

Abstract
The internal transcribed spacers of the nuclear ribosomal RNA gene cluster, termed ITS1 and ITS2, are the most frequently used nuclear markers for phylogenetic analyses across many eukaryotic groups including most plant families. The reasons for the popularity of these markers include: 1.) Ease of amplification due to high copy number of the gene clusters, 2.) Available cost-effective methods and highly conserved primers, 3.) Rapidly evolving markers (i.e. variable between closely related species), and 4.) The assumption (and/or treatment) that these sequences are non-functional, neutrally evolving phylogenetic markers. Here, our analyses of ITS1 and ITS2 for 50 species suggest that both sequences are instead under selective constraints to preserve proper secondary structure, likely to maintain complete self-splicing functions, and thus are not neutrally-evolving phylogenetic markers. Our results indicate the majority of sequence sites are co-evolving with other positions to form proper secondary structure, which has implications for phylogenetic inference. We also found that the lowest energy state and total number of possible alternate secondary structures are highly significantly different between ITS regions and random sequences with an identical overall length and Guanine-Cytosine (GC) content. Lastly, we review recent evidence highlighting some additional problematic issues with using these regions as the sole markers for phylogenetic studies, and thus strongly recommend additional markers and cost-effective approaches for future studies to estimate phylogenetic relationships.
Introduction
Molecular systematic approaches have traditionally relied on comparing a limited number of orthologous sequences to obtain estimates of species relationships across the tree of life. These phylogenetic markers are often selected based on a number of basic characteristics; including 1.) Ubiquitous presence across target taxa, 2.) Sufficient sequence or structural variation between taxa (i.e. synapomorphic characters), 3.) Ease of obtaining sequence data, 4.) Cost-effectiveness, and 5.) Having a fundamental understanding of the function of the locus and the possible selective forces acting on its sequence evolution. The internal transcribed spacers (ITS1 and ITS2) regions of the ribosomal RNA gene cluster are the most commonly used nuclear markers for estimating species relationships across plants based on the above criteria, including the assumption (or treatment) that these markers are non-functional and hence neutrally evolving. For example, the ITS regions are the most commonly used markers for estimating phylogenetic relationships across the mustard family (Brassicaceae) ( Figure 1 ) [1], [2], with sequences available for the majority of the 321 genera distributed across all 49 delimited tribes [3]. These studies have, in addition to delimiting tribes and estimating phylogenetic relationships among some tribes, also been successful at assigning many of the tribes into one of three monophyletic lineages ( Figure 1 ). Lineage I is comprised of twelve tribes including Camelineae that contains the model organism Arabidopsis thaliana. Lineage II has six tribes including the agronomically important Brassiceae that contains cruciferous vegetables (e.g cabbage, cauliflower, broccoli, and brussel sprouts). Lineage III includes six primarily Asian tribes. It is important to note that other phylogenetic markers have also been employed (see discussion)[4]–[8]; nonetheless more than 70 articles published since 2010 have used the ITS region to infer relationships across the Brassicaceae.
Figure 1. Phylogenetic Distribution of Hairpin Numbers for ITS Secondary Structures.

A tribal level phylogeny of the Brassicaceae, strict consensus tree of the 200 most parsimonious trees estimated with ITS sequences [2], was utilized to investigate the evolution of the number of hairpins present in the secondary structures of both ITS1 and ITS2. Bootstrap support values greater than 60% are shown above branches [2]. It is notable that the ITS tree does neither fully reflect the tribal phylogeny nor is at any deep node highly significantly supported, but overall-topology is in congruence with multi-locus phylogenies considering major lineages [7], [8]. Tribes not assigned to one of the three major lineages are actually combined with an "expanded lineage II" [46], which might have to be revised in future. The three major phylogenetic lineages are shown within colored blocks with Lineage I (orange), Lineage II (blue) and Lineage III (green). The number of hairpins for each secondary structure is shown at the phylogenetic tips with 3 (orange boxes), 4 (yellow boxes), 5 (green boxes), 6 (blue boxes), and 7 (purple boxes). Tribes with a lack of available complete ITS1 data are marked as 'NA'. Tribes with secondary structures with different number of total hairpins from different species are also indicated (e.g. Camelineae 6/7 for ITS1; 6 and 7 hairpin structures are observed) within the colored box of the fewest hairpined structure. Examples of secondary structures are shown (top-bottom order): 1. Anchonium billardierei ITS1 (Anchonieae), 2. Aethionema arabicum ITS1 (Aethionemeae), 3. Halimolobos lasiolaba ITS2 (Halimolobeae), and 4. Arabis scabra (Arabideae).
