of large chromatin remodeling complexes thought to be important in neural crest cell differentiation [49]. Overall, eight de novo truncating mutations were observed across 2597 cases in this gene; by contrast, no such mutations were observed in control siblings, or in over 6500 exomes in the ESP. The frequency of mutations in this gene is the highest of all genes screened thus far and nearly matches that of CNVs at 16p11.2, which is the most frequent recurrently deleted (0.5%) or duplicated (0.3%) locus in sporadic ASD [50,51].
The second strongest overall association, with two truncating mutations in ASD cases and three such mutations in ID cases, is SCN2A, a gene previously associated with epilepsy and seizure disorders [52,53]. SCN2A encodes a voltage-gated sodium channel (type II, alpha 1; Nav1.2) expressed throughout the brain, and is responsible for the generation and propagation of action potentials in neurons. The phenotype associated with this gene appears to be highly variable. Given the smaller number of ID cases, the prevalence of mutations in SCN2A is higher in ID – however, one of the ID cases also shows signs of autism. Lastly, only one of the five cases had a history of seizures [28], suggesting that mutations in SCN2A have highly variable phenotypic outcomes.
Another striking candidate is DYRK1A (dual-specificity tyrosine phosphorylation-regulated kinase 1A), for which three truncating mutations have been discovered in autism probands [24,44] and de novo structural variants in ID probands [54,55]. DYRK1A is a highly conserved gene whose dosage imbalance has been implicated in the cognitive deficits associated with Down syndrome. The gene interacts with the SWItch/Sucrose NonFermentable (SWI/SNF) complex [56] and is considered a master regulator of brain growth, affecting diverse aspects of neurogenesis, including neuronal proliferation, morphogenesis, differentiation, and maturation [57–59]. Mutations in the Drosophila ortholog (mnb) have been known for more than 20 years and result in a ‘minibrain phenotype’ where optic lobes and central brain hemispheres are reduced [60]. Similarly, heterozygous mice knockouts for Dyrk1A (+/−) show a reduction of brain volume in a region-specific manner as well as mental impairment [61,62]. Consistent with these models, all three human loss-of-function autism patients are cognitively impaired and microcephalic (Z-score < −2).
Three truncating mutations each of GRIN2B and SYNGAP1, and two mutations of TBR1, highlight the importance of excitatory/glutamatergic signaling in both ASD and ID – and are perhaps some of the most conclusive previously implicated genes to date. GRIN2B (found in an ASD case with ID) forms a subunit of an NMDA receptor associated with learning and memory, and targeted sequencing has linked it to neurodevelopmental disorders as well as its discovery in ASD [63]. This receptor participates in a larger postsynaptic complex with SYNGAP1, in which three mutations in ID patients have been observed in the present cohorts, as well as in multiple previous screens of ID [22,64]. Interestingly, although no mutations of SYNGAP1 were found in the Simons Simplex Collection (SSC) ASD cohorts, SYNGAP1 mutations were recently implicated in several cases of ID with ASD [65]. Finally, TBR1, together with the calcium/calmodulin-dependent serine protein kinase (MAGUK family, CASK) protein, regulates transcription of GRIN2B (as well as several other candidate ASD genes, such as RELN and AUTS2) [66].
