

Abstract
Multiple genetic studies have implicated the autophagy-related gene, ATG16L1, in the pathogenesis of Crohn disease (CD). While CD-related research on ATG16L1 has focused on the functional significance of ATG16L1 genetic variations, the mechanisms underlying the regulation of ATG16L1 expression are unclear. Our laboratory has described that microRNAs (miRNAs), key regulators of gene expression, are dysregulated in CD. Here, we report miRNA-mediated regulation of ATG16L1 in colonic epithelial cells as well as Jurkat T cells. Dual luciferase reporter assays following the transfection of vectors containing the ATG16L1 3′-untranslated region (3′UTR) or truncated 3′UTR fragments suggest that the first half of ATG16L1 3′UTR in the 5′ end is more functional for miRNA targeting. Of 5 tested miRNAs with putative binding sites within the region, MIR142-3p, upon transient overexpression in the cells, resulted in decreased ATG16L1 mRNA and protein levels. Further observation demonstrated that the luciferase reporter vector with a mutant MIR142-3p binding sequence in the 3′UTR was unresponsive to the inhibitory effect of MIR142-3p, suggesting ATG16L1 is a gene target of MIR142-3p. Moreover, the regulation of ATG16L1 expression by a MIR142-3p mimic blunted starvation- and L18-MDP-induced autophagic activity in HCT116 cells. Additionally, we found that a MIR142-3p inhibitor enhanced starvation-induced autophagy in Jurkat T cells. Our study reveals MIR142-3p as a new autophagy-regulating small molecule by targeting ATG16L1, implying a role of this miRNA in intestinal inflammation and CD.
Keywords: ATG16L1, autophagy, Crohn disease, microRNA, MIR142-3p
Introduction
Although the pathogenesis of inflammatory bowel disease (IBD) is poorly understood, IBD is generally considered to occur in genetically susceptible individuals resulting in an abnormal inflammatory response to commensal microbes and other environmental factors.1,2 Growing evidence indicates that autophagy may play a significant role in the pathogenesis of multiple diseases, including IBD.3-5 Genome-wide association studies have defined 2 autophagy-related genes, ATG16L1 (autophagy-related 16-like 1 [S. cerevisiae]) and IRGM (immunity-related GTPase family, M), as susceptibility genes for Crohn disease, a major subtype of IBD.6-8 Functional characterizations of the CD-associated genetic variations in ATG16L1 and IRGM have highlighted the importance of intact autophagic activity in the control of intestinal inflammation and CD. One study observed that suppressed autophagy or the presence of ATG16L1 genetic variations increases proinflammatory cytokine response in human primary immune cells after activation of NOD2 (nucleotide-binding oligomerization domain containing 2).9 Studies performed on mice with genetically engineered mutations in ATG16L1 confirmed a strong relationship between impaired autophagy and elevated inflammatory cytokine signaling associated with CD.10,11 This conclusion was supported by another study demonstrating that norovirus-infected hypomorphic ATG16L1 mice exhibit defects in autophagy and increased susceptibility to colitis induced by dextran sodium sulfate.12 ATG16L1 is absolutely required for autophagy machinery via participating in the formation of vesicular autophagosomes.4,13 However, the mechanisms governing the regulation of ATG16L1 expression are less known.
MicroRNAs are small noncoding RNA molecules that regulate up to 30% of human protein-coding genes by targeting the 3′-untranslated region of mRNAs, resulting in mRNA cleavage and/or translational arrest.14 The selectivity of miRNA/target gene pairs is determined mainly by the strict sequence complementarity between the seed region (nucleotides (nt) 2 to 7 at the 5′ end) of a miRNA and the miRNA recognition element in the target mRNA.15,16 The emergence of miRNAs as regulators in autophagy pathways has heightened the understanding of the role of autophagy in the pathogenesis of human diseases.17 Over a dozen miRNAs have recently been identified that directly regulate autophagic signaling and inhibit autophagic activity in certain cancer cell lines, including MIR30A (targets BECN1 and ATG5),18,19 MIR101-1 (targets STMN1, RAB5A, and ATG4D),20 MIR130A (targets ATG2B and DICER1),21 MIR375 (targets ATG7),22 MIR376B (targets BECN1 and ATG4C),23 MIR630 (targets ATG12 and UVRAG),24 and MIR885-3p (targets ULK2 and ATG16L2).25 We recently reported that MIR106B regulates multiple autophagy-related genes, including ATG16L1, in intestinal epithelial HCT116 cells.26 Interestingly, a single nucleotide polymorphism in the IRGM coding region alters MIR196 binding, leading to lower IRGM expression.27 In addition, MIR519A targets ATG16L1 in the cisplatin-sensitive SCC cell line.24 Taken together, these findings support the notion that miRNAs regulate autophagy-related genes and autophagy regulators, thereby largely influencing the efficacy of autophagy.
Our laboratory has demonstrated that miRNA dysregulation is of clinical significance in patients with IBD.28-30 Specifically, we have demonstrated that MIR192, an ulcerative colitis-associated miRNA, modulates the expression of macrophage inflammatory peptide-2α and NOD2 in intestinal epithelial cells.28,31 In the present study, we report a novel mature miRNA, MIR142-3p, that regulates autophagy by targeting ATG16L1 in human colonic epithelial cells. These MIR142-3p effects on ATG16L1 expression and autophagy were confirmed in Jurkat T cells.
Results
ATG16L1 3′UTR is functional
The ATG16L1 (RefSeq NM_030803) 3′UTR consists of a 1324 nt sequence. To determine whether the full-length 3′UTR of ATG16L1 or specific regions of the 3′UTR could modulate ATG16L1 gene expression, we generated a pMIR luciferase reporter vector bearing the ATG16L1 3′UTR and used the 3′UTR vector as a template to generate 3 additional vectors containing truncated 3′UTR fragments by PCR cloning (Fig. 1A). Fragment 1 (F1), F2, and F3 were 384, 697, and 1071 nt in length, respectively (all starting at the 5′ end of the wild-type (WT) 3′UTR). By transfecting these vectors into HCT116 cells, we found that the WT 3′UTR reduced luciferase activity by 75.7% when compared with the pMIR-GLO vector control (Fig. 1B), suggesting that the 3′UTR of ATG16L1 is possibly responsive to miRNA regulation and plays a significant role in regulating ATG16L1 gene expression. In contrast, the truncated F1, F2, and F3 3′UTR vectors reduced luciferase activities by 46.8%, 62%, and 71%, respectively (P values < 0.01 vs the pMIR-GLO control vector). With respect to the total reduction rate of the WT 3′UTR, the F1 sequence, which comprises less than one-third of the full-length 3′UTR, contributed 61.8% of the total reduction; the F2 sequence, which comprises about half of the full-length 3′UTR, contributed 81.9% of the total reduction. In addition, there were significant reductions in the luciferase activity between F1 and F2 (P < 0.01) and between F2 and F3 (P < 0.05), but no difference between F3 and the WT 3′UTR was found. These data suggest that the ATG16L1 3′UTR contains multiple functional miRNA binding sites and those binding sites locating within the promixal 5′ end of the 3′UTR play a more important regulatory role.