The ribosomal RNA gene cluster consists of seven components: the 5′ external transcribed spacer, the 18S rDNA exon, internal transcribed spacer 1 (ITS1), the 5.8S rDNA exon, internal transcribed spacer 2 (ITS2), the 28S rDNA exon, and the 3′ external transcribed spacer ( Figure 2 ) [9]. The rDNA exons are highly conserved across eukaryotes, but the ITS regions are variable in length due to point mutations and indels, resulting in regions varying in size from 500 to 700 bp across angiosperms [10] and from 1500–3700 bp in some gymnosperms [11]. The ITS regions are not incorporated into mature ribosomes, but undergo a specific cleavage during the maturation of the ribosomal RNAs that is catalysed by the secondary structure of ITS sequences themselves [12]–[15]. Despite this specific activity, these sequences have been treated as nearly neutrally evolving nuclear markers for phylogenetic reconstructions [16], for reviews see [17]–[19]. Here, we test whether nuclear encoded internal transcribed spacers (ITS1 and ITS2) are truly neutrally evolving, or if these regions are under selective constraints to maintain a functional self-splicing secondary structure across the Brassicaceae. In addition, we assess the phylogenetic utility of secondary structure data for inferring phylogenetic relationships.
Figure 2. Structure of the rDNA region in Plants.
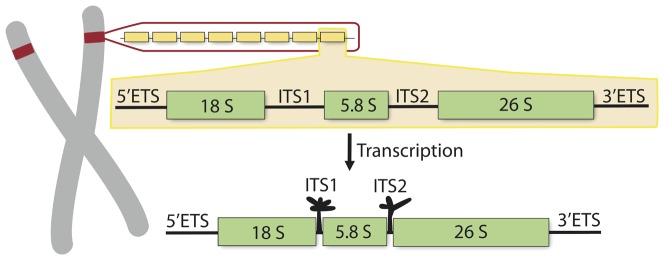
An illustration of the nucleolus organizing region (NOR), shown as red colored region on chromosome, is associated with forming the nucleolus and site for the biosynthesis of the components of the ribosome. The NOR region contains hundreds of tandem duplicated copies of rDNA gene clusters (depicted as yellow rectangles), and each gene cluster consists of seven main components including two internal transcribed spacers (i.e. ITS1 and ITS2). These ITS regions form self-splicing secondary structures as transcribed products. Shown is the 5 hairpin structure for ITS1 and the 3 hairpin structure for ITS2.
Methods
All possible ITS1 and ITS2 secondary structures for a total of 50 species (100 total structures) and 100 random sequences were modeled using RNAstructure Version 5.3 [20]. Sheet 1 in File S1 contains the list of species, NCBI GenBank accession numbers, lengths, total number of possible secondary structures, and a description of the lowest energy state structure for every species. All ITS sequences have been verified by taxonomic experts [2], annotated and deposited into BrassiBase (a comprehensive Brassicaceae database system; http://brassibase.cos.uni-heidelberg.de/) and within a proven phylogenetic context [21], [22]. The 100 random sequences, with an overall GC content and size variation identical to these 100 ITS sequences (Sheet 2 in File S1) were generated using a custom Perl script. A two-way ANOVA, using the R software package (Version 1.7.1) [23], was used to test the statistical significance between both the lowest energy state and total number of possible structures between the true ITS sequences and random sequences (Sheet 3 in File S1). The structures were further analyzed manually for the total number of paired bases and number of total hairpins (Sheet 4 & 5 in File S1). Hairpins were characterized as a complete, continuous loop formed by a set of closely paired nucleotides between two distant regions, with either single or branched structures that may include additional nested structures, while stems were characterized as structures that do not form immediate loops. A structure required a minimum of four nucleotide bonds (i.e. eight nucleotides) to be characterized as a hairpin. Single and branched structures were both treated as one hairpin. Sequence alignments for ITS1 and ITS2 can be found in Files S2 & S3, respectively.