Protein interaction networks converge on common pathways
Knowledge of molecular-level interaction between proteins has enabled the development of transcriptional networks [67] and protein–protein interaction (PPI) networks enriched for mutation in ASD and ID cases. These networks provide a powerful method to unify the landscape of mutations observed in genetically heterogeneous human disorders by leveraging regulatory interactions between genes and/or physical interactions between proteins. For example, Iossifov et al. found that 14/59 genes disrupted by de novo mutations were significantly enriched (P < 0.006) in a group of 842 genes previously defined [68] as regulated by fragile X mental retardation 1 (FMR1), the key protein disrupted in Fragile-X syndrome, and noted that this was not true for mutations found in siblings (2/28 part of FMR1-regulated genes), the general population, nor for all missense variants [26]. Neale et al. performed a similar analysis to previously identified ASD and ID risk genes – including a core set of 31 synaptic genes identified from previous proteomic studies – and found that genes with nonsynonymous de novo mutations had a significantly reduced network distance (i.e., they were more closely associated in the network) than was a set of ‘comparator’ genes derived from silent de novo mutations and sibling mutations [25]. Lastly, we developed a network for interactions between proteins corresponding to genes with de novo mutations, revealing a single connected component for 39% (49/126) of genes with disruptive or likely disruptive missense de novo mutations [24]. Notably, in follow-up MIP resequencing, we targeted ~50% network and ~50% non-network genes and found that 94% (16/17) of the newly discovered truncating mutations fell within the network (or a similar, expanded 74-gene network) – an observation unlikely to have occurred by chance (P = 0.0002). By contrast, the non-network genes had only six total mutations, only one of which was a truncating mutation.
We integrated the results from the six exome studies by forming PPI networks using experimentally verified interaction data from StringDB [69] (see the supplementary material online). We found that the PPI network based on all truncating and missense mutations in probands was significantly more clustered, had more edges, and created larger connected components than randomly sampled or permuted networks (P ≤ 0.009 for all tests; see the supplementary material online); by contrast, neither the genes with mutations in siblings, nor those with synonymous mutations (in either proband or siblings) showed any difference from the null distribution of networks (Table S1).
In order to summarize these PPI networks, we connected all truncating mutations as well as six genes with missense mutations with important roles in brain development (Figure 3; see the supplementary material online). The two largest connected components of this combined network encompass three broad functional pathways: the first connected component (13 proteins) forms a highly interconnected set of postsynaptic scaffolding proteins and receptors, including SYNGAP1, discs large homolog 4 (DLG4, Drosophila), GRIN2A/B, NLGN1, and NRXN1, whereas the second (nine proteins) contains both WNT signaling functions of CTNNB1, delta-like 1 (DLL1, Drosophila), and TBL1XR1 and chromatin remodeling functions, anchored by the CHD8 protein. We emphasize that while the nodes in the displayed network are partially based on a manually selected set of genes, the connected components formed are a strict subset of the unbiased PPI simulations described above and are larger than any connected component that can be formed using disruptive mutations found in siblings, synonymous changes, or randomly chosen genes.
Figure 3.

Predicted proteins disrupted by genic de novo mutations in autism spectrum disorder (ASD) and intellectual disability (ID) form a central connected network. Proteins corresponding to genes with de novo truncating mutations (red nodes) or selected missense mutations (blue nodes) in four ASD exome studies and two ID exome studies are connected using experimentally derived protein–protein interaction (PPI) data from StringDB [69]. Only medium- and high-confidence experimental interactions are shown, although we note that these may not always represent local interactions, protein–protein interactions, or interactions within the same subcellular compartment. Peripheral nodes (lighter shades) represent genes with additional truncating de novo mutations, which are separated from the central network by only a single node (white nodes; for this analysis we excluded SUMO1/SUMO2 and UBC, which are highly connected but nonspecific nodes).
In addition to the central networks in Figure 3, we also included 56 genes with truncating mutations in ASD or ID that are ‘one-step-removed’ (i.e., connected by one intermediate gene). Although these 56 nodes are in fact not significantly more connected to the network than a random set of genes (due to the high interconnectedness of the global PPI network), many of these nodes are promising ASD or ID candidates, and viewing them in the context of the central network may highlight new genes and pathways to study (see below for examples).
Interestingly, Gilman et al., using a novel probabilistic framework (NETBAG) in conjunction with CNV data from SSC families, highlighted several genes and pathways with remarkable premonition and overlap to those found in the present exome-based studies [70]. In particular, their model showed enrichment of the canonical WNT pathway, postsynaptic complexes, and dendritic spine development (e.g., DLG4, SYNGAP1) and several proteins involved in chromatin remodeling, including bromodomain adjacent to zinc finger domain 1B (BAZ1B) and SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 2 (SMARCA2), both of which interact with the central nodes of the chromatin remodeling network (Figure 3).