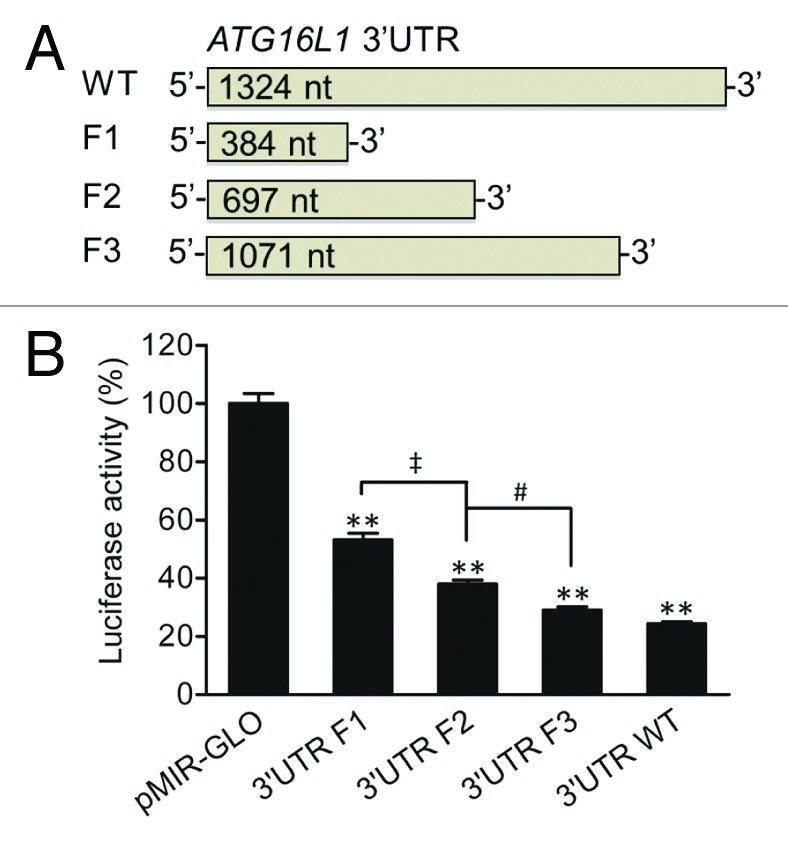
Figure 1.ATG16L1 3′UTR reduces firefly luciferase activity. (A) pMIR-GLO reporter vectors were constructed containing the ATG16L1 WT 3′UTR or truncated 3′UTR fragments, F1, F2, and F3, which were 384, 697, and 1071 nt in length, respectively. (B) HCT116 cells were transfected with the vectors containing the WT, F1, F2 or F3 ATG16L1 3′UTR or a pMIR-GLO vector control (200 ng/ml or 50 ng/well in 48-well plates). Firefly luciferase activity was measured and normalized to renilla luciferase activity. The data are expressed as the mean ± SEM (n = 4). **P < 0.01 vs the control pMIR-GLO vector. ‡P < 0.01 vs the 3′UTR F1. #P < 0.05 vs the 3′UTR F2.
MIR142-3p inhibits ATG16L1 expression
We performed an in silico analysis for putative miRNA binding sites spanning the F1 and F2 regions of the ATG16L1 3′UTR using several web-based bioinformatics tools including MicroCosm and PicTar, and identified 5 potential miRNA binding sites for MIR30B*, MIR142-3p, MIR505*, MIR548B-3p, and MIR770-5p (Fig. 2A). All 5 miRNAs were expressed in HCT116 cells at varying levels, with MIR770-5p being the highest expressed miRNA and MIR142-3p being the lowest expressed miRNA (Fig. 2B). Next, we examined the effects of these miRNAs on ATG16L1 expression by transfecting miRNA mimics into HCT116 cells. Real-time RT-PCR analysis showed that, when compared with the nonspecific mimic negative control (MIRNC), the transfection with the MIR142-3p mimic significantly reduced ATG16L1 mRNA expression by approximately 30% while the other 4 miRNA mimics had no significant inhibitory effects (Fig. 3A). Western blot analysis further demonstrated that the transfection of the MIR142-3p mimic resulted in decreased protein expression of the ATG16L1 isoform 1 and isoform 2 by 43% and 31%, respectively. In contrast, the mimics of 4 other miRNAs had weak or no effect on the ATG16L1 isoforms 1/2 protein expression (Fig. 3B). These data suggest that ATG16L1 might be a gene target for MIR142-3p.
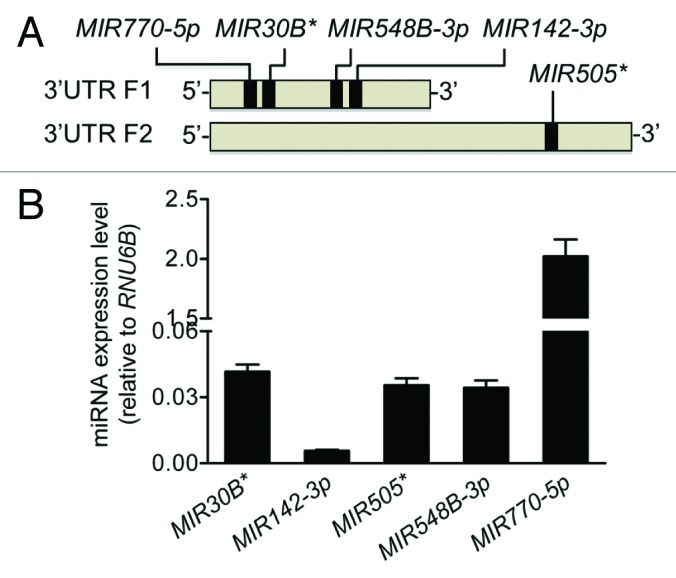
Figure 2. Predicted miRNAs are expressed in HCT116 cells. (A) An in silico analysis identified MIR30B*, MIR142-3p, MIR505*, MIR548B-3p, and MIR770-5p as having putative binding sites in the first half of the ATG16L1 3′UTR in the 5′ end. (B) The endogenous expression levels of the 5 putative miRNAs were analyzed by mature miRNA RT-PCR and normalized to RNU6B. The data are expressed as the mean ± SEM (n = 3).
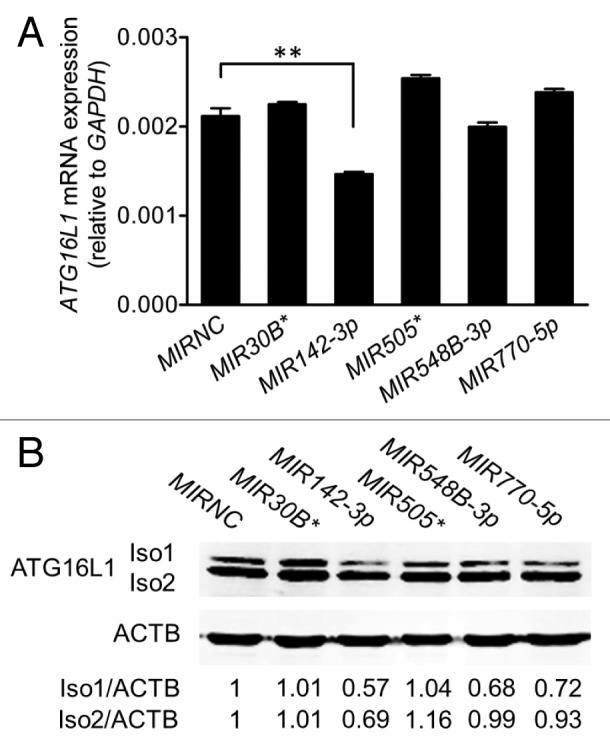
Figure 3.MIR142-3p regulates ATG16L1 expression. HCT116 cells were transfected with individual miRNA mimics or a mimic negative control (MIRNC) (50 nM). At 24 h post-transfection, (A) total RNA was extracted and assayed for ATG16L1 mRNA expression by real-time RT-PCR. The data are expressed as the mean ± SEM (n = 3). **P < 0.01 vs MIRNC; (B) whole cell lysates were assayed by western blot for ATG16L1 protein expression. The density of ATG16L1 isoform 1 (Iso1) and isoform 2 (Iso2) bands was quantitated using the Odyssey infrared imaging system and normalized to that of the corresponding loading control ACTB with the same treatment. Representative bands are shown.
In addition, MIR142-3p inhibitor was expected to upregulate ATG16L1 gene and protein expression levels in HCT116 cells, however, we did not detect visible influences (Fig. S1A and S1B), possibly due to low endogenous expression levels of MIR142-3p. Since MIR142-3p is mainly expressed and functionally active in the hematopoietic system,32 we demonstrated that MIR142-3p inhibitor transfection resulted in significant upregulation of ATG16L1 gene and protein expression in Jurkat T cells (Fig. S1C and S1D).
ATG16L1 is a gene target of MIR142-3p
To further assess the potential role of MIR142-3p in the modulation of ATG16L1 expression, we cotransfected the 5 individual miRNA mimics with the ATG16L1 3′UTR F1 vector. We found that in sharp contrast to the other 4 miRNA mimics, the MIR142-3p mimic significantly decreased the luciferase activity by 52.9% (Fig. 4A), indicating MIR142-3p specifically regulates the ATG16L1 3′UTR F1 function. To evaluate whether the effect of the MIR142-3p mimic is through its interaction with the predicted MIR142-3p binding sequence, the binding sequence of MIR142-3p at position 254 to 264 in the ATG16L1 3′UTR F1 was mutated and cloned in the pMIR-GLO vector (Fig. 4B). We cotransfected HCT116 cells with the WT or mutated ATG16L1 3′UTR F1 and the MIR142-3p mimic or MIRNC. We found that the inhibitory effect of MIR142-3p mimic transfection on luciferase activity was abrogated in the mutated 3′UTR F1 vector (Fig. 4C). In addition, even in the absence of MIR142-3p mimic transfection, the mutated 3′UTR F1 vector demonstrated an increased luciferase activity when compared with the WT 3′UTR F1 vector (Fig. 4C), strongly supporting the hypothesis that the MIR142-3p binding sequence in the ATG16L1 3′UTR F1 is involved in the direct interaction of MIR142-3p with the ATG16L1 3′UTR and is a contributor for the regulatory function of ATG16L1 3′UTR.