Results
All possible secondary structures for both ITS1 and ITS2 were modeled for 49 species distributed across 38 tribes in the family Brassicaceae and one outgroup species (Cleome lutea renamed Peritoma lutea Hook. [24]; Cleomaceae) ( Figure 3 ). The ITS1 and ITS2 regions are variable in length. ITS1 has a mean length of 263bp (max = 286bp; min = 238bp). ITS2 has a mean length of 184bp (max = 220bp; min = 177bp). The combined ITS1 and ITS2 sequences have a mean length of 224bp and a median length of 229bp. These ITS1 and ITS2 regions have a mean of 16.6 and 7.9 possible secondary structures, respectively. The combined ITS1 and ITS2 dataset has a mean of 10.5 possible secondary structures (median = 8.5; max = 30; min = 1). The mean for the lowest energy structure for all ITS1 and ITS2 sequences is −93.1 and −70.0 degrees, respectively. The combined ITS1 and ITS2 dataset has a mean of −81.6 degrees and median of −79.5 degrees for the lowest energy states (max = −58.2 degrees; min −114.5 degrees). The ITS1 structures have a mean of 153.8 paired bases (i.e. form bonds with other bases) (median = 154) and a mean of 5.8 total hairpins (median = 6). The ITS2 structures have a mean of 115 paired bases (median = 114) and a mean of 3.8 total hairpins (median = 4). The ITS1 sequences on average have ∼58.5% of all nucleotide positions paired, while ITS2 sequences on average have ∼62.5% of all nucleotide positions paired.
Figure 3. Hairpin Size Distributions for ITS1 and ITS2 Secondary Structures.

Panel A shows the frequency of the total percentage of paired bases for all ITS1 (Blue) and ITS2 (Red) sequences. The majority of both ITS regions have over 50% of positions paired with other sequence positions to form secondary structures. Panel B shows the frequency of the total number of hairpins for all the ITS1 (Blue) and ITS2 (Red) structures. These data are for the lowest energy state structure for each ITS sequence (see Supplemental File 1).
To test if the secondary structures of the lowest energy states for ITS sequences suggest selective constraint on the sequences encoding them, we generated 100 random sequences with an identical size range (286bp to 177bp) and overall average GC content (54.9%; see Sheet 2 in File S1). All possible secondary structures were modeled for each of the random sequences. These random sequences have a mean of 19.6 possible secondary structures (median = 17.5; max = 41; min = 5). The mean for the lowest energy structures for all random sequences is −69.9 (median = −70.7; max = −38.6; min = −92.8). We evaluated using a two-way ANOVA whether both the values of the lowest energy states and values for total possible secondary structures were significantly different between the 100 ITS and 100 random sequences ( Figure 4 ). The lowest energy state and total number of possible secondary structures were both highly significant, (P<2.2e-16) and (P<1.6e-07), respectively. The ratio of both these categories was not significantly different between ITS and random sequences (P = 0.1095), as would be predicted. Similarly, the lowest energy state and total number of possible secondary structures for the 50 ITS1 sequences alone compared to random sequences were also significantly different, (P<2e-16) and (P<1.2e-04), respectively. The 50 ITS2 sequences compared to random sequences were also significantly different for both the the lowest energy state (P<2.1e-07) and total number of possible secondary structures (P<6.5e-08).