The WNT pathway and chromatin remodeling modules of the network are linked by interaction between CHD8 and CTNNB1 (beta-catenin). Both of these proteins play important roles in neural development and growth. Beta-catenin, via downstream WNT pathways, influences neuronal migration, polarity, and synaptogenesis [71], and constitutive overexpression of beta-catenin in mice results in macrocephaly [72]. CHD8 negatively regulates beta-catenin via direct binding and, furthermore, downregulates beta-catenin-responsive genes by recruitment to their promoter regions [73]. Strikingly, ASD cases with truncating mutations in CHD8 have significant macrocephaly [44], whereas all three cases with truncating mutations in beta-catenin have microcephaly ([28]; B.J. O’Roak, unpublished). These reciprocal phenotypes suggest that CHD8 and beta-catenin form a regulatory network that controls head size by altering neuronal migration and growth during development (Figure 4). Other proteins with de novo mutations in this network include TBL1XR1, which binds beta-catenin [74], and DLL1, which is expressed in neural progenitor cells and part of the Delta/Notch signaling pathway [75].
Figure 4.
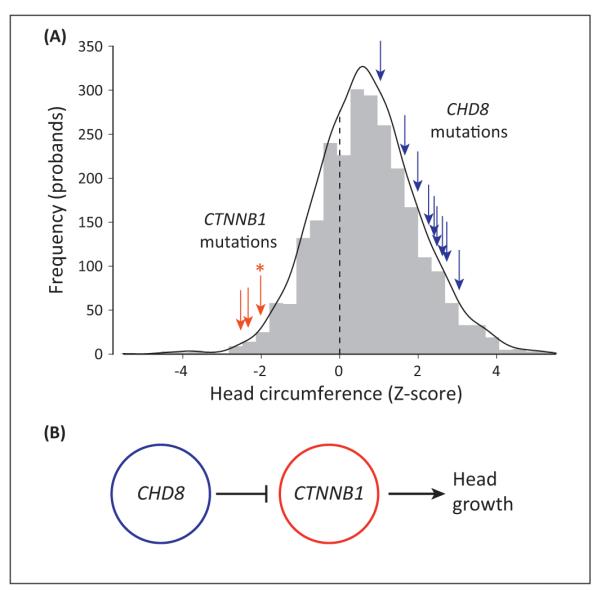
Chromodomain helicase DNA binding protein 8 (CHD8) and beta-catenin [catenin (cadherin-associated protein), beta 1, 88 kDa; CTNNB1] form putative regulatory networks for head size. (A) Truncating mutations in CTNNB1 (red arrows) and CHD8 (blue arrows) are found in patients with small and large head circumference, respectively. The gray histogram represents the background distribution of age- and sex-corrected head circumference Z-scores for 2446 probands from the Simons Simplex Collection (SSC) [36]. [The exact head circumference for one case (marked with *) with clinically reported microcephaly could not be determined, so the Z-score was estimated at −2.0, or the clinical threshold for microcephaly.] (B) A putative regulatory model of head growth where CHD8 negatively regulates CTNNB1 [73]; CTNNB1 promotes head growth and constitutive overexpression of CTNNB1 in mice results in macrocephaly [72].