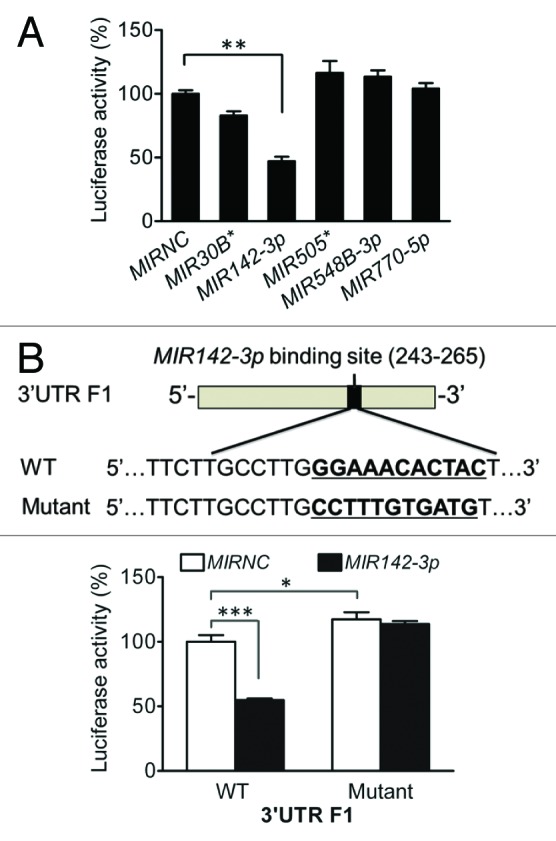
Figure 4.MIR142-3p targets the ATG16L1 3′UTR. (A) HCT116 cells were cotransfected with the F1 vector (200 ng/ml) and a miRNA mimic (10 nM), and luciferase reporter assays were performed 48 h after transfection. The data are expressed as the mean ± SEM (n = 4). **P < 0.01 vs the MIRNC. (B) To validate the predicted MIR142-3p binding sequence in the ATG16L1 3′UTR, 11 nucleotides at position 254 to 264 in the truncated F1 vector were substituted. (C) HCT116 cells were cotransfected with the WT or mutated F1 (200 ng/ml) vector and the MIR142-3p mimic or MIRNC (10 nM). At 48 h post-transfection, dual luciferase activities were determined. The data are expressed as the mean ± SEM (n = 4). *P < 0.05 and ***P < 0.001 vs the MIRNC.
To confirm ATG16L1 as a target gene of MIR142-3p, we reproduced the results in intestinal epithelial Caco-2 cells. Transfection of the vector bearing ATG16L1 3′UTR F1 reduced luciferase activity by 52% when compared with the pMIR-GLO vector control (Fig. S2A) and by cotransfecting Caco-2 cells with the WT or mutated ATG16L1 3′UTR F1 and the MIR142-3p mimic or MIRNC, we found that the MIR142-3p mimic further reduced luciferase activity of the WT 3′UTR F1 by 45.5% but had no effect on the mutated counterpart (Fig. S2B). Furthermore, MIR142-3p inhibited the basal levels of ATG16L1 gene and protein expression with respect to MIRNC (Fig. S2C and S2D).
MIR142-3p inhibits starvation-induced autophagy
In order to evaluate the functional consequence of MIR142-3p regulation of ATG16L1, we determined the effect of individual miRNA mimic transfection on the intracellular autophagic activity. Autophagy can be monitored at different steps, but most commonly used methods touch upon LC3 turnover and SQSTM1/p62 (sequestosome 1) degradation.33,34 Therefore, we used the conversion of LC3-I to LC3-II and the increasing number of LC3-II puncta as well as the decreased protein level of SQSTM1 as markers for autophagic activity. It should be noted that the transfection process itself is somewhat toxic to the cells and may induce autophagy.35 As indicated in Figure 5A, HCT116 cells exposed to MIRNC transfection showed increased ratio of LC3-II/LC3-I and decreased SQSTM1 protein expression when compared with the non-transfection control. Interestingly, increased autophagy by the transfection process was partially inhibited by the MIR142-3p mimic. Figure 5B shows that starvation-induced autophagy was readily apparent in transfected cells, as evidenced by a further increase in the LC3-II/LC3-I ratio and a decrease in SQSTM1 protein expression. Therefore, in this study, we compared the effects of several miRNA mimics on starvation-induced autophagy. When compared with MIRNC, the MIR142-3p mimic reduced the conversion of LC3-I to LC3-II and increased SQSTM1 protein expression as demonstrated by western blot analysis (Fig. 5B), and reduced the formation of LC3-II puncta as observed under fluorescence microscopy (Fig. 5C and D). In addition, the MIR142-3p mimic marginally reduced the number of puncta in unstarved transfected cells (P = 0.052). In contrast, the other 4 miRNA mimics showed no or weak inhibitory effects in both unstarved and starved cells. Although the MIR142-3p mimic affects the ratios of LC3-II/LC3-I, it showed no effect on total MAP1LC3B mRNA expression (Fig. 5E). These data suggest a causal relationship between the inhibition of ATG16L1 expression by MIR142-3p and the decreased autophagic activity induced by starvation.

Figure 5.MIR142-3p regulates starvation-induced autophagy in HCT116 cells. HCT116 cells were transfected with a miRNA mimic or MIRNC (50 nM). At 24 h post-transfection, cells were directly collected (A) or incubated in EBSS for autophagy induction for another 4 h (B). Autophagy was monitored based on the LC3 turnover and SQSTM1 degradation by western blot. The band densities were quantitated and the ratios of LC3-II/LC3-I and SQSTM1/GAPDH (or ACTB) were calculated and normalized. (C) LC3-II puncta were visualized by confocal imaging of cells immunostained for nucleus and LC3 using DAPI (blue) and Alexa Fluor 488 secondary antibody conjugates (green), respectively. Arrow indicates increasing LC3-II puncta. Representative imaging are shown. (D) LC3-II puncta per cell were quantitated by randomly counting 20 cells for each treatment group and the data are expressed as the mean ± SEM (n = 20). (E) Real-time RT-PCR analysis of MAP1LC3B mRNA expression. The data were expressed as the mean ± SEM (n = 6).
To confirm the relationship between MIR142-3p regulation of ATG16L1 expression and subsequent autophagic activity, we assessed the influence of a MIR142-3p inhibitor on autophagic activity in Jurkat T cells. Cells were transfected with a MIR142-3p inhibitor or negative control and starved for 2 h. After saponin-mediated release of soluble LC3-I, flow cytometry was performed for LC3-II, using a previously described method.36,37 The data demonstrate that inhibition of MIR142-3p in Jurkat T cells results in the increased autophagic activity induced by starvation (Fig. 6).

Figure 6.MIR142-3p regulates starvation-induced autophagy in Jurtak T cells. Jurkat T cells were transfected with a MIR142-3p inhibitor or control inhibitor (50 nM). After overnight culture, cells were incubated in EBSS for 2 h. Cell pellets were treated with 0.05% saponin to release soluble LC3-I and then incubated with anti-LC3B antibody or isotype control antibody. Flow cytometry was performed after incubation with an Alexa Fluor 488 anti-rabbit secondary antibody. Representative scatter plots and histograms of flow cytometry for the (A) isotype control antibody and for LC3-II in (B) MIR142-3p inhibitor-transfected cells and (C) control inhibitor-transfected cells are presented. LC3-II was increased in cells transfected with the MIR142-3p inhibitor as compared with the control inhibitor. Data represents 2 independent experiments.
