

Abstract
Phagophore-derived autophagosomes deliver cytoplasmic material to lysosomes for degradation and reuse. Autophagy mediated by the incompletely characterized actions of Atg proteins is involved in numerous physiological and pathological settings including stress resistance, immunity, aging, cancer, and neurodegenerative diseases. Here we characterized Atg17/FIP200, the Drosophila ortholog of mammalian RB1CC1/FIP200, a proposed functional equivalent of yeast Atg17. Atg17 disruption inhibits basal, starvation-induced and developmental autophagy, and interferes with the programmed elimination of larval salivary glands and midgut during metamorphosis. Upon starvation, Atg17-positive structures appear at aggregates of the selective cargo Ref(2)P/p62 near lysosomes. This location may be similar to the perivacuolar PAS (phagophore assembly site) described in yeast. Drosophila Atg17 is a member of the Atg1 kinase complex as in mammals, and we showed that it binds to the other subunits including Atg1, Atg13, and Atg101 (C12orf44 in humans, 9430023L20Rik in mice and RGD1359310 in rats). Atg17 is required for the kinase activity of endogenous Atg1 in vivo, as loss of Atg17 prevents the Atg1-dependent shift of endogenous Atg13 to hyperphosphorylated forms, and also blocks punctate Atg1 localization during starvation. Finally, we found that Atg1 overexpression induces autophagy and reduces cell size in Atg17-null mutant fat body cells, and that overexpression of Atg17 promotes endogenous Atg13 phosphorylation and enhances autophagy in an Atg1-dependent manner in the fat body. We propose a model according to which the relative activity of Atg1, estimated by the ratio of hyper- to hypophosphorylated Atg13, contributes to setting low (basal) vs. high (starvation-induced) autophagy levels in Drosophila.
Keywords: Atg1, Atg13, autophagy, Drosophila, Atg17/FIP200, lysosome, Ref(2)P/p62, TOR
Introduction
During autophagy, cytoplasmic material including organelles is transported to lysosomes for degradation and recycling of building blocks. The main pathway, macroautophagy (simply referred to as autophagy hereafter) is characterized by the formation of double-membrane autophagosomes, which then fuse with lysosomes to deliver their contents for breakdown. Autophagy is an ancient starvation survival pathway promoting stress resistance and longevity in all eukaryotes. In addition, autophagy acquired numerous additional functions in multicellular organisms and protects from various diseases including cancer and neurodegeneration.1
The evolutionarily conserved MTOR (mechanistic target of rapamycin) kinase promotes cell growth in part by phosphorylating RPS6K (ribosomal protein S6 kinase) and EIF4EBP1 (eIF-4E binding protein 1) to enhance protein synthesis, and inhibits autophagy through direct phosphorylation of Atg1 (or its mammalian orthologs ULK [unc 51-like autophagy activating kinase] 1 and 2) when bound to actively digesting lysosomes.2-4 Recent in vitro experiments suggest that MTOR activity depends on intralysosomal amino acids.5 Starvation leads to dissociation of MTOR from lysosomes and autophagy induction, which has been suggested to be required for reactivation of MTOR, potentially by restoring the amino acid pool in lysosomes through the breakdown of autophagic cargo.2,6 Thus, the regulation of cell growth and autophagy is integrated at the level of lysosomes.
Formation of autophagosomes is controlled by ~20 conserved autophagy-related (Atg) gene products, most of which were originally discovered in yeast.7 Most of these proteins assemble into complexes to promote autophagosome formation at a site commonly referred to as the PAS (phagophore assembly site). Various potential sources including ER, mitochondria, Golgi, and plasma membrane have been suggested to contribute lipids to sustain the growth of initially appearing membrane cisterns (called phagophores), which engulf cytoplasmic material and give rise to autophagosomes after sealing of the edges.8,9 It is widely accepted that initiation of autophagy is usually triggered by activation of an Atg1 kinase complex (consisting of Atg1, 13, 17, 29, and 31 in yeast), followed by phagophore nucleation and assembly steps that require an Atg14-containing autophagy-specific lipid kinase complex, the transmembrane protein Atg9, and 2 ubiquitin-like protein conjugation systems mediating the C-terminal lipidation of Atg8 family proteins. Atg7 acts as an E1-like enzyme for both Atg8 and Atg12, followed by actions of the E2-like enzymes Atg3 and Atg10, respectively. Atg12 is then covalently attached to Atg5 and subsequently assembles into a large complex that also contains Atg16. This complex facilitates Atg8 lipidation and anchoring into phagophore and autophagosome membranes through a phosphatidyl-ethanolamine tail. Atg8 is recycled from the outer membrane, while the pool bound to the inner membrane is degraded after fusion with a lysosome.9
Low-level, basal autophagic degradation contributes to the turnover of orgenelles and proteins in metazoan cells, thus maintaining cellular homeostasis. We and others have shown that ubiquitinated protein aggregates accumulate in autophagy gene mutant brains in fly and mouse models.1,10,11 These aggregates also contain the multidomain protein SQSTM1/p62 (sequestosome 1) in mammals, or its fly ortholog Ref(2)P (refractory to sigma P). SQSTM1/Ref(2)P acts as a selective receptor for cargo recognition during autophagy. Binding of Atg8 homologs to SQSTM1/Ref(2)P ensures autophagic degradation of ubiquitinated protein aggregates, as SQSTM1/Ref(2)P contains an Atg8-interacting motif and a C-terminal ubiquitin-binding domain.12-14 Consequently, SQSTM1/Ref(2)P is also degraded in autolysosomes.
Recent studies characterized the mammalian ULK complex in cultured cells, which contains the multidomain adaptor protein RB1CC1 (RB1-inducible coiled-coil 1)/FIP200 (200 kDa FAK family kinase-interacting protein), ATG13, the Atg1 orthologs ULK1 and ULK2, and the animal-specific subunit C12orf44 (chromosome 12 open reading frame 44)/ATG101.3,15-18 As loss of RB1CC1 blocks autophagy, it has been suggested to play a role similar to Atg17, a scaffold protein required for starvation-induced autophagy as part of the yeast Atg1 complex. These studies revealed dynamic changes in the electrophoretic mobility of multiple subunits in nutrient-replete vs. autophagy-inducing conditions. In vitro kinase assays in these reports suggest that MTOR and ULK1/2 kinases are responsible for most of these phosphorylation events.3,16 Importantly, cell type-specific differences have been seen, but overall the phosphorylation status of endogenous ATG13 decreases during starvation.3 The regulation of the endogenous Atg1 kinase complex in an in vivo setting is still uncharacterized.
Here we explore the role of the single Drosophila ortholog of RB1CC1, Atg17/FIP200 in autophagy (also known by the annotation symbol CG1347).19,20 It is referred to as Atg17 in Flybase, the Drosophila genome homepage, as Banreti et al. suggested this symbol even though they also show that this Drosophila protein is an ortholog of mammalian RB1CC1, and it is not homologous to yeast Atg17.20 We use both loss- and gain-of-function studies to show that Atg17 activates endogenous Atg1 kinase to facilitate autophagy, and that Atg17 localizes to perilysosomal aggregates of the selective autophagy cargo Ref(2)P in polyploid larval Drosophila tissues.