Figure 4. Comparison of Secondary Structures of 100 ITS and 100 Random Sequences.

A scatter plot of the lowest energy state values (x-axis) and all possible secondary structures (y-axis) for 50 ITS1 (Blue Diamonds), 50 ITS2 (Red Squares) and 100 randomly generated sequences (Green Triangles) (Supplemental File 1) estimated using RNAstructure 5.3 (Reuter and Mathews, 2010).
Lastly, we evaluated the utility of secondary structure data to resolve phylogenetic relationships at the tribal level across the Brassicaceae ( Figure 1 ). ITS1 sequences are highly variable, ranging between 4 to 8 hairpins, with some conserved phylogenetic patterns within tribes but limited phylogenetic conservation between closely related tribes. Six hairpin structures for ITS1 are the most frequent for Lineage I (min = 5; max = 7) and Lineage II species (min = 4; max = 7), while 5 hairpin structures were most frequent among Lineage III species (Min = 4; Max = 7). ITS2 sequences are more slowly evolving, ranging between 3 to 5 hairpins, with most species having 4 hairpin secondary structures. Lineage I consists largely of 4 hairpin structures (min = 3; max = 5) and Lineage III consists of both 3 and 4 hairpin structures for ITS2. Lineage II species consist mostly of 4 hairpin structure ITS2 sequences, except for one species (Sisymbrium irio) with a 3 hairpin structure within the tribe Sisymbrieae. The ancestral state for the most common recent ancestor of the Brassicaceae is likely a 5 or 6 hairpin structure for ITS1, and a 3 or 4 hairpin structure for ITS2.
Discussion
The results of this study show that the majority of sequence sites for both ITS1 and ITS2 are not independently evolving, but rather are co-evolving with at least one other position to preserve the molecule's secondary structure. Our analyses also revealed that these secondary structures have a significantly lower energy state and significantly fewer possible alternate secondary structures compared to random sequences with a similar guanine-cytosine (GC) content and length distribution. Collectively, these results do not support the neutrality of these sequences across the Brassicaceae (as commonly assumed and/or implemented for phylogenetic analyses), but rather strongly suggest that these sequences are under selective constraint to maintain functional self-splicing secondary structures. Thus, the majority of mutations that occur within these regions must likely undergo compensatory mutations, since most sites are co-evolving, to maintain properly functioning secondary structures.
Many Brassicaceae studies to date have used ITS as a phylogenetic marker to estimate relationships and delimit new taxonomic groups (e.g. tribes). Based on our results, this fact means that many phylogenetically informative sites were unintentionally treated as independent despite the fact that they were co-evolving. At a minimum, such assumptions will tend to overstate the confidence in phylogenetic hypotheses as inferred with methods such as the bootstrap. Thus, we advise future studies to identify all co-evolving sites and make appropriate adjustments prior to employing these regions as phylogenetic markers. However, we also want to emphasize the importance of proper consistent annotation prior to modeling secondary structures, particularly if structures are to be compared across species [25], [26]. For this reason, we generated random sequences with an identical length variation and overall GC content to actual ITS sequences to permit accurate comparisons of secondary structure characteristics. For the Brassicaceae community, the BrassiBase database has nearly 2,000 annotated ITS sequences and alignments are available for species distributed across all family tribes. For the broader eukaryotic community, the ITS2 Database [27] is an outstanding publicly available resource for the annotation, secondary structure prediction, and estimating phylogenetic relationships of ITS2 sequences.