MIR142-3p modulates NOD2-dependent autophagy and IL8 mRNA expression
NOD2 is another identified susceptibility gene for CD.38,39 Recent studies suggest that the NOD2 pathway and autophagy are functionally cross-regulated.40-42 NOD2 signaling activates autophagy, with the latter proven to play an active role in intracellular bacterial clearance.40-42 Therefore, we investigated the effect of MIR142-3p mimic transfection on NOD2-dependent autophagic activity. HCT116 cells were treated with L18-MDP, a synthetic derivative of NOD2 ligand muramyl dipeptide, for 2 h. We found that L18-MDP induced autophagy as indicated by increased LC3-II/LC3-I ratio and decreased SQSTM1 amount, but the degradation of LC3-II and SQSTM1 could be blocked by the lysosome inhibitor bafilomycin A1 (Fig. 7A). As expected, MIR142-3p mimic transfection inhibited the autophagic response to L18-MDP, as indicated by the decreased LC3-II/LC3-I ratio. Furthermore, it is know that autophagy could negatively regulate the production of certain inflammatory cytokines (e.g., IL1B and IL18), but positively regulate the secretion of other cytokines (e.g., IL8 and TNF).43 Our another manuscript observes that the IL8 gene is significantly upregulated in HCT116 cells upon MDP stimulation,31 so we determined L18-MDP-induced IL8 gene expression in HCT116 cells transfected with MIR142-3p. The results showed that the MIR142-3p mimic significantly inhibited IL8 mRNA expression (Fig. 7B), supporting the positive regulation of autophagy for IL8.
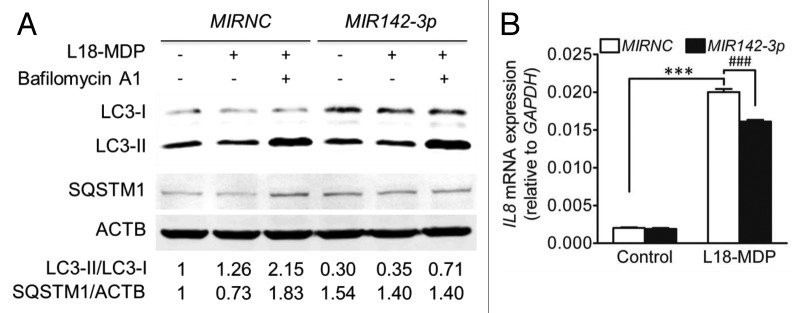
Figure 7.MIR142-3p regulates L18-MDP-induced autophagy and IL8 gene expression. (A) HCT116 cells were transfected with the MIR142-3p mimic or MIRNC (50 nM) and at 48 h post-transfection, cells were treated L18-MDP (100 ng/ml) in the presence or absence of bafilomycin A1 (100 nM) for 2 h. Autophagy was monitored by western blot analysis of LC3 turnover and SQSTM1 degradation. (B) As in A, cells were transfected with the MIR142-3p mimic or MIRNC (10 nM) and at 24 h post-transfection, cells were treated with L18-MDP (100 ng/ml) in fresh complete medium for 4 h. L18-MDP-induced inflammatory response was measured using increasing IL8 mRNA expression as an indicator. The data are expressed as the mean ± SEM (n = 3). ***P < 0.001 vs the baseline control transfected with MIRNC. ###P < 0.001 vs the L18-MDP-treated control transfected with MIRNC.
MIR142-3p overexpression increases starvation-induced cell death
It is known that starvation induces cell death and under starvation conditions, autophagy is an important cell survival mechanism by which cytoplasmic materials are degraded for energy metabolism and protein synthesis essential for cell survival.44,45 Therefore, we investigated the influence of MIR142-3p regulation of ATG16L1 and autophagy on cell death and apoptosis induced by starvation. HCT116 cells were transfected with the MIR142-3p mimic and then deprived of amino acids and serum for 24 h. Figure 8A illustrates that starvation resulted in significantly reduced cell viability and MIR142-3p overexpression further decreased cell viability. Similarly, starvation induced apoptosis and MIR142-3p mimic transfection further enhanced apoptosis (Fig. 8B). In addition, we further determined cell death and apoptosis by using ANXA5/annexin V and 7-AAD (7-aminoactinomycin D) staining. The results show that MIR142-3p transfection increased the percentage of ANXA5-positive cells and 7-AAD-positive cells (Fig. S3). These data suggest that, in agreement with previous studies,44,45 autophagy is a cell survival mechanism under starvation conditions and suppressed autophagy may exacerbate starvation-induced cell death and apoptosis.
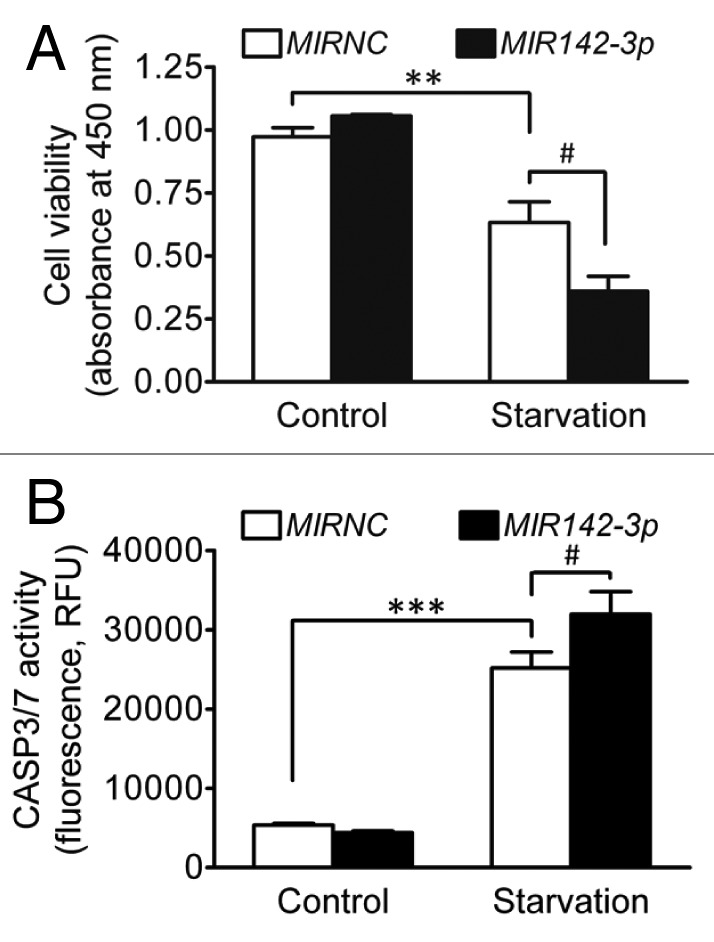
Figure 8.MIR142-3p increases cell death induced by starvation. HCT116 cells were seeded in 96-well plates. After transfection with the MIR142-3p mimic or MIRNC (50 nM), cells were incubated in normal complete medium or in EBSS for starvation induction for 24 h. (A) Cell viability was determined using a colorimetric assay kit. (B) Apoptosis was determined by assaying the CASP3 and CASP7 activities. RFU indicates relative fluorescence units. The data are expressed as the mean ± SEM (n = 3). **P < 0.01 and ***P < 0.001 vs the unstarved control transfected with MIRNC. #P < 0.05 vs the starved control transfected with MIRNC.
MIR142-3p has no regulatory effects on other putative target genes associated with autophagy
In addition to ATG16L1, there are several other autophagy-related genes and autophagy regulators that are predicted target genes of MIR142-3p, including ATG5, ATG14, GABARAPL1, RICTOR, and TP53INP2.17 In order to rule out the possibility of the MIR142-3p regulation of multiple autophagy-related or -regulating genes that results in a lower autophagic activity, we assessed the effect of MIR142-3p overexpression on the mRNA expression levels of these putative target genes in HCT116 cells and our data did not find a significant influence (Fig. S4A–S4E). ATG5 participates in autophagy by conjugating to ATG12 and then to ATG16L1. Therefore we also determined if the MIR142-3p mimic affects ATG5 protein expression and found that it has no negative regulatory effect (Fig. S4F).
Starvation coinduces ATG16L1 and MIR142-3p
To understand the physiological changes of ATG16L1 gene expression and MIR142-3p expression following a short-term of starvation, ATG16L1 mRNA expression and endogenous mature MIR142-3p expression were assessed in HCT116 cells that were deprived of nutrients for different time points (0 to 6 h) (Fig. 9). The results showed that there was a modest but statistically significant increase in the ATG16L1 gene expression shortly after starvation induction, which peaked at about 1 h and was followed by a gradual decrease. ATG16L1 gene expression returned to the baseline levels at about 6 h. Interestingly, starvation also induced MIR142-3p expression. However, the increased MIR142-3p expression lagged behind the increased ATG16L1 mRNA expression. The increased MIR142-3p corresponded with the decreasing ATG16L1 expression, suggesting that the delayed increase in the expression of MIR142-3p may help control the magnitude of ATG16L1 gene expression.