Results
Generation of Atg17-null mutants, overexpression lines, and antibodies
Taking advantage of a transposon insertion (EY03045) in the first intron of Atg17, we generated a chromosomal deletion that removes practically the entire coding sequence (Fig. 1A and B). Loss of Atg17 resulted in pharate adult lethality in homozygotes, and escapers were very rarely seen in hemizygotes (Fig. S1A), similar to previously described Atg1 and Atg13 mutants.21,22 We generated polyclonal antibodies against Atg17, and used these to show that no protein product is detected in null mutants (Fig. 1C). We also established inducible transgenic Atg17 lines, the expression of which can be triggered by the presence of appropriate driver transgenes (Fig. 1C). Low-level expression of transgenic Atg17-GFP restored viability of Atg17-null mutant animals in genetic rescue experiments, confirming that lethality was due to loss of Atg17 (Fig. S1A).
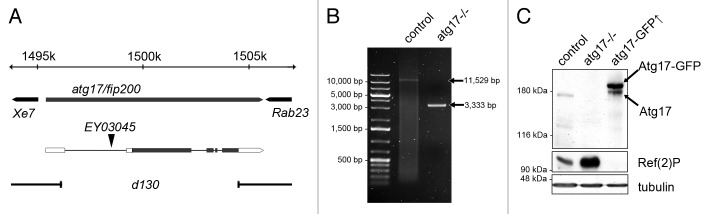
Figure 1. Generation of atg17-null mutants, overexpression lines, and antibodies. (A) atg17[d130]-null mutants harbor a 8,196 bp chromosomal deletion that leaves behind only the last 55 nucleotides of the 4,071-nucleotide open reading frame (black bars), after improper excision of the P element EY03045 located in the first intron. Open bars: untranslated regions. (B) PCR using primers flanking the atg17 locus demonstrates the extent of the genomic deletion in homozygous atg17[d130] mutant larvae compared with controls. (C) Western blots detect endogenous Atg17 of 170 kDa in control larvae, and also show the overexpressed protein in larvae expressing Atg17-GFP. No protein product is seen in mutants. The selective autophagy cargo Ref(2)P accumulates in atg17-null mutants and disappears in larvae constitutively overexpressing Atg17-GFP.
Atg17 is required for starvation-induced and basal autophagy upstream of Atg1
Starvation leads to the formation of autophagosomes and autolysosomes in fat bodies of control larvae. Both structures are labeled by mCherry-Atg8a, as this transgenic reporter is specifically bound to autophagosomes, and the mCherry tag remains fluorescent and accumulates in autolysosomes (Fig. 2A).21,23 Loss of Atg17 completely blocked the punctate mCherry-Atg8a response to starvation (Fig. 2B and I). LysoTracker Red (LTR) is a commonly used staining for autolysosomes in the larval fat body. Atg17 was cell-autonomously required for starvation-induced formation of LTR-positive autolysosomes in fat body cell clones (Fig. 2C and J). We also raised polyclonal antibodies against Atg8a, enabling the identification of nascent autophagosomes in immunostaining experiments (Fig. S1B, S1C, and S1E). Anti-Atg8a staining revealed severely impaired autophagosome formation in Atg17-null mutant or RNAi cells in mosaic experiments, compared with neighboring control cells that contained numerous Atg8a-positive dots after starvation (Fig. 2D and K; Fig. S1D and S1E). Finally, ultrastructural analysis confirmed the absence of autophagosomes and autolysosomes in fat bodies of starved Atg17-null mutant larvae (Fig. 2E, F, and L).

Figure 2.atg17 is required for starvation-induced and basal autophagy. (A and B) mCherry-Atg8a-positive autophagosomes and autolysosomes form in fat body cells of starved control (A) but not in atg17-null mutant larvae (B). (C) No LTR (LysoTracker Red)-positive autolysosomes form in GFP-positive atg17-null mutant cells, compared with surrounding control cells in the same fat body tissue in starved animals. (D) Endogenous Atg8a-positive autophagosomes are missing and Ref(2)P aggregates accumulate in starved atg17-null mutant cells (these are marked by the lack of GFP in this image, unlike in C), compared with neighboring control cells. (E and F) Electron microscopy shows that autophagosomes (1–4) and autolysosomes (AL) form in fat body cells of control (E) but not in atg17-null mutant larvae during starvation (F). (G) Depletion of atg17 in GFP-positive cells results in large-scale accumulation of Ref(2)P in fat bodies of well-fed larvae. (H) Depletion of atg17 prevents starvation-induced mCherry-Atg1 dot formation. (I–N) Quantification of data from (A and B) (I, n = 12/genotype), (C) (J, n = 14), (D) (K, n = 18), (E and F), showing the ratio of autophagosomes and autolysosomes relative to total cytoplasm (L, n = 4/genotype), (G) (M, n = 10), (H) (N, n = 10). Scale bar: (A–D, G, and H) 20 µm; (E and F) 1 µm. Error bars: s.d., *P < 0.05, **P < 0.01.
Basal autophagic degradation occurs at a low level in many tissues, making it difficult to estimate by conventional autophagy reporters. As the specific autophagy cargo Ref(2)P accumulates upon reduction of basal autophagy, determining its levels is a common test in these experiments.23-25 Indeed, loss of Atg17 resulted in large-scale accumulation of endogenous Ref(2)P in western blots (Fig. 1C), and in null mutant and RNAi cells (Fig. 2D, G, K, and M).
The Atg1 kinase complex, and its subunit Atg17 in yeast and the corresponding proposed ortholog RB1CC1 in mammalian cells in particular, is suggested to act most upstream in the hierarchy of Atg protein complexes.26,27 In line with that, we found that knockdown of Atg17 prevented starvation-induced punctate mCherry-Atg1 localization (Fig. 2H and N). We and others have shown previously that overexpression of Atg1 activates autophagy, and also reduces cell size at least in part by negative feedback on TOR (target of rapamycin).22,28,29 High-level expression of Atg1 could still trigger punctate Atg8a labeling in Atg17-null mutant fat body cells and reduced cell size in epistasis experiments (Fig. S2A and S2B).
Atg17 is required for developmental autophagy and programmed removal of salivary glands during metamorphosis
Increased level of the molting hormone ecdysone at a low juvenile hormone concentration leads to initiation of wandering behavior, when L3 stage larvae leave the food in search of an appropriate place for pupariation. At the same time, ecdysone triggers autophagy in polyploid larval tissues including fat body, presumably facilitating the removal of these organs which become obsolete during metamorphosis.30 Loss of Atg17 blocked developmental autophagy in fat body cells, based on lack of punctate LTR and mCherry-Atg8a signals (Fig. 3A–E). In addition, programmed elimination of larval salivary glands during metamorphosis was impaired in Atg17-null mutants (Fig. 3F–I), similar to previous observations in animals carrying mutations in genes that act downstream of the Atg1 kinase complex during autophagy.31 The volume of prepupal midguts strongly decreases during the onset of metamorphosis in an autophagy-dependent manner.10,32 Midgut shrinkage was also attenuated by loss of Atg17 in our experiments (Fig. S2C–S2F).

Figure 3.atg17 is necessary for developmental autophagy and proper salivary gland histolysis. (A and B) Punctate LTR staining is observed in controls (A), but not in atg17-null mutant fat bodies of L3 stage wandering larvae prior to metamorphosis (B). (C) Quantification of data from (A and B); n = 20/genotype. (D) Depletion of atg17 in GFP-positive cells blocks the formation of mCherry-Atg8a positive autophagosomes and autolysosomes in fat bodies of wandering larvae. (E) Quantification of data from (D); n = 26. (F–I) Larval salivary glands (SG) are visible in hematoxylin-eosin stained paraffin sections of both control (F, n = 3) and atg17-null mutant (G, n = 5) white prepupae (0 h relative to puparium formation, rpf). Salivary gland histolysis is completed by 24 h rpf in controls (H, n = 7), but numerous large, persisting salivary gland cells (arrowheads) are visible in all similarly aged atg17-null mutant pupae (I, n = 9). Scale bars: (A, B, and D) 20 µm, (F–I) 60 µm. Error bars: s.d., **P < 0.01.