For estimates of tribal relationships across the Brassicaceae (or likely for similar family level phylogenies), ITS1 and ITS2 contains insufficient signal to obtain a robust, well-resolved phylogeny ( Figure 1 ). Thus, ITS must be combined with other markers to estimate deep level phylogenetic relationships. Additionally, the ITS marker is known to often undergo gene conversion following hybridization and allopolyploidization events, in which the sequence from one subgenome replaces those of the other subgenome [28]–[30]. For example, this homogenization process of rDNA repeats (i.e. concerted evolution) has been shown to occur very rapidly, within less than 100 years, in two allopolyploid Cardamine (Brassicaceae) species [31], and multiple populations of allopolyploid Tragopogon mirus and Tragopogon miscellus [32]. Also, the process of non-concerted evolution and the origin of paralogous copies have been described in the Brassicaceae [30]. Thus, these features and evolutionary histories of ITS sequences are not ideal for estimating species relationships alone, especially for groups like the Brassicaceae that have multiple documented ancient and recent polyploidization events [33]–[35]. Although reliance on ITS as the sole source of phylogenetic evidence can be criticized for reasons given here, it remains a highly efficient locus for generating species-level phylogenetic inferences in most plant groups. At least across the Brassicaceae, the phylogenetic estimates obtained from the ITS markers for within various subfamilial units have largely been congruent with other markers and data [36]–[38]. On the other hand, at the entire family level (i.e. tribes and major lineages), the phylogenetic signal from this marker has been insufficient to resolve major relationships.
Single-marker approaches are known to produce misleading phylogenetic estimates for species relationships [39], but incongruence between gene phylogenies and species phylogenies can be identified and resolved using multiple independently evolving markers [40]–[43]. More recent family-wide Brassicaceae studies are utilizing multi-locus datasets, which have yielded improved phylogenetic estimates for many clades but still limited resolution for the relationship among the three major lineages (shown in Figure 1 ) and majority of tribes [44]–[46]. Therefore future studies should survey additional markers with sufficient phylogenetic signal, preferably with different patterns of inheritance (e.g. mitochondrial, plastid, and nuclear), to estimate species relationships, identify incongruent markers with unique evolutionary histories, and ultimately obtain better insights into more complex evolutionary processes. A large-scale data approach has already been demonstrated to have sufficient signal to resolve some difficult phylogenetic relationships across the Brassicaceae [47], [48], and will serve as a valuable resource to address a range of fundamental questions in evolution remaining for the family including understanding the mechanisms responsible for shifts in speciation rates [49], evolution of chemical defenses against herbivores [50], and improve our understanding of novel morphological varation [51].
Supporting Information
Secondary Structure data for ITS1, ITS2, and random sequences. Spreadsheet contains the list of species, NCBI GenBank accession numbers, lengths, total number of possible secondary structures, and a description of the lowest energy state structure for every ITS1, ITS2, and random sequence.
(XLSX)
Sequence alignments for ITS1. Sequence alignments for ITS1 for 49 Brassicaceae and one outgroup Cleomaceae found in File S1.
(TXT)
Sequence alignments for ITS2. Sequence alignments for ITS2 for 49 Brassicaceae and one outgroup Cleomaceae found in File S1.
(TXT)
Funding Statement
PPE and JCP acknowledge support from the USA National Science Foundation (DEB 1146603, DBI 1110443, and NPGI 1202793). M.A.K. acknowledged funding from the German Research Foundation (DFG) under the code KO2302-11/1. 1. http://www.nsf.gov/; 2. http://www.dfg.de/en/. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. German DA, Friesen N, Neuffer B, Al-Shehbaz IA, Hurka H (2009) Contribution to ITS phylogeny of the Brassicaceae, with special reference to some Asian taxa. Plant Syst. and Evol. 283: 33–56. [Google Scholar]