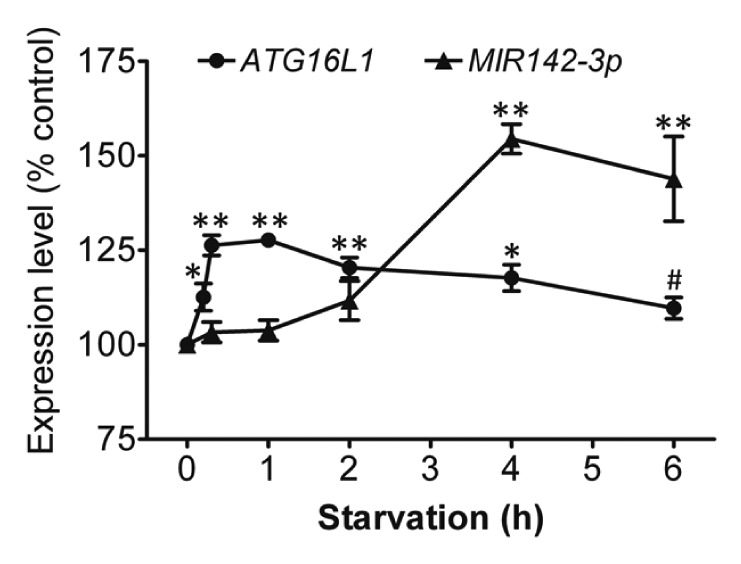
Figure 9. Starvation coinduces ATG16L1 and MIR142-3p. HCT116 cells were incubated in EBSS for different time points (0 to 6 h), then the relative expression levels of ATG16L1 mRNA and endogenous mature MIR142-3p were assessed by qRT-PCR and normalized with GAPDH and RNU6B, respectively. The expression levels of both ATG16L1 and MIR142-3p for the unstarved controls are considered as 100%. The data are expressed as the mean ± SEM (n = 3). *P < 0.05 and **P < 0.01 vs the corresponding unstarved control. #P < 0.05 vs the ATG16L1 mRNA expression at 1 h.
Discussion
It has been shown that impaired autophagy is intimately associated with the pathogenesis of many human diseases.3-5 For example, decreased expression of BECN1 is associated with the development or progression of human malignancies.46-48 Autophagy is a complex signaling process involving over 35 autophagy-related gene products and numerous autophagy-regulating molecules that act in concert.13 ATG16L1 is required for autophagy through the involvement of ATG12-ATG5-ATG16L1 complexes in the formation of autophagosomes.13 ATG16L1 has drawn special attention recently due to the identification of ATG16L1 as a risk gene for CD.6,7 Human and animal studies have demonstrated that defects in ATG16L1 function are closely associated with the elevated intestinal inflammation and aberrant Paneth cell function usually seen in CD.9-12 Autophagy may represent an important mechanism in the interaction of host genetic susceptibility and immune dysregulation that contribute to the pathogenesis of CD.49 Therefore, exploring how ATG16L1 and autophagy pathways are regulated may increase our understanding of the pathogenesis of CD. miRNAs represent a novel mechanism for the post-transcriptional regulation of gene expression. Our laboratory has reported that miRNAs were differentially expressed in the peripheral blood and intestinal tissues of IBD patients.28-30 Herein, we provided the first evidence of miRNA regulation of human ATG16L1 expression in intestinal epithelial cell lines as well as Jurkat T cells.
Of the 5 tested miRNAs that have putative binding sites in the ATG16L1 3′UTR, MIR142-3p was found to have the most significant effect on ATG16L1 expression. The negative regulation of ATG16L1 expression by MIR142-3p was confirmed by transfecting HCT116 and Caco-2 cells with a MIR142-3p mimic as well as transfecting Jurkat T cells with a MIR142-3p inhibitor. miRNAs are known to affect target mRNA stability and/or mRNA translation, either case ultimately leading to a decreased protein expression and function. Translational inhibition may occur if the complementary sequences between the paired miRNA and mRNA do not match well;15 mRNA degradation will possibly occur if there is a relatively perfect complementarity between the pair.16 Our data suggest that MIR142-3p regulates ATG16L1 expression possibly via the mRNA degradation mechanism or both mRNA degradation and translational inhibition mechanisms. To validate the binding sequence in the 3′UTR for MIR142-3p, the predicted binding site at position 254 to 264 was substituted. We found that the mutated 3′UTR F1 vector showed increasing luciferase activity and was refractory to the inhibitory effect of MIR142-3p, indicating that the binding site is involved in the MIR142-3p regulatory function.
Moreover, the regulation of ATG16L1 expression by MIR142-3p was evaluated by determining the effect of mimic transfection on the autophagic activity in HCT116 cells. We used the formation of LC3-II and SQSTM1 degradation as markers for autophagy.33,34 The decreased formation of LC3-II (or the decreased LC3-II/LC3-I ratio) and the elevated amount of SQSTM1 in starved HCT116 cells following MIR142-3p mimic transfection suggests that the decreased expression of ATG16L1 by MIR142-3p leads to inhibition of autophagic activity. The influence of MIR142-3p on autophagic activity was confirmed by demonstrating MIR142-3p inhibitor enhancement of starvation-induced autophagy in Jurkat T cells. MIR142-3p regulation of ATG16L1 also affected L18-MDP-induced autophagy and inflammatory response. These are very interesting findings in the context of CD because ATG16L1 and NOD2 are 2 important genes associated with the pathogenesis of CD. Our findings suggest that a dysregulated miRNA may affect both autophagy and NOD2 signaling, consequently compromising the removal of invading bacteria and the control of inflammatory inflammation. Additionally, because autophagy has been implicated in aspects of cell death and apoptosis,44,50 we evaluated the effect of the MIR142-3p mimic on cell viability and apoptosis of HCT116 cells exposed to starvation for 24 h. We found that inhibition of ATG16L1 and autophagy by MIR142-3p enhanced starvation-induced cell death and apoptosis, confirming earlier reports that autophagy is beneficial for cell survival under starvation condition.44,45 Although several other autophagy-related genes, i.e., ATG5, ATG14, and GABARAPL1, and autophagy-regulating genes, i.e., RICTOR and TP53INP2, are also predicted to be targets of MIR142-3p,17 our data suggest that the possibility of MIR142-3p regulation of autophagy through these several autophagy-related and -regulating genes are small, and so far, no other autophagy-related or -regulating genes have been identified as target genes of MIR142-3p.
Luciferase reporter assays with the ATG16L1 3′UTR vectors indicate that ATG16L1 may harbor multiple miRNA binding sites and are therefore subject to the regulation by several miRNAs simultaneously and coordinately. In addition, it is possible that other 3′UTR regulatory elements may play a role in ATG16L1 expression. It has been recently reported in SCC-wt-ΔNp63α cells that MIR519A regulates ATG16L1.24 Hsa-MIR519A has a putative binding site in the ATG16L1 3′UTR with the seed region at the position 1035–1041 near the 3′ end. Our data suggest that the miRNA binding sites at the 5′ end of ATG16L1 3′UTR contribute much more to the 3′UTR regulatory function than do those at the 3′ end. Our present study focuses on MIR142-3p which has a binding site close to the 5′ end of the ATG16L1 3′UTR.
MIR142-3p is broadly conserved among mammals and is crucial for the maturation of hematopoietic cells.51 Growing evidence supports an active involvement of MIR142-3p in the modulation of chronic inflammatory response.52-54 It is upregulated in immune-related disorders including psoriasis,52 systemic lupus erythematosus,55 and systemic sclerosis.56MIR142-3p has also been implicated in the cell differentiation process associated with the development of human cancers such as T cell leukemia,57,58 acute myeloid leukemia,59 and squamous cell carcinoma.60,61 In addition to ATG16L1, several other target genes for MIR142-3p have been identified recently such as Rock2 (Rho-associated, coiled-coil containing protein kinase 2),51Adcy9 (adenylyl cyclase 9) in CD4+ T cells,62NR3C1 [nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor)] in T-leukemic cells,58RAC1 in hepatocellular carcinoma cell lines,63IL6 in dendritic cells,64 and a clock gene, ARNTL/BMAL1, in 293ET and NIH3T3 cell lines.65 In particular, the regulation of dendritic cell responses to endotoxin by targeting IL6 provides robust evidence for MIR142-3p as an anti-inflammatory miRNA.64 Nevertheless, MIR142-3p may exhibit distinct properties in inflammation via the regulation of autophagy. Although until now, miRNA microarray analyses have not disclosed an aberrant expression of MIR142-3p in patients with active vs. inactive CD, this miRNA indeed is dysregulated in IBD-associated inflammation.66 In addition, it was found that MIR142-3p is significantly elevated in chemically induced acute colitis.67 These cumulative data indicate MIR142-3p is involved in various dysregulated inflammatory responses, including IBD-associated intestinal inflammation.