Atg17 localizes to Ref(2)P aggregates near lysosomes
Atg17-GFP dots showed 52% colocalization with endogenous Atg8a, which is recruited to the PAS and remains bound to autophagosomes (n = 611 dots counted, Fig. 4A). Atg5 is a commonly used marker for PAS.9,23,33 Stalled PASs containing upstream Atg proteins accumulate in yeast and mammalian cells lacking downstream ATG genes.26,27 We used this strategy to analyze the colocalization of endogenous Atg17 and Atg5 in Atg8a-null mutants, in which PAS formation is stalled and no autophagosomes form.22,26 We did not detect cytoplasmic dots positive for endogenous Atg17 or Atg5 in gastric ceca cells of well-fed larvae (Fig. 4B). Punctate structures appeared in response to starvation, and Atg17 showed 81% colocalization with Atg5 dots (n = 97, Fig. 4C). This colocalization experiment was performed in gastric ceca, a tissue that produces similar starvation-induced autophagic response to fat bodies,10,30 because endogenous Atg5-positive structures were difficult to visualize in fat body cells of starved larvae. 89% of Atg17-GFP dots colocalized with the selective cargo Ref(2)P in control fat cells, and 93% of endogenous Atg17 colocalized with Ref(2)P aggregates in Atg7-null mutants (n = 391 and 148, respectively, Fig. 4D and E). As expected, no cytoplasmic Atg17 dots were seen in starved Atg17-null mutants, whereas most likely nonspecific nuclear staining remained (Fig. 4F).

Figure 4. Atg17 is associated with lysosomes and colocalizes with Atg8a, Atg5, and Ref(2)P. (A) Atg17-GFP colocalizes with endogenous Atg8a in fat cells of starved larvae. (B) No punctate Atg17 or Atg5 structures are seen in well-fed Atg8a-null mutant gastric ceca. (C) Endogenous Atg17 colocalizes with Atg5-positive PAS in starved Atg8a-null mutant gastric ceca. (D) Atg17-GFP localizes to Ref(2)P aggregates in control larvae. (E) Endogenous Atg17 colocalizes with endogenous Ref(2)P in Atg7-null mutants. (F) No cytoplasmic Atg17 dots are seen in starved atg17-null mutants, while nonspecific nuclear staining is observed. (G) Transient expression of Atg17-GFP rescues punctate LTR staining in fat body cells of starved atg17-null mutants. Note that Atg17 dots are tightly associated with LTR-positive autolysosomes. (H) Endogenous Atg17 accumulates near Cp1-positive lysosomes in Atg7-null mutant fat cells. (I) Punctate Ref(2)P-GFP structures are observed near LTR-positive autolysosomes. Scale bar in (A): 20 µm for (A, B, E, F, H, and I); in (C): 20 µm for (C, D, and G). Boxed areas are shown enlarged with merged image (top), red channel in grayscale (middle) and green channel in grayscale (bottom). FB, fat body; GC, gastric ceca.
In genetic rescue experiments, transient expression of Atg17-GFP fully rescued the autophagy defect of Atg17-null mutants (Fig. 4G). We noticed that Atg17-GFP exhibited a punctate pattern, and practically all dots clustered around LTR- and mCherry-Atg8a-positive autolysosomes (Fig. 4G; Fig. S3A). Moreover, Venus-tagged Atg1 also formed punctate structures closely associated with LTR-positive autolysosomes (Fig. S3B). In line with these, endogenous Atg17 dots closely associated with, but did not colocalize with the resident lysosomal hydrolase Cp1 (Cysteine proteinase-1)/cathepsin L, both in Atg7 or Atg8a-null mutant fat body or gastric ceca cells of starved larvae (Fig. 4H; Fig. S3C).
Previous studies in yeast showed that phagophores assemble at cytoplasmic areas excluding ribosomes and organelles.34 We were able to identify ribosome-free areas with associated 40- to 60-nm vesicles, which is the reported size of Atg9 vesicles in yeast,35 and these structures could be observed next to autolysosomes in ultrastructural images (Fig. S3D and S3E). We speculate that these may represent the phagophore formation site in Drosophila, although alternative interpretations are also possible.
Aggregates of the specific autophagy cargo Ref(2)P-GFP also frequently clustered around LTR-positive autolysosomes in starved Drosophila larvae (Fig. 4I). MTOR, an upstream negative regulator of autophagy, is bound to lysosomes in growing cells, and starvation results in dispersion of MTOR and its dissociation from lysosomes. SQSTM1 is associated with MTOR complex 1.12 We found that FLAG-TOR displayed a punctate distribution in fat bodies of well-fed Drosophila larvae that showed 95% overlap with Ref(2)P dots (n = 77), and starvation led to a complete dispersion of FLAG-TOR but not Ref(2)P (Fig. S3F and S3G).
Overexpression of Atg17 enhances autophagy in an Atg1-dependent manner
Constitutive expression of Atg17-GFP dramatically reduced the levels of endogenous Ref(2)P in well-fed larvae, indicating enhanced autophagic degradation (Fig. 1C). Similarly, overexpression of Atg17 in GFP-marked fat cells led to the formation of mCherry-Atg8a and mCherry-Atg1 positive autophagic structures in well-fed mosaic animals (Fig. 5A, B, J, and K). Heat shock-mediated transient expression of Atg17-GFP also induced punctate LTR staining (Fig. 5C, D, and L). Coexpression of a kinase-dead, dominant-negative (DN) Atg1 attenuated Atg17-induced LTR staining, but not punctate Atg17 localization (Fig. 5E and L). Similarly, overexpression of Atg17 increased the number of Atg8a-positive autophagosomes in an Atg1-dependent manner in fat bodies of well-fed larvae (Fig. 5F–I, and M). These results suggest that Atg17-induced autophagy requires Atg1.

Figure 5. Overexpression of Atg17 enhances autophagy in well-fed larvae. (A and B) Overexpression of Atg17 in GFP-positive cells promotes mCherry-Atg8a (A) and mCherry-Atg1 (B) puncta formation. (C) Practically no LTR dots are seen in fat body cells of well-fed control larvae expressing GFP. (D) Transient expression of Atg17-GFP induces punctate LTR staining. (E) Coexpression of dominant-negative Atg1 blocks Atg17-induced LTR staining. (F) Only a few Atg8a puncta are seen in heat-shocked control larvae. (G) Heat shock-induced transient expression of Atg17-GFP enhances autophagosome-associated Atg8a dot formation. (H) Coexpression of GFP has no effect on punctate Atg8a staining induced by Atg17-GFP. (I) Coexpression of dominant-negative Atg1 prevents Atg17-induced Atg8a dot formation. (J–M) Quantification of data from (A) (J, n = 22), (B) (K, n = 18) and (C–E) (L, n = 14/genotype), (F–I) (M, n = 10/genotype). Scale bar: (A–I) 20 µm. Error bars: s.d., **P < 0.01.