- 2. Warwick SI, Mummenhoff K, Sauder CA, Koch MA, Al-Shehbaz IA (2010) Closing the gaps: Phylogenetic relationships in the Brassicaceae based on DNA sequence data of nuclear ribosomal ITS region. Plant Syst. and Evol. 285: 1–24. [Google Scholar]
- 3. Al-Shehbaz IA (2012) A generic and tribal synopsis of the Brassicaceae (Cruciferae). Taxon 61: 931–954. [Google Scholar]
- 4. Koch M, Haubold B, Mitchell-Olds T (2001) Molecular systematics of the Brassicaceae: evidence from coding plastidic matK and nuclear Chs sequences. Am. J. Bot. 88: 534–544. [PubMed] [Google Scholar]
- 5. Bailey CD, Koch MA, Mayer M, Mummenhoff K, O'Kane SL Jr, et al. (2006) Toward a global phylogeny of the Brassicaceae. Mol. Biol. Evol. 23: 2142–2160. [DOI] [PubMed] [Google Scholar]
- 6. Beilstein MA, Al-Shehbaz IA, Kellogg EA (2006) Brassicaceae phylogeny and trichome evolution. Am. J. Bot. 93: 607–619. [DOI] [PubMed] [Google Scholar]
- 7. Beilstein MA, Nagalingum NS, Clements MD, Manchester SR, Mathews S (2010) Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana . Proc. Natl. Acad. Sci. U.S.A. 107: 18724–18728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Couvreur TL, Franzke A, Al-Shehbaz IA, Bakker FT, Koch MA, et al. (2010) Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae). Mol. Biol. Evol. 27: 55–71. [DOI] [PubMed] [Google Scholar]
- 9. Wheeler WC, Honeycutt RL (1988) Paired sequence difference in ribosomal RNAs: evolutionary and phylogenetic implications. Mol. Biol. Evol. 5: 90–96. [DOI] [PubMed] [Google Scholar]
- 10. Baldwin BG, Sanderson MJ, Porter JM, Wojciechowski MF, Campbell CS, et al. (1995) The ITS region of nuclear ribosomal DNA: A valuable source of evidence on angiosperms phylogeny. Ann. Mo. Bot. Gard. 82: 247–277. [Google Scholar]
- 11. Alvarez I, Wendel JF (2003) Ribosomal ITS sequences and plant phylogenetic inference. Mol. Phylogenet. Evol. 29: 417–434. [DOI] [PubMed] [Google Scholar]
- 12. Venema J, Tollervey D (1999) Ribosome synthesis in Saccharomyces cerevisiae . Annu. Rev. Genet. 33: 261–311. [DOI] [PubMed] [Google Scholar]
- 13. Dron M, Rahire M, Rochaix JD (1982) Sequence of the chloroplast 16S rRNA gene and its surrounding regions of Chlamydomonas reinhardii . Nucleic Acids Res. 10: 7609–7620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morgan JA, Blair D (1998) Trematode and monogenean rRNA ITS2 secondary structures support a four-domain model. J. Mol. Evol. 47: 406–419. [DOI] [PubMed] [Google Scholar]
- 15. Mai JC, Coleman AW (1997) The internal transcribed spacer 2 exhibits a common secondary structure in green algae and flowering plants. J. Mol. Evol. 44: 258–271. [DOI] [PubMed] [Google Scholar]
- 16. Hillis DM, Dixon MT (1991) Ribosomal DNA: molecular evolution and phylogenetic inference. Q. Rev. Biol. 66: 411–453. [DOI] [PubMed] [Google Scholar]
- 17.Koch M, Al-Shehbaz IA (2009) Molecular Systematics and Evolution of "wild" crucifers (Brassicaceae or Cruciferae). In: Biology and Breeding of Crucifers (ed S Gupta) Pp 1–18 Taylor and Francis Group. ISBN 978-1-4200-8608-9. [Google Scholar]
- 18.Lysak MA, Koch M (2011) Phylogeny, Genome and Karyotype Evolution of Crucifers (Brassicaceae). In: Genetics and Genomics of the Brassicaceae (eds R Schmidt, I Bancroft) New York Springer Verlag Pp 1–18. ISBN 978-1-4419-7117-3. [Google Scholar]
- 19. Poczai P, Hyvönen J (2010) Nuclear ribosomal spacer regions in plant phylogenetics: Problems and prospects. Mol. Biol. Rep. 37: 1897–1912. [DOI] [PubMed] [Google Scholar]
- 20. Reuter JS, Mathews DH (2010) RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koch MA, Kiefer M, German DA, Al-Shehbaz I, Franzke A, et al. (2012) BrassiBase: Tools and Biological Resources to Study Characters and Traits in the Brassicaceae - Version 1.1. Taxon 61: 1001–1009. [Google Scholar]
- 22. Kiefer M, Schmickl R, German DA, Lysak MA, Al-Shehbaz I, et al. (2014) BrassiBase: Introduction to a Novel Knowledge Database on Brassicaceae Evolution. Plant Cell Physiol. 55(1): e3. [DOI] [PubMed] [Google Scholar]
- 23.Team RDC (2008) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria ISBN 3-900051-07-0.