Convergence on a second common pathway – chromatin remodeling – has primarily been driven by overlap between genetic syndromes and de novo mutations in sporadic ASD and ID. As discussed, CHD8 possesses ATP-dependent chromatin remodeling activity and directly interacts with CHD7 [47], which is responsible for CHARGE syndrome, a complex syndrome in which up to two-thirds of patients have been found to have ASD [48]. Several de novo missense mutations in ASD cases have been noted in genes encoding for chromodomain helicase proteins, including CHD7 and CHD3, and a de novo frame-shift in CHD2 was found by Rauch et al. in an ID case [29]. A second syndrome, Coffin-Siris syndrome (OMIM 135900), characterized by ID and severe speech delays, was recently attributed to truncating mutations or disruptive CNVs in ARID1B [encoding a subunit of the SWI/SNF chromatin remodeling complex [76], AT rich interactive domain 1B (SWI1-like)], and one de novo frameshift of ARID1B was found in a sporadic ASD case [24]. Additional disruptive de novo mutations recognized in ASH1L [ash1 (absent, small, or homeotic)-like (Drosophila)], KDM6B [lysine (K)-specific demethylase 6B], and MLL5 [myeloid/lymphoid or mixed-lineage leukemia 5 (trithorax homolog, Drosophila)] suggest that the chromatin remodeling activity of these proteins may be an underlying pathway implicated in ASD and ID [26]. Finally, we note that mutations in KANSL1 (KAT8 regulatory NSL complex subunit 1, né KIAA1267), a histone acetyltransferase with similar p53 regulatory activity to CHD8, were recently found to underlie 17q21.31 microdeletion syndrome, in which ID is a characteristic feature [77]. However, no mutations in KANSL1 have been found in ASD cases, although this is likely to be due to exclusion of known clinical syndromes from these cohorts.
In addition to these newly proposed pathways, de novo mutations also highlight the importance of genes with roles in synaptic function and localization – a pathway previously suspected to be disrupted in ASD [78]. Many of these genes with de novo mutations form a closely related network of postsynaptic proteins, including the GTPase activating protein SYNGAP1, NMDA receptor subunits GRIN2B and GRIN2A, the scaffolding proteins DLG4 and CASK (the underlying mutation in CASK syndrome, OMIM 300749), and NRXN1, which has been previously associated with ASD [42]. In conjunction with TBR1, CASK also transcriptionally activates several known neurodevelopmental genes, such as RELN (reelin), a gene with critical roles in neuronal development, synaptogenesis, and plasticity [79]. Finally, this pathway is closely linked to SHANK3 (SH3 and multiple ankyrin repeat domains 3), a previously identified ASD protein with up to 1% mutation frequency in ASD cases [80,81], although no mutations in this gene have been identified in the six studies presented here. Although the reasons for this are not fully clear, it is likely that the high GC content of the gene impedes current short-read sequencing platforms (Box 1).
Interaction networks (Figure 3) can also suggest novel targets for mutation screens or functional studies. For example, although discs large (Drosophila) homolog-associated protein 1 (DLGAP1) plays a central role in connecting the ‘synaptic function’ component to beta-catenin, no mutations have been observed in DLGAP1. Similarly, SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily a, member 4 (SMARCA4) connects bromodomain and WD repeat domain containing 1 (BRWD1) to the in-network activity-dependent neuroprotector homeobox (ADNP) protein. These proteins, as well as other ‘nearby’ proteins suggested by PPI networks, can provide novel targets for mutation screens and deeper functional/pathway study. It is likely that sequencing studies of patients will identify novel candidates for PPI networks, creating a reiterative process by which networks and genetics mutually inform.
Despite their widespread role in the current study of ASD and ID, PPI networks have several important limitations. First, protein interactions are difficult to assay experimentally and often are not at a proteomic scale, resulting in false negatives and false positives in databases. In addition, the extent to which the temporal and spatial nature of interactions is captured also limited, and in our network we do not distinguish between different interaction types (regulatory or physical) or cellular compartments. For example, whereas CASK binds NRXN1 presynaptically [82], binding to the transcription factor TBR1 is in the nucleus [83]. Second, although our PPI network only uses experimentally verified interactions, the impact and weight of interactions can vary considerably for different nodes, especially for ‘hub’ nodes, which can interact with hundreds of other proteins. Finally, current PPI networks do not take into account the functional impact of mutations on the proteins or the interactions themselves.