Atg17 is a subunit of the Atg1 kinase complex
To further characterize the role of Drosophila Atg17, we performed coimmunoprecipitations with Atg1 kinase complex subunits in cultured Drosophila cells. Atg17-GFP readily coprecipitated with FLAG-tagged Atg13 and FLAG-tagged Atg101 (Fig. 6A). Atg17-GFP was clearly detectable in precipitates of kinase-dead or wild-type myc-Atg1, although in lower amounts than in case of Atg13 or Atg101 (Fig. 6B). Note that overexpression of wild-type Atg1 strongly reduces the expression of tagged proteins including its own expression, which is likely due to inhibition of TOR-dependent translation by Atg1.29 In these experiments, coexpression of myc-Atg1 but not kinase-dead myc-Atg1 also decreased the electrophoretic mobility of Atg17-GFP. This is consistent with mammalian studies showing that ULK1/2 can phosphorylate RB1CC1, which is facilitated by ATG13.16 Tagged overexpressed proteins also coprecipitated from larval extracts (Fig. S4A).

Figure 6. Interactions of Atg17, Atg1 and Atg13. (A and B) Immunoprecipitation experiments in cultured Drosophila cells. Atg17-GFP coprecipitates with Atg13-FLAG and Atg101-FLAG, but not with FLAG-GFP (A). Atg17-GFP binds weakly to kinase-dead and wild-type myc-Atg1, but not to anti-myc beads (B). Note the decreased mobility of Atg17-GFP upon coexpression of myc-Atg1. (C) Of the truncated Atg13 constructs (see left panel), a fragment containing the putative unstructured regions (231 to 523) binds strongly to Atg17-GFP. (D) Myc-Atg1 binds to the C-terminus (393 to 523) of Atg13. (E) Expression of myc-Atg1 strongly decreases and active TOR (amino acids 1333 to 2470) slightly increases the mobility of HA-Atg13 in cultured cells. (F) Expression of myc-Atg1 results in a shift (indicated by arrowheads) of Atg13 fragments containing the C terminus (393 to 523 and 231 to 523). IN, input, IP, immunoprecipitation, SN, supernatant.
ATG13 has been reported to directly bind to both RB1CC1 and the Atg1 orthologs ULK1 and ULK2 in mammalian cells, which prompted us to map the regions involved in these interactions in Drosophila.3,16,17 All Atg13 homologs share an N-terminal domain, followed by poorly conserved regions (Fig. S5).17,36 We generated HA-GFP-tagged truncated fragments of Atg13 for interaction studies (Fig. 6C). The highest amount of Atg17-GFP was observed in precipitates of the putative unstructured regions (amino acids 231 to 523) of Atg13 (Fig. 6C). Drosophila Atg13 has recently been shown to interact with Atg1, but the region involved has remained unknown.21 We found that Atg1 was coprecipitated with the C-terminal region (amino acids 393 to 523) of Atg13 (Fig. 6D). Although our data do not rule out the possibility that the observed interactions reflect indirect binding, these results are consistent with previous mammalian reports, which show the direct binding of ATG13 to both ULK1/2 and RB1CC1.16,17
Atg17 activates Atg1 in vivo
Atg13 phosphorylation levels are influenced by TOR and Atg1 kinases in yeast, flies, and mammals.17 In agreement with these, coexpression of Atg1 strongly increased, while an N-terminally truncated active fragment of TOR appeared to slightly reduce the overall phosphorylation level of HA-Atg13 in cultured Drosophila cells (Fig. 6E). In line with our interaction data, co-expression of Atg1 decreased the mobility of Atg13 fragments containing the C-terminus (Fig. 6F), suggesting that Atg1phosphorylated these fragments.
To gain more insight into the regulation of endogenous Atg1 kinase complex, we raised polyclonal antisera against Drosophila Atg13 and Atg1, which specifically recognized endogenous proteins in western blots (Fig. 7A; Fig. S4B). It was striking that endogenous Atg13 appeared as multiple bands in fat body extracts prepared from well-fed animals, and starving larvae for 3 or 24 h strongly increased the ratio of slower mobility to faster mobility bands (Atg13high/Atg13low, Fig. 7B). Endogenous Atg1 was difficult to visualize using our antibody in fat body extracts of well-fed animals, although the bands became clearly visible upon starvation (Fig. S4C). Epitope mapping suggested that this antibody may recognize the middle region of Atg1, which is rich in prolines and serines (Fig. S4D). As the sites phosphorylated by TOR (a proline-directed Ser/Thr kinase) are found in this region of Atg1 homologs,37 TOR-mediated phosphorylation may be responsible for the reduced affinity of our antibody in nutrient-replete fat body extracts. The usefulness of this reagent is thus limited, therefore anti-Atg1 counterparts of western blots are shown in supplemental data.

Figure 7. Atg17 regulates Atg13 phosphorylation in vivo. (A) Western blots using our novel antibody recognize endogenous Atg13, which is absent from atg13-null mutant larvae. Asterisk denotes a nonspecific band that serves as loading control. (B) Starvation strongly decreases the mobility of Atg13 in larval fat body extracts. (C) Phosphatase treatment of starved fat body lysates demonstrates that Atg13 is phosphorylated in vivo. (D) Transient overexpression of myc-Atg1, or Tsc1 and Tsc2 decreases Atg13 mobility in fat bodies of fed larvae. (E) Transient overexpression of dominant-negative Atg1 reduces the ratio of slower mobility Atg13 forms relative to higher mobility forms in fat bodies of well-fed larvae. Dominant-negative Atg1 expression practically eliminates all Atg13 phospho-forms in starved fat bodies. (F) Transient (low) or constitutive (high) expression of Atg17-GFP decreases Atg13 mobility in fat bodies of well-fed animals. The starvation-induced shift of Atg13 to slower mobility forms is lost in atg17-null mutants. (G) Coexpression of dominant-negative Atg1 prevents Atg17-induced shift of Atg13 to slower mobility forms in fat body extracts of well-fed larvae. Numbers indicating Atg13 high/low ratio are calculated by densitometric evaluation of blots as described in Materials and Methods.
Phosphatase treatment practically reduced both Atg13 and Atg1 to single bands, suggesting that these endogenous proteins are phosphorylated (Fig. 7C; Fig. S4E). In agreement with that, overexpression of Atg1 led to a shift of endogenous Atg13 to slower mobility forms in fat bodies of well-fed animals, similar to genetic suppression of TOR by co-overexpressing Tsc1 and Tsc2 (Fig. 7D; Fig. S4F). Expression of dominant-negative Atg1 in fat bodies of fed larvae reduced the amount of slower mobility forms of endogenous Atg13, and in starved larvae practically eliminated all Atg13high forms (Fig. 7E; Fig. S4G). Thus, Atg1 kinase appears to be responsible for starvation-induced hyperphosphorylation of Atg13, and likely also contributes to phosphorylation of Atg13 in nutrient-replete conditions in vivo.
In line with the essential role of Atg17 in autophagy, the starvation-induced shift from Atg13low to Atg13high forms was blocked in Atg17-null mutants (Fig. 7F). Constitutive or transient expression of Atg17-GFP increased the Atg13high/Atg13low ratio even in well-fed larvae (Fig. 7F; Fig. S4H). In agreement with our microscopy data that Atg17-induced autophagy requires Atg1, coexpression of kinase-dead Atg1 blocked Atg17-induced Atg13 hyperphosphorylation in fat bodies of well-fed larvae (Fig. 7G; Fig. S4I), suggesting that Atg17 induces autophagy at least in part by activating Atg1.