Our data strongly indicate that MIR142-3p may fine-tune the amplitude of ATG16L1 gene expression and the overall autophagic response. However, the role of MIR142-3p in regulating ATG16L1 expression and autophagic activity may depend on the endogenous and inducible levels of MIR142-3p in specific cell populations. For cells with endogenously low expression levels, slight upregulation may cause a significant effect on the target gene expression. This may explain why the exogenous MIR142-3p mimic could produce significant regulatory effects on ATG16L1 expression and autophagic activity. Similarly, for cells with endogenously high MIR142-3p expression levels, a slight reduction may also produce significant regulatory effects on ATG16L1 expression and autophagic activity. Of note, further studies are warranted to confirm the significance of MIR142-3p regulation of ATG16L1 and the autophagy process in vivo. Additionally, due to the dual role of autophagy in controlling cell viability,5 the consequences of MIR142-3p regulation of ATG16L1 and autophagy at different stages of IBD progression need to be determined. Taken together, we demonstrated that MIR142-3p negatively regulates ATG16L1 and autophagic activity, implying a role of this miRNA in the intestinal inflammation and CD. These data may help develop miRNA-based therapeutics and diagnostics for CD.
Materials and Methods
Cell culture
HCT116, Caco-2, and Jurkat cells were previously obtained from American Type Culture Collection and grown in McCoy 5A, DMEM, and RPMI 1640 medium (Cellgro, 10-050-CV, 10-013-CV, and 10-040-CV), respectively, supplemented with 10% fetal bovine serum (Atlanta Biologicals, S11550) at 37 °C in a 5% CO2 incubator. Cells were split regularly and cells between passages 5 and 20 were used in this study. For autophagy induction by starvation, cells were rinsed twice and then incubated in Earle’s balanced salt solution (EBSS; Sigma, E2888) for indicated time. For autophagy induction by L18-MDP (InvivoGen, tlrl-lmdp), cells were incubated in fresh complete medium.
3′UTR construct
The pMIR-GLO reporter vector (Promega, E133A) containing ATG16L1 3′UTR downstream of the firefly luciferase gene was designed then generated from GenScript. This ATG16L1 3′UTR-containing vector was used as a template to create 3 more vectors containing truncated ATG16L1 3′UTR fragments (Fig. 1A) by PCR cloning with the cloning sites SacI and SalI. All 3 fragments started at the 5′ end of the ATG16L1 3′UTR. The forward primer for all 3 fragments was 5′-AATGAGCTCG GGCTCTCAGG G-3′. The reverse primers for F1, F2, and F3 were 5′-AATGTCGACG ACGAGAGAAA GCACAGA-3′, 5′-AATGTCGACC ATTTCCCTGG AAAAGAT-3′, and 5′-AATGTCGACC CAGAGAACAA GCATGGA-3′, respectively. The 3′UTR fragments were amplified using Platinum Taq DNA polymerase (Invitrogen, 10966), and the PCR products were purified using QuickClean II PCR purification kit (GenScript, L00419) and confirmed by agarose gel electrophoresis. Following restriction enzyme digestion and purification, sticky-end ligation into the pMIR-GLO vector was performed using T4 ligase (New England Biolabs, M0202), and then the ligation product was transformed into DH5α competent cells, and screened on LB plates. The accuracy of the truncated 3′UTR fragment inserts was confirmed by complete sequencing analysis (UChicago DNA Sequencing and Genotyping Facility). A mutant of the truncated 3′UTR F1 at position 254 to 264 was created by GenScript.
Transfection
The miRNA mimic negative control (HMC0003) and mimics of MIR30B* (HMI0457), MIR142-3p (HMI0219), MIR505* (HMI0625), MIR548B-3p (HMI0736), and MIR770-5p (HMI0923) were obtained from Sigma. MIR142-3p inhibitor was from Ambion (4464084). HCT116 and Caco-2 cells grown at 30% confluence in appropriate culture plates were transfected with a miRNA mimic, MIR142-3p inhibitor, pMIR-ATG16L1 3′UTR vector or corresponding negative control using Lipofectamine 2000 reagent (Invitrogen, 11668) in OPTI-MEM1 reduced serum medium (Invitrogen, 51985) for 4.5 h according to the manufacturer’s protocol, then the transfection medium was replaced with normal complete medium. Jurkat cells were seeded at 2 × 106 cells per well in 6-well plate and transfected with the MIR142-3p mimic or inhibitor for 24 h, then normal complete medium added. At 24 or 48 h post-transfection, cells were incubated in EBSS or treated with L18-MDP plus bafilomycin A1 (LC Laboratories, B-1080), a lysosome inhibitor, for autophagy induction or directly harvested for evaluating the transfection effects of miRNA mimics, MIR142-3p inhibitor, and pMIR-ATG16L1 3′UTR vectors on the luciferase activity, ATG16L1 expression, or autophagic activity.
Luciferase reporter assay
Cells were lysed in passive lysing buffer and then analyzed for the firefly and renilla luciferase activities using the commercial Dual-Luciferase reporter assay system (Promega, E1910) on the GloMax-multi Detection Luminometer (Promega, E7031, Madison, WI), and the firefly luciferase activity was normalized to the renilla luciferase activity.
Real-time RT-PCR for mRNA
Total RNA was extracted using TRIzol reagent (Invitrogen, 15596018). The integrity, quantity, and purity of RNA were examined using Nano-Drop 1000 Spectrophotometer (Thermo Scientific, Wilmington, DE). For each sample, 500 ng of total RNA was converted to cDNA using the SuperScript VILO cDNA synthesis kit (Invitrogen, 11754) on the C1000 thermal Cycler system (Bio-Rad, 185-1048). The relative amount of tested mRNA to the housekeeping gene GAPDH or ACTB was determined using QuantiFast SYBR Green PCR kit (Qiagen, 204054). Quantitative PCR amplifications were performed on the LightCycler 480 real-time PCR system (Roche Diagnostics, Indianapolis, IN). The thermal profile was 95 °C for 5 min followed by 45 cycles of 95 °C for 10 s, 56 °C for 20 s, and 60 °C for 30 s. The following primer pairs were used: ATG16L1, 5′-CAGTTACGTG GCGGCAGGCT-3′ (forward) and 5′-ACAACGTGCG AGCCAGAGGG-3′ (reverse); ATG5, 5′-GCCATAGCTT GGAGTAGGTT TGGCT-3′ (forward) and 5′-GCGTGAAACA AGTTGGAATT CGTCC-3′ (reverse); ATG14, 5′-GAACACCTAG GCAGGTCAGG-3′ (forward) and 5′- AAAACCGGGG ACTAGGCAAG-3′ (reverse); MAP1LC3B, 5′-ACCATGCCGT CGGAGAAG-3ʹ (forward) and 5ʹ-ATCGTTCTAT TATCACCGGG ATTTT-3′ (reverse); GABARAPL1, 5′-AGGGTCCCCG TGATTGTAGA-3′ (forward) and 5′-TAAGGCGTCC TCAGGTCTCA-3′ (reverse); RICTOR, 5′-GGCCTTGGGA AATATCGGCT-3′ (forward) and 5′-CCTCACAGCA TCCCAGTTGT-3′ (reverse); TP53INP2, 5′-CACCCCAGCA TGTCCGTTTA-3′ (forward) and 5′-CAATTCCCCT TCGCTGAGGT-3′ (reverse); IL8, 5′-CTGCGCCAAC ACAGAAATTA-3′ (forward) and 5′-TGAATTCTCA GCCCTCTTCA A-3′ (reverse); GAPDH, 5′-CGACCACTTT GTCAAGCTCA-3′ (forward) and 5′-AGGGGAGATT CAGTGTGGTG-3′ (reverse); and ACTB, 5′-CTCTTCCAGC CTTCCTTCCT-3′ (forward) and 5′-AGCACTGTGT TGGCGTACAG-3′ (reverse).