Comparing and contrasting mutations in ASD and ID
In examining data presented in this review, several observations regarding the genetic or etiologic differences between ASD and ID diagnoses emerge, although the significant imbalance in the number of available exomes for ASD (n = 593) and ID (n = 151) advises caution in these comparisons. First, whereas the statistical significance of the clustering of the PPI network does not depend on the inclusion of the two ID studies (Table S1), some genes in the network have been observed only in ID studies (e.g., SYNGAP1 and DLG4; marked as half-filled nodes in Figure 3). Although these genes are closely linked in the PPI network to other well-characterized ASD genes, there may be subtle differences and divisions in this network based on phenotype. By contrast, mutations in some of the top genes result in heterogeneous phenotypes: of eight truncating de novo CHD8 mutations in ASD probands, five had ID and three had IQs above 90 [44]. Whether such heterogeneity is due to genetic background, epistatic effects, or even differences in environmental exposure during development is not yet clear.
Concluding remarks and future directions
New sequencing technology and the establishment of large well-phenotyped family-based cohorts, such as the SSC, have enabled the systematic discovery of mutations that underlie the genetic etiology of ASD and ID. As a measurable indicator of progress, we note that in 2005, approximately 10% of the genetic etiology of autism was understood. Within 7 years advances in genomics technology facilitated the rapid discovery of de novo SNVs and CNVs leading to the discovery of disruptive genetic variants that may account for another ~25% of cases. Although the extent of locus heterogeneity in ASD and ID was initially underestimated, the development of low-cost/high-throughput MIP-based resequencing has strongly implicated a half-dozen novel genes accounting for 1% of disease based on a limited survey of 44 genes [44]. If the yield of de novo loss-of-function mutation continues, there will be several hundred additional candidates available for testing by 2014 because it is anticipated that more than 4000 autism exomes will have been generated. Many of these genes may in fact define distinct clinical ‘subtypes’ of ASD upon detailed examination of patients with a common genetic etiology, consistent with the hypothesis that autism is an umbrella term underlying many different and distinct ‘autisms’. This is reminiscent of the work with CNVs, where the identification of recurrent mutations and patient follow-up led to the identification of novel syndromes and subtypes from idiopathic cases of disease [14]. There is already compelling evidence for this based on an assessment of multiple patients with DYRK1A and CHD8 mutations, which appear to define microcephalic and macrocephalic subtypes, respectively. Alternatively, the ‘genotype first’ approach may also reveal phenotypic variability of genic mutations across a diverse array of neuropsychiatric and neurodevelopmental disorders. Similar to CNVs of 16p11.2 and 15q13.3, which are associated with several disorders, there is evidence for this already for mutations associated with SETBP1 (SET binding protein 1) [84] and SCN2A, resulting in very different outcomes. Establishment of cohorts with different types of mutations and careful study of their phenotypes and comorbidities may reveal specific protein domains and mutation types associated with different diseases.
The knowledge of specific genes, loci, and pathways now spurs the development of functional experiments (Box 2). These include using novel methods with induced pluripotent stem cells to assay specific mutations in a patient with Timothy syndrome [85,86], as well as established model systems, such as mouse and zebrafish models to explore the roles of DYRK1A [87] and PTEN [88] in brain volume. Even more encouraging is the emergence of therapies designed to correct specific pathways disrupted in Fragile-X; these have shown promise in mouse models [89] and are currently undergoing Phase II clinical trials. Improving knowledge of ASD genetic and neurobiological etiologies will aid in diagnosis of ASD/ID subtypes, allowing for specific recruitment for clinical trials and the development of targeted therapeutics for each subtype. This model is akin to the heterogeneity seen in other broad categories of human disorders and disease and has proven to be successful in many cases (e.g., specific therapeutics for a particular mutation in cystic fibrosis or specific form of cancer). Integrating the genetics, neurology, and pathophysiology of these disorders holds considerable promise not only for our understanding of the biology of the human brain but also for potential treatments.
Box 2. Outstanding questions.
Variant discovery and interpretation
Which mutations are necessary and sufficient for, as opposed to simply increasing the risk of, developing ID or ASD? What constitutes proof of a genetic cause of autism/ID?