Discussion
Basal autophagy primarily functions as a housekeeping mechanism, ensuring proper turnover and quality control of macromolecules and organelles. High levels of autophagy are induced by starvation, and degradation of dispensable cellular constituents provides nutrients and building blocks for synthetic and energy producing reactions. This prosurvival function of autophagy is conserved in all eukaryotes including yeast, and probably represents its most ancient role. In contrast, autophagy was found to promote cell death during metamorphosis, as proper programmed elimination of polyploid larval salivary glands and midguts is suggested to depend on autophagy.31,32 All 3 pathways (basal, starvation-induced, and developmental autophagy) serving various cellular and organismal functions require Atg17, indicating that its protein product plays an equally critical role in autophagy in these different contexts. A related study published while our manuscript was in preparation also reports that Drosophila Atg17 is required for starvation-induced and developmental autophagy.38
Previous reports establish that RB1CC1 is an essential subunit of the Atg1/ULK kinase complex in mammalian cells.3,15,16 RB1CC1 directly binds the C-terminal region of human ATG13, which facilitates its interaction with, and phosphorylation by, ULK1 and ULK2.16 ULK1 and ULK2 also bind to the C-terminal region of ATG13, and phosphorylate multiple serine and threonine residues during autophagy induction. In addition, mammalian ATG13 is phosphorylated by MTOR in vitro in kinase assays.16 Thus, a model was proposed, according to which ATG13 is inhibited by MTOR-mediated phosphorylation under basal conditions. Starvation results in activation of ULK1/2, and then it directly phosphorylates mammalian ATG13, which is thought to be important for the induction of autophagy. The Atg1 mammalian orthologs ULK1 and ULK2 are inhibited at least in part by direct MTOR-mediated phosphorylation and undergo autophosphorylation during autophagy induction.17 Interestingly, the Atg1 complex appears to be regulated differently in yeast: Atg13 is phosphorylated by TOR in up to 8 different residues in well-fed cells, and Atg13 undergoes practically complete dephosphorylation during autophagy induction.39 Six of these phosphorylation events occur in or near the central region of yeast Atg13, and are thought to destabilize the Atg1 complex. An unphosphorylatable Atg13 mutant triggers autophagy in nutrient-replete conditions independent of TOR in yeast.39 A recent paper shows that the assembled Atg1 kinase complex can be detected in yeast cells under nutrient-replete conditions as well.40 This pool may be important to sustain the Cvt (cytoplasm-to-vacuole targeting) pathway, an ATG gene-dependent vesicular route transporting certain hydrolases to the vacuole in well-fed yeast cells. The Cvt pathway shares certain similarities with basal autophagy observed in metazoan cells, such as specific recognition of aggregated cargo proteins. In fact, a double-membrane Cvt vesicle resembles a small autophagosome, which only contains specific cargo and no bulk cytosol.41
Atg1 and Atg17 have been suggested to bind to more central regions of yeast Atg13, unlike in mammals.36 These differences are reflected at the sequence level as well: only the N-terminal domain appears to be conserved, although the double arginine motifs thought to function as a phosphorylation sensor in yeast Atg13 are missing in higher eukaryotes (Fig. S5). Drosophila Atg13 has also been reported to bind to Atg1, but the region involved has not been determined.21 We found that Atg1 interacts with the C-terminal part of Atg13, similar to mammals. Our results also reveal that the interaction of Atg13 and RB1CC1 is conserved, but the C-terminal region may not be sufficient: only a fragment containing both the middle and C-terminus of Atg13 elicited strong binding to Atg17, although we did not test whether this interaction is also direct in Drosophila.
There are striking differences between yeast, fly, and mammalian cells in the overall phosphorylation status of Atg13 during autophagy induction. The phosphorylation level of yeast Atg13 strongly decreases upon starvation, it does not change much in mammalian cells, whereas it strongly increases in polyploid larval fat bodies in starved Drosophila larvae. The shift of Atg13 to more phosphorylated forms during starvation agrees with the findings of a recent report that relied on cooverexpressed proteins.21 Although other kinases may also phosphorylate and thus modulate endogenous Atg13, our data strongly suggest that Atg1 is mostly responsible for starvation-induced hyperphosphorylation of Atg13 in Drosophila. First, overexpression of Atg1 results in a remarkable decrease in the mobility of both tagged and endogenous Atg13, both in cultured cells and in well-fed larvae. Second, overexpression of dominant-negative Atg1 leads to a near-complete block of starvation-induced Atg13 phosphorylation in vivo, practically eliminating all slower mobility bands in fat body lysates. Drosophila Atg17 appears to be necessary for starvation-induced phosphorylation of endogenous Atg13, similar to its mammalian ortholog RB1CC1.3 Finally, transient or constitutive overexpression of Atg17 induces autophagy and also shifts Atg13 to slower mobility forms, both in an Atg1-dependent manner. Thus, endogenous Atg17 appears to be required for the Atg1-mediated hyperphosphorylation of Atg13 during starvation in vivo, and its overexpression likely enhances basal autophagy at least in part by promoting Atg1 kinase activity toward Atg13.
We recently reported that Atg genes including Atg1 and Atg13 are required for basal autophagy in Drosophila.24 Therefore, Atg1 must have some residual kinase activity in nutrient-replete conditions as well. A pool of active Atg13 (phosphorylated by Atg1) likely exists even in well-fed cells, which may be important for sustaining basal autophagy. In line with these, we found that overexpression of dominant-negative Atg1 reduces the amount of hyperphosphorylated Atg13 observed in well-fed cells. Based on the correlation of changes in Atg13 phosphorylation patterns with autophagic status, we propose a model according to which the ratio, and not simply the presence or absence of different pools of Atg1 complexes may regulate the level of autophagy in Drosophila (Fig. 8). Currently there is no evidence for the number and exact sites of Atg13 phosphorylation events in Drosophila. Thus, our model will need to be tested by the identification of all phosphosites in Atg13 and determination of the percentage of phosphorylated vs. nonphosphorylated states for each site in different conditions, followed by functional analysis of phospho-mimetic and nonphosphorylatable mutant forms of Atg13 in vivo.

Figure 8. A hypothetical model of Atg13 function during basal and induced autophagy. Based on correlation of overall Atg13 phosphorylation level with autophagic status, Atg13 is likely activated at least in part by Atg1-mediated phosphorylation on multiple residues. Question marks near phosphogroups of Atg13 highlight the fact that the number and exact sites of these phosphorylation events is currently unknown. The ratio of this active Atg13 appears to be lower during nutrient-replete conditions relative to the inactive form, which is potentially phosphorylated by TOR or other kinases, or not phosphorylated at all. The presence of the active form at a lower level under basal conditions may be necessary to support low-level autophagic activity. Starvation, genetic inactivation of TOR, and overexpression of Atg1 or Atg17 appear to increase the ratio of active Atg13, which may contribute to increased autophagosome formation observed in response to these autophagy-inducing treatments.
Previous reports propose that Atg17 and its proposed functional counterpart RB1CC1 acts most upstream of all Atg/ATG proteins in yeast and mammals, respectively.26,27 Our genetic epistasis results also support the upstream role of Atg17 in autophagy in Drosophila. In contrast, a recent report suggests that Drosophila Atg17 functions downstream of Atg1.38 This conclusion was based on: i) failure of overexpressed Atg17 to influence the mobility of overexpressed Atg1 in western blots of cultured cell lysates, ii) Atg1 phosphorylation level remained responsive to cellular energy status in cultured atg17 siRNA cells and starved hypomorphic Atg17 mutants, iii) Atg17 RNAi reduced myc-Atg1 overexpression-induced LTR and acridin-orange staining in eye disks, and iv. coexpression of Atg17 with Atg1 did not induce eye degeneration. Importantly, no loss-of-function tests were performed in Atg17-null mutants, so interpretation of the above phenotypes may be complicated by the presence of residual protein. Another potential explanation is that Atg17 and Atg1 are subunits of the same complex and appear to function together during autophagy induction, which makes it difficult to clearly establish their hierarchical relationship.