Real-time RT-PCR for endogenous mature miRNAs
cDNA was synthesized from total RNA using the NCode VILO miRNA cDNA synthesis kit (Invitrogen, A11169) followed by qPCR using the NCode EXPRESS SYBR GreenER miRNA qRT-PCR kit (Invitrogen, A11193). The thermal profile was 95 °C for 5 min followed by 40 cycles of 95 °C for 15 s, 56 °C for 1 min, and 60 °C for 20 s. The expression of miRNAs was calculated relative to RNU6B, a ubiquitous small nuclear RNA. The forward primers for MIR30B*, MIR142-3p, MIR505*, MIR548B-3p, MIR770-5p, and RNU6B were 5′-CTGGGAGGTG GATGTTTACT TC-3′, 5′-TGTAGTGTTT CCTACTTTAT GGA-3′, 5′-GGGAGCCAGG AAGTATTGAT GT-3′, 5′-CAAGAACCTC AGTTGCTTTT GT-3′, 5′-TCCAGTACCA CGTGTCAGGG CCA-3′, and 5′-CGCAAGGATG ACACGCAAAT TCG-3′, respectively. A universal reverse primer provided in the qRT-PCR kit was used.
Western blot analysis
Cells were lysed in CelLytic MT mammalian tissue lysis/extraction reagent (Sigma, C3228) containing 1% protease inhibitor cocktail (Sigma, P8340) and then mixed with the same volume of Laemmli sample buffer (Bio-Rad, 161-0737) containing 2-mercaptoethanol. After heat denaturation, equal amounts (15 μg) of cellular proteins were separated by SDS-PAGE, followed by electrotransfer onto a PVDF membrane (Bio-Rad, 162-0177). After blocking with 5% nonfat milk, the immunoblot was performed by incubation with primary antibodies (1:1,000 dilution), then Alexa Fluor 680 (Invitrogen, A21084) and IRDye 800CW (LI-COR Biosciences, 926-32213) conjugated secondary antibodies (1:10,000 dilution). The immunocomplexes were visualized and band intensities were quantified using an Odyssey infrared imaging system (LI-COR Biosciences, Lincoln, NE). The following primary antibodies were used: rabbit anti-ATG16L1, ATG5, SQSTM1, and LC3B (Cell Signaling, 8089, 8540, 5114, and 2775, respectively) as well as goat anti-ACTB and GAPDH (Santa Cruz Biotechnology, SC-1615 and SC-20357, respectively).
Confocal imaging
Cells grown on cover glasses were transfected with a mimic and then starved for 4 h as mentioned above. Cells were fixed in 4% paraformaldehyde and permeabilized by 0.5% Triton X-100. Following blocking with 3% bovine serum albumin (Sigma, A9647), cells were serially incubated in rabbit anti-LC3B (1:200 dilution) and donkey anti-rabbit Alexa Fluor 488 (1:250 dilution; Invitrogen, A11034). Finally, cells were rinsed and mounted on cover glasses with Prolong Gold anti-fade reagent with 4’-6-diamidino-2-phenylindole (DAPI; Invitrogen, P36931), and the immunostaining was observed under an Olympus DSU spinning disk confocal microscope (Olympus America Inc., Center Valley, PA) with a 60 × oil lens.
Flow cytometry
Flow cytometry for LC3 was performed as previously described.36,37 Briefly, Jurkat T cells were transfected with the MIR142-3p inhibitor and negative control (50 nM) by using Lipofectamine LTX and Plus reagents (Invitrogen, 15338-100) according to the manufacturer’s protocol specific for Jurkat cells. After transfection for 4 h, equal amount of complete medium was added. On the next day, the cultured medium was removed and the cells were incubated in EBSS containing 15 mM NH4Cl for 2 h. The cells were pelleted and resuspended in phosphate-buffered saline (PBS) containing 0.05% saponin for 5 min to release the soluble LC3-I from cells. Cells were then incubated with anti-LC3B primary antibody (MBL International, PM036, 1:200) for 20 min, rinsed with PBS, incubated with Alexa Fluor 488-anti-rabbit secondary antibody (1:1000) for 15 min, and rinsed twice with PBS. FACS data were collected using a FACSCanto flow cytometer (Becton Dickinson) with FASCDiva software. More than 30,000 events were captured for a sample. Data analysis was performed with FlowJo.
Cell viability and apoptosis assay
HCT116 cells were seeded in transparent or white 96-well plates. After transfection with the MIR142-3p mimic or MIRNC (50 nM), cells were incubated in normal complete medium or in EBSS for 24 h. Cell viability was assayed using the Cell Counting Kit-8 (Dojindo, CK04-11) and quantified by reading absorbance at 450 nm with a 630 nm reference on a microplate reader (Tecan, Infinite M200 PRO, Research Triangle Park, NC). Alternatively, apoptosis was determined by assaying the activities of caspase-3 and -7 using the Apo-ONE Homogeneous CASP3 and CASP7 (Caspase-3/7) assay kit (Promega, G7791) according to the manufacturer’s specifications or analyzed using the PE ANXA5 apoptosis detection kit I (BD PharMingen, 559763) and a flow cytometer.
Statistical analysis
GraphPad Prism 4.0 (GraphPad Software) was used for the statistical analysis. For 2 treatment groups, the Student t test was used. For 3 or more treatment groups, one-way ANOVA was used with Bonferroni post-test for the comparison of selected 2 treatment groups as well as Dunnett post-test for comparing all other treatment groups to the corresponding control. A value P < 0.05 was considered statistically significant.
Supplementary Material
Disclosure of Potential Conflicts of Interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This publication was made possible by the National Institute of Health (RO3 DK086549 to JHK), NIH T32 Gastrointestinal training grant (DK07074 to UChicago for ZZ), and the NIH DDRCC core grant (P30 DK42086). Its contents are solely the responsibility of the authors and do not represent the official views of the NIH. The authors would like to thank Dr Eugene Chang and Dr Joel R Pekow for their advice.
Glossary
Abbreviations:
- 7-AAD
7-aminoactinomycin D
- Adcy9
adenylyl cyclase 9
- ANXA5
annexin V, ATG16L1, autophagy-related 16-like 1 (S. cerevisiae)
- CASP3/7
caspase 3/7, apoptosis-related cysteine peptidase
- CD
Crohn disease
- DAPI
4′-6-diamidino-2-phenylindole
- EBSS
Earle’s balanced salt solution
- IBD
inflammatory bowel disease
- IRGM
immunity-related GTPase family, M
- L18-MDP
muramyl dipeptide with a C18 fatty acid chain
- LC3
microtubule-associated protein 1 light chain 3
- miRNA
microRNA
- MIRNC
miRNA mimic negative control
- NOD2
nucleotide-binding oligomerization domain containing 2
- NR3C1
nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor)
- nt
nucleotide
- PBS
phosphate-buffered saline
- RFU
relative fluorescence unit
- ROCK2
Rho-associated, coiled-coil containing protein kinase 2
- RT-PCR
reverse transcription-polymerase chain reaction
- SQSTM1/p62
sequestosome 1
- 3′UTR
3′-untranslated region
- WT
wild-type
References
- 1.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–17. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eskelinen EL, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009;1793:664–73. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 5.Rosenfeldt MT, Ryan KM. The role of autophagy in tumour development and cancer therapy. Expert Rev Mol Med. 2009;11:e36. doi: 10.1017/S1462399409001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 7.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al. Wellcome Trust Case Control Consortium Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–2. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plantinga TS, Crisan TO, Oosting M, van de Veerdonk FL, de Jong DJ, Philpott DJ, van der Meer JW, Girardin SE, Joosten LA, Netea MG. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut. 2011;60:1229–35. doi: 10.1136/gut.2010.228908. [DOI] [PubMed] [Google Scholar]
- 10.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 11.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–63. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell. 2010;141:1135–45. doi: 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 14.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 15.Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–11. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A. 2006;103:4034–9. doi: 10.1073/pnas.0510928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jegga AG, Schneider L, Ouyang X, Zhang J. Systems biology of the autophagy-lysosomal pathway. Autophagy. 2011;7:477–89. doi: 10.4161/auto.7.5.14811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, Liu CG, Yang JM. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy. 2009;5:816–23. doi: 10.4161/auto.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu Y, Yang L, Zhao M, Zhu S, Kang R, Vernon P, Tang D, Cao L. Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia. 2012;26:1752–60. doi: 10.1038/leu.2012.65. [DOI] [PubMed] [Google Scholar]