- 24. Feodorova TA, Voznesenskaya EV, Edwards GE, Roalson EH (2010) Biogeographic patterns of diversification and the origins of C4 in Cleome (Cleomaceae). Syst. Bot. 35: 811–826. [Google Scholar]
- 25. Gottschling M, Hilger HH, Wolf M, Diane N (2001) Secondary Structure of the ITS1 Transcript and its Application in a Reconstruction of the Phylogeny of Boraginales. Plant Biol. 3: 629–636. [Google Scholar]
- 26. Goertzen LR, Cannone JJ, Gutell RR, Jansen RK (2003) ITS Secondary Structure derived from comparative analysis: implications for sequence alignment and phylogeny of the Asteraceae. Mol. Phylogenet. Evol. 2: 216–234. [DOI] [PubMed] [Google Scholar]
- 27. Koetschan C, Hackl T, Mueller T, Wolf M, Foerster F, et al. (2012) ITS2 database IV: Interactive Taxon Sampling for Internal Transcribed Spacer 2 Based Phylogenies. Mol. Phylogenet. Evol. 63: 585–588. [DOI] [PubMed] [Google Scholar]
- 28. Wendel JF, Schnabel A, Seelanan T (1995) An unusual ribosomal DNA sequence from Gossypium gossypioides reveals ancient, cryptic, intergenomic introgression. Mol. Phylogenet. Evol. 4: 298–313. [DOI] [PubMed] [Google Scholar]
- 29. Wendel JF, Schnabel A, Seelanan T (1995) Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc. Natl. Acad. Sci. U.S.A 92: 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koch MA, Dobes C, Mitchell-Olds T (2003) Multiple hybrid formation in natural populations: concerted evolution of the internal transcribed spacer of nuclear ribosomal DNA (ITS) in North American Arabis divaricarpa (Brassicaceae). Mol. Biol. Evol. 20: 338–350. [DOI] [PubMed] [Google Scholar]
- 31. Franzke A, Mummenhoff K (1999) Recent hybrid speciation in Cardamine (Brassicaceae). Conversion of nuclear ribosomal ITS sequences in statu nascendi . Theor. Appl. Genet. 98: 831–834. [Google Scholar]
- 32. Kovarik A, Pires JC, Leitch AR, Lim KY, Sherwood AM, et al. (2005) Rapid concerted evolution of nuclear ribosomal DNA in two Tragopogon allopolyploids of recent and recurrent origin. Genetics 169: 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lysak MA, Koch MA, Pecinka A, Schubert I (2005) Chromosome triplication found across the tribe Brassiceae. Genome Res. 15: 516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blanc G, Hokamp K, Wolfe KH (2003) A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 13: 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simillion C, Vandepoele K, Van Montagu MC, Zabeau M, Van de Peer Y (2002) The hidden duplication past of Arabidopsis thaliana . Proc. Natl. Acad. Sci. U.S.A 99: 13627–13632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jordon-Thaden I, Hase I, Al-Shehbaz I, Koch MA (2010) Molecular phylogeny and systematics of the genus Draba (Brassicaceae) and identification of its most closely related genera. Mol. Phylogenet. Evol. 55: 524–540. [DOI] [PubMed] [Google Scholar]
- 37. German DA, Grant JR, Lysak MA, Al-Shehbaz IA (2011) Molecular phylogeny and systematics of the tribe Chorisporeae (Brassicaceae). Plant Syst. Evol. 294: 65–86. [Google Scholar]
- 38. Heenan PB, Goeke DF, Houliston GJ, Lysak MA (2012) Phylogenetic analyses of ITS and rbcL DNA sequences for sixteen genera of Australian and New Zealand Brassicaceae result in the expansion of the tribe Microlepidieae. Taxon 61: 970–979. [Google Scholar]