Atg17 colocalizes with markers of early autophagic structures both in controls and atg7 and Atg8a-null mutants, in which autophagosome formation is impaired and stalled PAS dots accumulate. The aggregate-forming protein SQSTM1 is a specific cargo for autophagy and is recruited to the PAS in cultured cells and mouse tissues.14,25 SQSTM1 interacts with MTOR complex 1, which is found on lysosomes in growing mammalian cells. This localization is facilitated by the binding of MTOR to RRAG (Ras-related GTP binding) proteins and the Ragulator complex. These latter components are anchored to lysosomes partly through their physical interaction with subunits of the lysosomal proton pump v-ATPase complex.5 Ref(2)P aggregates also colocalize with TOR in fat body cells of well-fed Drosophila larvae, and starvation results in dispersion of TOR. As Ref(2)P puncta appear to remain associated with lysosomes and colocalize with Atg17 during starvation, these protein aggregates near lysosomes may be specifically captured into forming autophagosomes after TOR is inactivated. To our knowledge, this localization has not been reported in animal models or cells yet, so it remains to be seen if it is a characteristic of these large polyploid cells and tissues, which primarily function as nutrient storing and metabolic organs in the fly larva. It is important to note that the PAS always assembles at aggregates of the selective cargo prApe1 (precursor aminopeptidase I) near the vacuole, the equivalent of the metazoan lysosomal network in yeast.42 The functional significance of this location is not known yet. A recent report argues that vesicles containing the transmembrane protein Atg9 may be recycled from vacuole/autolysosome membranes to fuel ongoing cycles of autophagosome generation in yeast.35 If Atg9 plays similar roles in Drosophila, then the putative perilysosomal localization of early autophagic structures may give instant access to these membrane-recycling vesicles. The perilysosomal localization of PAS suggested by our data may also couple upstream signals such as TOR with robust induction of autophagy at lysosome-associated Ref(2)P aggregates in polyploid larval Drosophila cells. It will be interesting to see if the association of SQSTM1 and Atg proteins near lysosomes is also observed in a mammalian tissue in future studies.
Materials and Methods
Fly strains and genetics
Flies were maintained on standard yeast/cornmeal/agar media. P element excisions and fat body cell clones were generated as described earlier in detail.10,22,23,33 Briefly, RNAi cell clones were generated spontaneously in larvae carrying hs-Flp, Act > CD2 > Gal4 and UAS-RNAi (and UAS-GFPnls as a knockdown cell marker). For mutant cell clones, 2 to 4 h embryos were heat shocked in a 38 °C water bath for 1 h to generate loss-of-function cells (marked either by lack of GFP expression, or vice versa, by GFP expression in case of the positively marked mutant clone generation system).10,23,33,43 Overexpression experiments were routinely performed using standard binary expression systems. In these, Gal4 and QF transcription factors drive expression of transgenes under the influence of UAS or QUAS promoters, respectively.43 For transient expression, UAS-Atg17-GFP was induced by hs-Gal4 and a 30 to 60 min heat shock at 37 °C, followed by 3 h of recovery before analysis. For constitutive expression, QUAS-Atg17-GFP was driven by ET49-QF.43 The following stocks were used in this study: atg17[KK104864], atg8a[KK109654] (both obtained from the Vienna Drosophila RNAi Center), atg12[JF02704],24 UAS-Atg17[EY03045], w[1118] used as control, FRT82B, Ubi-GFP, UAS-GFP, cg-Gal4, ET49-QF, QUAS-GFP, tub-QS, Df(3R)BSC464 (all obtained from the Bloomington Drosophila Stock Center), atg17[d130] (referred to as atg17−/− or null mutant throughout the paper), UAS-Atg17-GFP, QUAS-Atg17-GFP, r4-mCherry-Atg1 (all described in this study), UAS-Atg1[6B], UAS-Atg1[DN],22 UAS-Lamp1-GFP, hsFlp[22], Atg13[d81], r4-mCherry-Atg8a, UAS-myc-Atg1, UAS-Atg13-FLAG, UAS-FLAG-tor, UAS-Ref(2)P-GFP, r4-Gal4, UAS-Tsc1, UAS-Tsc2 (kindly provided by Tom Neufeld),21 UAS-Venus-Atg1 (kindly provided by Toshifumi Tomoda),44 atg7[d77], atg7[d14],10 atg8a[d4],24 atg1[KG07993] (obtained from the Drosophila Genetic Resource Center).
Generation of polyclonal antibodies
Full-length Atg8a and partial Atg17 (amino acids 1 to 420) coding sequences were amplified by PCR and cloned into pEV (kindly provided by Peter Rapali) as NdeI-XhoI fragments. The full Atg1 and partial TOR (amino acids 2,345–2,470) coding regions were PCR amplified, blunt cloned into XmnI-EcoRV-digested pENTR1A (Invitrogen, 11813011) and subsequently recombined into pDEST17 (Invitrogen, 11803012). The Atg13 entry clone (kindly provided by Tom Neufeld) was also recombined into pDEST17. All these N-terminally His-tagged proteins were expressed in E. coli Rosetta strain and purified using Ni-NTA agarose beads (Qiagen/Biomarker, 30210). Recombinant proteins were used to immunize rats in-house following standard procedures with Freund’s adjuvants (Sigma, F5881 and F5506, respectively). Recombinant Atg8a was also used to immunize rabbits (Immunogenes).
Generation of transgenic lines
Coding sequences were amplified with specific primers from cDNA and cloned into pR4-mCherry-Atg8a (kindly provided by Tom Neufeld) by replacing the BsrGI fragment (Atg8a coding region) to generate pR4-mCherry-Atg1. An 1xHA-EGFP-Sirup coding sequence was chemically synthesized (Genscript) and cloned into pUAST (DGRC 1000) with EcoRI and XhoI to generate pUAS-GFP-Sirup. A pUAS-C-GFP plasmid was generated by amplifying the HA-EGFP sequence from pUAS-GFP-Sirup and cloning it as an XbaI-NheI fragment into the XbaI site of pUAST. This vector was subsequently used to generate pUAS-Atg17-GFP by cloning the Atg17 coding region amplified from cDNA as an XbaI fragment upstream of GFP. Atg17-GFP sequences were amplified from this plasmid and cloned into pQUAST43 as a NotI fragment to generate pQUAS-Atg17-GFP. Coding sequences for pUAS-3xHA-Atg101 were chemically synthesized (Genscript). Transgenic flies were established by standard embryo injection techniques (Bestgene).