What is the role of missense mutations in the pathogenesis of ASD and ID? What features distinguish pathogenic missense variants from those that are more benign?
What is the value of whole-exome versus whole-genome sequencing data? How can variants in nonexonic regions be interpreted, and what is their pathogenic effect?
How will the 8–10% of protein-coding sequence being missed by next-generation technologies be surveyed?
How can knowledge of PPI networks guide functional studies and additional genotyping for ASD and ID genes?
ASD/ID etiology
To what extent does the impact of de novo variants depend on the underlying genetic background of the individuals?
In what contexts, and to what extent, do differing proposed genetic etiology models of ASD and ID interact? What is the relative contribution of rare variants, syndromic causes, and common variants to the overall gestalt of ASD? Is there a fraction of the heritable risk that will never be explained?
What role does epigenetics and environment play? Will the identification of hundreds of ASD genes help to identify new environmental or gene-by-environment components?
Clinical questions
How and when should research discoveries be applied clinically?
What is the value of returning results of specific mutations to families?
Will the definition of specific subtypes lead to clinically distinguishable forms of autism? How will these data inform future molecular therapies?
How will clinical cohorts of tens to hundreds of thousands of patients be amassed and research studies coordinated to resolve the heterogeneity of these disorders?
Supplementary Material
Glossary
- CNV (copy number variant)
loss or insertion of DNA, typically larger than 50 bp and often up to several megabases.
- Connected component
a set of connected nodes that are part of a PPI network and can represent a pathway, complex protein structure, or cellular function.
- GC bias
the tendency for sequencing reactions to produce fewer reads in regions of the genome with a high fraction of GC base pairs.
- Hidden species problem
a method for estimating an unknown number of classes (species) from a distribution of observed counts.
- Indel (insertion/deletion)
loss or insertion of DNA, between 1 and 50 bp in length.
- Loss-of-function or truncating mutation
a nonsense, frameshift, or splice-site mutation that prevents complete translation of a functional protein.
- Missense mutation
a mutation that alters the amino acid composition of a protein but does not prohibit its complete translation.
- PPI network
a protein–protein interaction network that defines ‘nodes’ as proteins and ‘edges’ as interactions (which may be physical, expression-based, or computationally predicted).
- Sequence coverage
the average or median number of sequence reads per genomic base pair in a sequencing experiment. Higher coverage enables more accurate discovery of variants.
- SNP/SNV (single nucleotide polymorphism/variant)
single-base changes in DNA. Typically, SNPs are higher frequency and refer to alleles observed to be segregating in a population.
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.tins.2013. 11.005.
References
- 1.Fu Y-H, et al. Variation of the CGG repeat at the fragile X site results in genetic instability: resolution of the Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 2.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 3.Matsuura T, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 4.Steffenburg S, et al. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J. Child Psychol. Psychiatry. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- 5.Bailey A, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol. Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 6.Hallmayer J, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry. 2011;68:1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Constantino JN, et al. Autism recurrence in half siblings: strong support for genetic mechanisms of transmission in ASD. Mol. Psychiatry. 2013;18:137–138. doi: 10.1038/mp.2012.9. [DOI] [PubMed] [Google Scholar]
- 8.Marshall CR, et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Vries BBA, et al. Diagnostic genome profiling in mental retardation. Am. J. Hum. Genet. 2005;77:606–616. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levy D, et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 12.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cooper GM, et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharp AJ, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 15.Sharp AJ, et al. A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat. Genet. 2008;40:322–328. doi: 10.1038/ng.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller DT, et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J. Med. Genet. 2009;46:242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helbig I, et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat. Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ben-Shachar S, et al. Microdeletion 15q13.3: a locus with incomplete penetrance for autism, mental retardation, and psychiatric disorders. J. Med. Genet. 2009;46:382–388. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vissers LELM, et al. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 23.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Roak BJ, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neale BM, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanders SJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 29.Rauch A, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 30.Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Neurosci. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 32.Ku CS, et al. A new paradigm emerges from the study of de novo mutations in the context of neurodevelopmental disease. Mol. Psychiatry. 2012;18:141–153. doi: 10.1038/mp.2012.58. [DOI] [PubMed] [Google Scholar]
- 33.Bamshad MJ, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 34.Berg JM, Geschwind DH. Autism genetics: searching for specificity and convergence. Genome Biol. 2012;13:247. doi: 10.1186/gb-2012-13-7-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devlin B, et al. Do common variants play a role in risk for autism? Evidence and theoretical musings. Brain Res. 2011;1380:78–84. doi: 10.1016/j.brainres.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newschaffer CJ, et al. The epidemiology of autism spectrum disorders. Annu. Rev. Public Health. 2007;28:235–258. doi: 10.1146/annurev.publhealth.28.021406.144007. [DOI] [PubMed] [Google Scholar]
- 37.Hertz-Picciotto I, et al. The CHARGE study: an epidemiologic investigation of genetic and environmental factors contributing to autism. Environ. Health Perspect. 2006;114:1119–1125. doi: 10.1289/ehp.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poultney CS, et al. Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am. J. Hum. Genet. 2013;93:607–619. doi: 10.1016/j.ajhg.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krumm N, et al. Transmission disequilibrium of small CNVs in simplex autism. Am. J. Hum. Genet. 2013;93:595–606. doi: 10.1016/j.ajhg.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klei L, et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol. Autism. 2012;3:9. doi: 10.1186/2040-2392-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gratten J, et al. Interpreting the role of de novo protein-coding mutations in neuropsychiatric disease. Nat. Genet. 2013;45:234–238. doi: 10.1038/ng.2555. [DOI] [PubMed] [Google Scholar]
- 42.Kim H-G, et al. Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 2008;82:199–207. doi: 10.1016/j.ajhg.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Talkowski ME, et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Roak BJ, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishiyama M, et al. Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-β-catenin signaling pathway. Mol. Cell. Biol. 2012;32:501–512. doi: 10.1128/MCB.06409-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nishiyama M, et al. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol. 2009;11:172–182. doi: 10.1038/ncb1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Batsukh T, et al. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum. Mol. Genet. 2010;19:2858–2866. doi: 10.1093/hmg/ddq189. [DOI] [PubMed] [Google Scholar]
- 48.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 49.Bajpai R, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463:958–962. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar RA, et al. Association and mutation analyses of 16p11.2 autism candidate genes. PLoS ONE. 2009;4 doi: 10.1371/journal.pone.0004582. http://dx.doi.org/10.1371/journal.pone.0004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Walsh KM, Bracken MB. Copy number variation in the dosage-sensitive 16p11.2 interval accounts for only a small proportion of autism incidence: a systematic review and meta-analysis. Genet. Med. 2011;13:377–384. doi: 10.1097/GIM.0b013e3182076c0c. [DOI] [PubMed] [Google Scholar]
- 52.Kamiya K, et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J. Neurosci. 2004;24:2690–2698. doi: 10.1523/JNEUROSCI.3089-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogiwara I, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009;73:1046–1053. doi: 10.1212/WNL.0b013e3181b9cebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Bon BWM, et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet. 2011;79:296–299. doi: 10.1111/j.1399-0004.2010.01544.x. [DOI] [PubMed] [Google Scholar]
- 55.Moller RS, et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 2008;82:1165–1170. doi: 10.1016/j.ajhg.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lepagnol-Bestel A-M, et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum. Mol. Genet. 2009;18:1405–1414. doi: 10.1093/hmg/ddp047. [DOI] [PubMed] [Google Scholar]