- 39. Maddison WP (1997) Gene Trees in Species Trees. Syst. Biol. 46: 523–536. [Google Scholar]
- 40. Hillis DM (1995) Approaches for Assessing Phylogenetic Accuracy. Syst. Biol. 44: 3–16. [Google Scholar]
- 41.Wendel JF, Doyle JJ (1998) Phylogenetic incongruence: window into genomes history and molecular evolution. In: Soltis DE, Soltis PS, Doyle JJ, editors. Molecular Systematics of Plants II. Boston: Kluwer Academy Publications. ISBN: 978-0-412-11131-0. [Google Scholar]
- 42. Edwards SV (2009) Is a new and general theory of molecular systematics emerging? Evolution 63: 1–19. [DOI] [PubMed] [Google Scholar]
- 43. Edwards SV (2009) Natural Selection and Phylogenetic Analysis. Proc. Natl. Acad. Sci. U.S.A 106: 8799–8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koch MA, Dobeš C, Kiefer C, Schmickl R, Klimeš L, et al. (2007) Supernetwork identifies multiple events of plastid trnF(GAA) pseudogene evolution in the Brassicaceae. Mol. Biol. Evol. 24: 63–73. [DOI] [PubMed] [Google Scholar]
- 45. Couvreur TLP, Franzke A, Al-Shehbaz IA, Bakker FT, Koch MA, et al. (2010) Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae). Mol. Biol. Evol. 27: 55–71. [DOI] [PubMed] [Google Scholar]
- 46. Franzke A, Lysak MA, Al-Shehbaz IA, Koch MA, Mummenhoff K (2011) Cabbage family affairs: The evolutionary history of Brassicaceae. Trends Plant Sci. 16: 108–116. [DOI] [PubMed] [Google Scholar]
- 47. Duarte JM, Wall PK, Edger PP, Landherr LL, Ma H, et al. (2010) Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol. Biol. 10: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnson MTJ, Carpenter EJ, Tian Z, Bruskiewich R, Burris JN, et al. (2012) Evaluating Methods for Isolating Total RNA and Predicting the Success of Sequencing Phylogenetically Diverse Plant Transcriptomes. PLoS One 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schranz ME, Mohammadin S, Edger PP (2012) Ancient whole genome duplications, novelty and diversification: The WGD Radiation Lag-Time Model. Curr. Opin. Plant Biol. 15: 147–153. [DOI] [PubMed] [Google Scholar]
- 50. Hofberger JA, Lyons E, Edger PP, Chris Pires J, Eric Schranz M (2013) Whole genome and tandem duplicate retention facilitated glucosinolate pathway diversification in the mustard family. Genome Biol. Evol. 5: 2155–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mayfield-Jones D, Washburn JD, Arias T, Edger PP, Pires JC, et al. (2013) Watching the grin fade: Tracing the effects of polyploidy on different evolutionary time scales. Semin. Cell Dev. Biol. 24: 320–331. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Secondary Structure data for ITS1, ITS2, and random sequences. Spreadsheet contains the list of species, NCBI GenBank accession numbers, lengths, total number of possible secondary structures, and a description of the lowest energy state structure for every ITS1, ITS2, and random sequence.
(XLSX)
Sequence alignments for ITS1. Sequence alignments for ITS1 for 49 Brassicaceae and one outgroup Cleomaceae found in File S1.
(TXT)
Sequence alignments for ITS2. Sequence alignments for ITS2 for 49 Brassicaceae and one outgroup Cleomaceae found in File S1.
(TXT)