Cell culture
D.Mel-2 cells (a serum-independent S2 line) were maintained in Express Five Serum-Free Medium with penicillin and streptomycin (Invitrogen, 10486025 and 15140122, respectively). Cells were transfected with UAS constructs and metallothionein-Gal4 plasmid using TransIT-2020 reagent (Mirus/BioTec, MIR5405) according to the manufacturer’s recommendations. 48 h later, protein expression was induced by adding 1mM CuSO4 for overnight incubation. The following DNA constructs were used in cell culture experiments: pUAS-atg17-GFP, pUAS-myc-atg1 (generated by recombination of the Atg1 entry clone into pTMW [DGRC 1107]), pUAS-atg13-FLAG (generated by recombination of the Atg13 entry clone into pTWF [DGRC 1116]), pUAS-FLAG-tor, pUAS-myc-atg1[KD] (kindly provided by Tom Neufeld), pUAS-Atg101-FLAG (generated by PCR amplifying and blunt cloning the Atg101 coding region into XmnI-EcoRV-digested pENTR1A, followed by recombination into pTWF). Atg101 coding sequences were replaced with GFP in pUAS-3xHA-Atg101, and PCR amplified Atg13 and Atg1 fragments were cloned into this pUAS-3xHA-GFP plasmid using NotI-Acc65I to generate the various truncated forms. An active TOR fragment (amino acids 1333 to 2470)16 was amplified from pUAS-FLAG-tor and ligated as an XbaI fragment into pUAST. Gateway vectors and ESTs used for cloning were obtained from the Drosophila Genomics Resource Center (DGRC). Primer and synthetic gene sequences used in molecular cloning experiments are available on request.
Histology and imaging
For starvation treatments, early L3 larvae were floated in 20% sucrose for 3 h, dissected and mounted in 50% glycerol/PBS (phosphate-buffered saline) with 1 µg/µl DAPI (4',6-diamidino-2-phenylindole, Sigma, D9542) for transgenic reporters or stained in 100 µM LTR (Invitrogen, L7528) in PBS before mounting. Images were obtained on a fluorescent microscope (Carl Zeiss, Axioimager M2) equipped with a grid confocal unit (Carl Zeiss, Apotome2) using Plan-NeoFluar 40× 0.75 NA air or 100× 1.30 NA oil immersion objectives (Carl Zeiss), Axiocam Mrm camera (Carl Zeiss) and Axiovision software (Carl Zeiss), or an laser scanning microscope (Olympus, FV500) with a 63× 1.45 NA oil immersion objective (Olympus) in sequential scanning mode. Primary images were processed in Photoshop (Adobe) to produce final figures. Tissues were processed for conventional electron microscopy as described previously.10,23
Statistics
For statistical analyses, either 300 × 300 pixel areas were selected randomly from different genotypes, or randomly selected GFP-positive cells together with neighboring control cells from the same images were evaluated in clonal studies. Dot number and size data was extracted by setting an appropriate threshold for the relevant channel in ImageJ (National Institutes of Health). Data were imported into SPSS Statistics version 20 (IBM), which was used to test for normality and calculate P values. In pairwise comparisons, normal distribution data were analyzed with 2-tailed, 2-sample Student t test, whereas Mann-Whitney u-tests were used when one or both data sets showed a non-normal distribution, respectively.23,45
Primary antibodies
The following primary antibodies were used in this study: rat anti-Atg8a (IF 1:300, this study), rat anti-GFP (WB 1:5,000),24 mouse anti-FLAG (IF 1:100, WB 1:2,000, Sigma, F1804), mouse anti-myc (WB 1:5,000, Sigma, M4439), rabbit anti-HA (WB 1:2,000, Sigma, H6908), rat anti-Atg17 (IF 1:100, WB 1:5,000, this study), rabbit anti-Ref(2)P/p62 (IF 1:2,000, WB 1:5,000),24 mouse anti-tubulin (WB 1:1,000, DSHB AA4.3 s), rat anti-Atg1 (WB 1:5,000, this study), rat anti-Atg13 (WB 1:5,000, this study), rat anti-TOR (WB 1:5,000, this study), rabbit anti-Atg5 (IF 1:100, Sigma, A0856), rabbit anti-cathepsin L/Cp1 (IF 1:150, Abcam, ab58991), chicken anti-GFP (IF 1:1,500, Invitrogen, A10262). Note that the specificity of our rat and rabbit anti-Atg8a antisera described here has recently been shown elsewhere as well.23,45
Immunostaining of whole-mount larval tissues
Bisected third instar larvae were inverted and fixed with 3.7% paraformaldehyde in PBS overnight at 4 °C. Next, samples were rinsed twice and washed for 2 h in PBS, permeabilized for 15 min in PBTX-DOC (PBS with 0.1% Triton X-100 and 0.05% sodium deoxycholate) and blocked for 3 h in 3% goat serum in PBTX-DOC. Samples were then incubated overnight at 4 °C with primary antibodies in 1% goat serum in PBTX-DOC. After 3 × 30 min washes in PBTX-DOC, samples were incubated with secondary antibodies diluted 1:1,500 in 1% goat serum in PBTX-DOC for 4 h at room temperature. Finally, after 3 × 15 min washes in PBTX-DOC and 1 × 15 min in PBS, fat bodies and midguts were dissected and mounted in 50% glycerol/PBS with 0.2 μM DAPI. The following secondary antibodies were used: Alexa 488 anti-chicken, Alexa 488 anti-rabbit, Alexa 546 anti-rabbit, Alexa 568 anti-rat, Alexa 568 anti-mouse, Alexa 647 anti-rabbit (all used in 1:1,500, Invitrogen A11039, A11034, A11035, A11077, A1104, A21245, respectively).
Immunoprecipitation
Cultured cells were collected by centrifugation, washed twice in PBS and lysed on ice in lysis buffer (0.5% Triton-X100, 150 mM NaCl, 1 mM EDTA, 20 mM TRIS-HCl pH 8) containing complete protease and phosphatase inhibitor cocktails (Sigma P8340 and Roche 04906845001, respectively), spun for 10 min at 10,000 g in an Eppendorf 5430R at 4 °C, followed by the addition of 20 µl anti-HA or anti-FLAG slurry (Sigma A2095 and A2220, respectively) to the cleared supernatant fraction. After incubation at 4 °C for 2 h, beads were collected by centrifugation at 5,000 g for 30 s at 4 °C followed by extensive washes in lysis buffer, and finally boiled in 20 µl 2× Laemmli sample buffer (Sigma S3401).
Western blots and densitometry
Equal amounts of proteins were loaded and separated by denaturing SDS-PAGE and transferred to Immobilon-P PVDF membrane (Millipore/Biocenter IPVH00010) in transfer buffer (0.025 M Trizma base, 0.2 M glycine, 10% methanol). Membranes were blocked in CBB (casein blocking buffer, 0.5% casein [Sigma C3400] dissolved in 0.1 M Trizma base, followed by adjusting pH to 7.5 with HCl) for 1 h at room temperature, washed 3 × 5 min in TBST (0.025 M Trizma base pH adjusted to 7.5 with HCl, 0.9% NaCl, 0.1% Tween 20), incubated with primary antibodies in 1:1 CBB (casein blocking buffer)/TBST for 1 h at room temperature, followed by 3 × 10 min washes in TBST. Blots were then incubated with AP (alkaline phosphatase) conjugated goat anti-mouse, anti-rabbit (Millipore/Biocenter, AP124A and AP132A, respectively) or rabbit anti-rat (Sigma, A6066) secondary antibodies diluted 1:5,000 in 1:1 CBB (casein blocking buffer)/TBST for 1 h at room temperature. Blots were washed 3 × 10 min in TBST and 1 × 5 min in AP (alkaline phosphatase) buffer (100 mM Trizma base, 100 mM NaCl, 5 mM MgCl2, 0,05% Tween 20, pH adjusted to 9.5 with HCl) and then developed using NBT-BCIP (Sigma, 72091) diluted 1:50 in AP (alkaline phosphatase) buffer.