- 57.Mazur-Kolecka B, et al. Effect of DYRK1A activity inhibition on development of neuronal progenitors isolated from Ts65Dn mice. J. Neurosci. Res. 2012;90:999–1010. doi: 10.1002/jnr.23007. [DOI] [PubMed] [Google Scholar]
- 58.Guedj F, et al. DYRK1A: a master regulatory protein controlling brain growth. Neurobiol. Dis. 2012;46:190–203. doi: 10.1016/j.nbd.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 59.Yabut O, et al. Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J. Neurosci. 2010;30:4004–4014. doi: 10.1523/JNEUROSCI.4711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tejedor F, et al. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14:287–301. doi: 10.1016/0896-6273(95)90286-4. [DOI] [PubMed] [Google Scholar]
- 61.Fotaki V, et al. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002;22:6636–6647. doi: 10.1128/MCB.22.18.6636-6647.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song WJ, et al. Isolation of human and murine homologues of the Drosophila minibrain gene: human homologue maps to 21q22.2 in the Down syndrome “critical region”. Genomics. 1996;38:331–339. doi: 10.1006/geno.1996.0636. [DOI] [PubMed] [Google Scholar]
- 63.Endele S, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- 64.Hamdan FF, et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 2009;360:599–605. doi: 10.1056/NEJMoa0805392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berryer MH, et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum. Mutat. 2012;34:385–394. doi: 10.1002/humu.22248. [DOI] [PubMed] [Google Scholar]
- 66.Bedogni F, et al. Tbr1 regulates regional and laminar identity of postmitotic neurons in developing neocortex. Proc. Natl. Acad. Sci. U.S.A. 2010;107:13129–13134. doi: 10.1073/pnas.1002285107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Voineagu I, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jensen LJ, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gilman SR, et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salinas PC, Zou Y. Wnt signaling in neural circuit assembly. Annu. Rev. Neurosci. 2008;31:339–358. doi: 10.1146/annurev.neuro.31.060407.125649. [DOI] [PubMed] [Google Scholar]
- 72.Chenn A, Walsh CA. Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb. Cortex. 2003;13:599–606. doi: 10.1093/cercor/13.6.599. [DOI] [PubMed] [Google Scholar]
- 73.Thompson BA, et al. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol. Cell. Biol. 2008;28:3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cadigan KM. Wnt/β-catenin signaling: turning the switch. Dev. Cell. 2008;14:322–323. doi: 10.1016/j.devcel.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 75.Barton A, Fendrik AJ. Sustained vs. oscillating expressions of Ngn2, Dll1 and Hes1: a model of neural differentiation of embryonic telencephalon. J. Theor. Biol. 2013;328:1–8. doi: 10.1016/j.jtbi.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 76.Santen GWE, et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 2012;44:379–380. doi: 10.1038/ng.2217. [DOI] [PubMed] [Google Scholar]
- 77.Koolen DA, et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat. Genet. 2012;44:639–641. doi: 10.1038/ng.2262. [DOI] [PubMed] [Google Scholar]
- 78.Glessner JT, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang T-F, et al. Identification of Tbr-1/CASK complex target genes in neurons. J. Neurochem. 2004;91:1483–1492. doi: 10.1111/j.1471-4159.2004.02845.x. [DOI] [PubMed] [Google Scholar]
- 80.Durand CM, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moessner R, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fairless R, et al. Polarized targeting of neurexins to synapses is regulated by their C-terminal sequences. J. Neurosci. 2008;28:12969–12981. doi: 10.1523/JNEUROSCI.5294-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hsueh Y-P, et al. Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature. 2000;404:298–302. doi: 10.1038/35005118. [DOI] [PubMed] [Google Scholar]
- 84.Hoischen A, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat. Genet. 2010;42:483–485. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- 85.Yazawa M, et al. Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature. 2011;471:230–234. doi: 10.1038/nature09855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paşca SP, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat. Med. 2011;17:1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ahn K-J, et al. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol. Dis. 2006;22:463–472. doi: 10.1016/j.nbd.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 88.Backman SA, et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat. Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- 89.Michalon A, et al. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron. 2012;74:49–56. doi: 10.1016/j.neuron.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.