All western blots were repeated on independent biological samples with similar results, and representative examples are shown. For fat body lysates, 25 to 30 fat bodies were dissected and pooled per genotype. The ratio of slower vs. faster mobility Atg13 forms (upper bands vs. lowest band) was calculated by densitometry using ImageJ (National Institutes of Health). Briefly, lanes were selected using the Rectangular Selection tool, and plots were drawn using Plot Lanes. A Straight Line tool was used to enclose the peak corresponding to the bands of interest, peaks were highlighted by the Wand tool, and Label Peaks was used to collect data.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Sarolta Pálfia, Zsófia Kováts, Eszter Papp, and Eszter Vágó for technical assistance, Attila L Kovács for comments on the manuscript, colleagues and public repositories listed in the methods section for reagents, and the Hungarian Scientific Research Fund (OTKA K83509), Wellcome Trust (087518/Z/08/Z) and Hungarian Academy of Sciences (BO/00552/11) for support.
Glossary
Abbreviations:
- Atg
autophagy-related
- C12orf44
chromosome 12 open reading frame 44
- Cp1
cysteine proteinase-1
- Cvt
cytoplasm-to-vacuole targeting
- DAPI
4',6-diamidino-2-phenylindole
- DN
dominant-negative
- FIP200
200 kDa FAK family kinase-interacting protein
- Gal4
galactose metabolism 4
- Lamp1
lysosomal-associated membrane protein 1
- LTR
LysoTracker Red
- PAS
phagophore assembly site
- MTOR
mechanistic target of rapamycin
- PBS
phosphate-buffered saline
- PBTX-DOC
PBS with Triton X-100 and sodium deoxycholate
- QF
Q factor
- QUAS
Q factor upstream activation sequence
- RB1CC1
RB1-inducible coiled-coil 1
- Ref(2)P
refractory to sigma P
- SQSTM1
sequestosome 1
- TBST
tris-buffered saline and Tween 20
- TOR
target of rapamycin
- Tsc
tuberous sclerosis complex
- UAS
upstream activation sequence
- ULK
unc-51 like autophagy activating kinase
References
- 1.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Juhász G. Interpretation of bafilomycin, pH neutralizing or protease inhibitor treatments in autophagic flux experiments: novel considerations. Autophagy. 2012;8:1875–6. doi: 10.4161/auto.21544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13:453–60. doi: 10.1038/ncb2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–83. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–6. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klionsky DJ, Cregg JM, Dunn WA, Jr., Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–45. doi: 10.1016/S1534-5807(03)00296-X. [DOI] [PubMed] [Google Scholar]
- 8.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Klionsky DJ. The regulation of autophagy - unanswered questions. J Cell Sci. 2011;124:161–70. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Juhász G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–6. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, Finley KD. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy. 2008;4:176–84. doi: 10.4161/auto.5269. [DOI] [PubMed] [Google Scholar]
- 12.Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci. 2012;37:230–6. doi: 10.1016/j.tibs.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, Rusten TE, Stenmark H, Brech A. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180:1065–71. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 15.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi: 10.1083/jcb.200712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–9. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Chan EY, Longatti A, McKnight NC, Tooze SA. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29:157–71. doi: 10.1128/MCB.01082-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erdi B, Nagy P, Zvara A, Varga A, Pircs K, Ménesi D, Puskás LG, Juhász G. Loss of the starvation-induced gene Rack1 leads to glycogen deficiency and impaired autophagic responses in Drosophila. Autophagy. 2012;8:1124–35. doi: 10.4161/auto.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bánréti Á, Lukácsovich T, Csikós G, Erdélyi M, Sass M. PP2A regulates autophagy in two alternative ways in Drosophila. Autophagy. 2012;8:623–36. doi: 10.4161/auto.19081. [DOI] [PubMed] [Google Scholar]
- 21.Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. 2009;20:2004–14. doi: 10.1091/mbc.E08-12-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takáts S, Nagy P, Varga Á, Pircs K, Kárpáti M, Varga K, Kovács AL, Hegedűs K, Juhász G. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol. 2013;201:531–9. doi: 10.1083/jcb.201211160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pircs K, Nagy P, Varga A, Venkei Z, Erdi B, Hegedus K, Juhasz G. Advantages and limitations of different p62-based assays for estimating autophagic activity in Drosophila. PLoS One. 2012;7:e44214. doi: 10.1371/journal.pone.0044214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itakura E, Mizushima N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011;192:17–27. doi: 10.1083/jcb.201009067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 2007;12:209–18. doi: 10.1111/j.1365-2443.2007.01050.x. [DOI] [PubMed] [Google Scholar]
- 27.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–76. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SB, Kim S, Lee J, Park J, Lee G, Kim Y, Kim JM, Chung J. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007;8:360–5. doi: 10.1038/sj.embor.7400917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7:1212–21. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rusten TE, Lindmo K, Juhász G, Sass M, Seglen PO, Brech A, Stenmark H. Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev Cell. 2004;7:179–92. doi: 10.1016/j.devcel.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–48. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–6. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juhász G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, Neufeld TP. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol. 2008;181:655–66. doi: 10.1083/jcb.200712051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, Noda T, Ohsumi Y. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol. 1999;147:435–46. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198:219–33. doi: 10.1083/jcb.201202061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jao CC, Ragusa MJ, Stanley RE, Hurley JH. A HORMA domain in Atg13 mediates PI 3-kinase recruitment in autophagy. Proc Natl Acad Sci U S A. 2013;110:5486–91. doi: 10.1073/pnas.1220306110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dorsey FC, Rose KL, Coenen S, Prater SM, Cavett V, Cleveland JL, Caldwell-Busby J. Mapping the phosphorylation sites of Ulk1. J Proteome Res. 2009;8:5253–63. doi: 10.1021/pr900583m. [DOI] [PubMed] [Google Scholar]
- 38.Kim M, Park HL, Park HW, Ro SH, Nam SG, Reed JM, Guan JL, Lee JH. Drosophila Fip200 is an essential regulator of autophagy that attenuates both growth and aging. Autophagy. 2013;9:1201–13. doi: 10.4161/auto.24811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010;30:1049–58. doi: 10.1128/MCB.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, Stoffel I, Brezovich A, Verma M, Hansmann I, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J. 2012;31:3691–703. doi: 10.1038/emboj.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki K. Selective autophagy in budding yeast. Cell Death Differ. 2013;20:43–8. doi: 10.1038/cdd.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott SV, Nice DC, 3rd, Nau JJ, Weisman LS, Kamada Y, Keizer-Gunnink I, Funakoshi T, Veenhuis M, Ohsumi Y, Klionsky DJ. Apg13p and Vac8p are part of a complex of phosphoproteins that are required for cytoplasm to vacuole targeting. J Biol Chem. 2000;275:25840–9. doi: 10.1074/jbc.M002813200. [DOI] [PubMed] [Google Scholar]
- 43.Potter CJ, Tasic B, Russler EV, Liang L, Luo L. The Q system: a repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell. 2010;141:536–48. doi: 10.1016/j.cell.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toda H, Mochizuki H, Flores R, 3rd, Josowitz R, Krasieva TB, Lamorte VJ, Suzuki E, Gindhart JG, Furukubo-Tokunaga K, Tomoda T. UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 2008;22:3292–307. doi: 10.1101/gad.1734608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nagy P, Varga A, Pircs K, Hegedűs K, Juhász G. Myc-driven overgrowth requires unfolded protein response-mediated induction of autophagy and antioxidant responses in Drosophila melanogaster. PLoS Genet. 2013;9:e1003664. doi: 10.1371/journal.pgen.1003664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.