- 20.Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, Jäättelä M, Lund AH. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2011;30:4628–41. doi: 10.1038/emboj.2011.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kovaleva V, Mora R, Park YJ, Plass C, Chiramel AI, Bartenschlager R, Döhner H, Stilgenbauer S, Pscherer A, Lichter P, et al. miRNA-130a targets ATG2B and DICER1 to inhibit autophagy and trigger killing of chronic lymphocytic leukemia cells. Cancer Res. 2012;72:1763–72. doi: 10.1158/0008-5472.CAN-11-3671. [DOI] [PubMed] [Google Scholar]
- 22.Chang Y, Yan W, He X, Zhang L, Li C, Huang H, Nace G, Geller DA, Lin J, Tsung A. miR-375 inhibits autophagy and reduces viability of hepatocellular carcinoma cells under hypoxic conditions. Gastroenterology. 2012;143:177–87, e8. doi: 10.1053/j.gastro.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Korkmaz G, le Sage C, Tekirdag KA, Agami R, Gozuacik D. miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1. Autophagy. 2012;8:165–76. doi: 10.4161/auto.8.2.18351. [DOI] [PubMed] [Google Scholar]
- 24.Huang Y, Guerrero-Preston R, Ratovitski EA. Phospho-ΔNp63α-dependent regulation of autophagic signaling through transcription and micro-RNA modulation. Cell Cycle. 2012;11:1247–59. doi: 10.4161/cc.11.6.19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, Chuang AY, Ratovitski EA. Phospho-ΔNp63α/miR-885-3p axis in tumor cell life and cell death upon cisplatin exposure. Cell Cycle. 2011;10:3938–47. doi: 10.4161/cc.10.22.18107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhai Z, Wu F, Chuang AY, Kwon JH. miR-106b fine tunes ATG16L1 expression and autophagic activity in intestinal epithelial HCT116 cells. Inflamm Bowel Dis. 2013;19:2295–301. doi: 10.1097/MIB.0b013e31829e71cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hébuterne X, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–5. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 28.Wu F, Zikusoka M, Trindade A, Dassopoulos T, Harris ML, Bayless TM, Brant SR, Chakravarti S, Kwon JH. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology. 2008;135:1624–35, e24. doi: 10.1053/j.gastro.2008.07.068. [DOI] [PubMed] [Google Scholar]
- 29.Wu F, Zhang S, Dassopoulos T, Harris ML, Bayless TM, Meltzer SJ, Brant SR, Kwon JH. Identification of microRNAs associated with ileal and colonic Crohn’s disease. Inflamm Bowel Dis. 2010;16:1729–38. doi: 10.1002/ibd.21267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu F, Guo NJ, Tian H, Marohn M, Gearhart S, Bayless TM, Brant SR, Kwon JH. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis. 2011;17:241–50. doi: 10.1002/ibd.21450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chuang AY, Chuang JC, Zhai Z, Wu F, Kwon JH. NOD2 expression is regulated by microRNAs in colonic epithelial HCT116 cells. Inflamm Bowel Dis. 2014;20:126–35. doi: 10.1097/01.MIB.0000436954.70596.9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–6. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 33.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 34.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Man N, Chen Y, Zheng F, Zhou W, Wen LP. Induction of genuine autophagy by cationic lipids in mammalian cells. Autophagy. 2010;6:449–54. doi: 10.4161/auto.6.4.11612. [DOI] [PubMed] [Google Scholar]
- 36.Eng KE, Panas MD, Karlsson Hedestam GB, McInerney GM. A novel quantitative flow cytometry-based assay for autophagy. Autophagy. 2010;6:634–41. doi: 10.4161/auto.6.5.12112. [DOI] [PubMed] [Google Scholar]
- 37.Kaminskyy V, Abdi A, Zhivotovsky B. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy. 2011;7:83–90. doi: 10.4161/auto.7.1.13893. [DOI] [PubMed] [Google Scholar]
- 38.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 39.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 40.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–7. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 41.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 42.Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology. 2010;139:1630–41, e1-2. doi: 10.1053/j.gastro.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harris J. Autophagy and cytokines. Cytokine. 2011;56:140–4. doi: 10.1016/j.cyto.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 44.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16:1040–52. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 46.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–6. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 47.Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I, Mannucci S, De Nisi MC, Toscano M, Malagnino V, et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol. 2007;30:429–36. [PubMed] [Google Scholar]
- 48.Cheng HY, Zhang YN, Wu QL, Sun XM, Sun JR, Huang X. Expression of beclin 1, an autophagy-related protein, in human cervical carcinoma and its clinical significance. Eur J Gynaecol Oncol. 2012;33:15–20. [PubMed] [Google Scholar]
- 49.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323–35. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berry DL, Baehrecke EH. Autophagy functions in programmed cell death. Autophagy. 2008;4:359–60. doi: 10.4161/auto.5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishiyama T, Kaneda R, Ono T, Tohyama S, Hashimoto H, Endo J, Tsuruta H, Yuasa S, Ieda M, Makino S, et al. miR-142-3p is essential for hematopoiesis and affects cardiac cell fate in zebrafish. Biochem Biophys Res Commun. 2012;425:755–61. doi: 10.1016/j.bbrc.2012.07.148. [DOI] [PubMed] [Google Scholar]
- 52.Joyce CE, Zhou X, Xia J, Ryan C, Thrash B, Menter A, Zhang W, Bowcock AM. Deep sequencing of small RNAs from human skin reveals major alterations in the psoriasis miRNAome. Hum Mol Genet. 2011;20:4025–40. doi: 10.1093/hmg/ddr331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Danger R, Pallier A, Giral M, Martínez-Llordella M, Lozano JJ, Degauque N, Sanchez-Fueyo A, Soulillou JP, Brouard S. Upregulation of miR-142-3p in peripheral blood mononuclear cells of operationally tolerant patients with a renal transplant. J Am Soc Nephrol. 2012;23:597–606. doi: 10.1681/ASN.2011060543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perri R, Nares S, Zhang S, Barros SP, Offenbacher S. MicroRNA modulation in obesity and periodontitis. J Dent Res. 2012;91:33–8. doi: 10.1177/0022034511425045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, Xu ZM, Yin YB. Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus. 2007;16:939–46. doi: 10.1177/0961203307084158. [DOI] [PubMed] [Google Scholar]
- 56.Makino K, Jinnin M, Kajihara I, Honda N, Sakai K, Masuguchi S, Fukushima S, Inoue Y, Ihn H. Circulating miR-142-3p levels in patients with systemic sclerosis. Clin Exp Dermatol. 2012;37:34–9. doi: 10.1111/j.1365-2230.2011.04158.x. [DOI] [PubMed] [Google Scholar]
- 57.Bellon M, Lepelletier Y, Hermine O, Nicot C. Deregulation of microRNA involved in hematopoiesis and the immune response in HTLV-I adult T-cell leukemia. Blood. 2009;113:4914–7. doi: 10.1182/blood-2008-11-189845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lv M, Zhang X, Jia H, Li D, Zhang B, Zhang H, Hong M, Jiang T, Jiang Q, Lu J, et al. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-α and cAMP/PKA pathways. Leukemia. 2012;26:769–77. doi: 10.1038/leu.2011.273. [DOI] [PubMed] [Google Scholar]
- 59.Wang XS, Gong JN, Yu J, Wang F, Zhang XH, Yin XL, Tan ZQ, Luo ZM, Yang GH, Shen C, et al. MicroRNA-29a and microRNA-142-3p are regulators of myeloid differentiation and acute myeloid leukemia. Blood. 2012;119:4992–5004. doi: 10.1182/blood-2011-10-385716. [DOI] [PubMed] [Google Scholar]
- 60.Mascaux C, Laes JF, Anthoine G, Haller A, Ninane V, Burny A, Sculier JP. Evolution of microRNA expression during human bronchial squamous carcinogenesis. Eur Respir J. 2009;33:352–9. doi: 10.1183/09031936.00084108. [DOI] [PubMed] [Google Scholar]
- 61.Lin RJ, Xiao DW, Liao LD, Chen T, Xie ZF, Huang WZ, Wang WS, Jiang TF, Wu BL, Li EM, et al. MiR-142-3p as a potential prognostic biomarker for esophageal squamous cell carcinoma. J Surg Oncol. 2012;105:175–82. doi: 10.1002/jso.22066. [DOI] [PubMed] [Google Scholar]
- 62.Huang B, Zhao J, Lei Z, Shen S, Li D, Shen GX, Zhang GM, Feng ZH. miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep. 2009;10:180–5. doi: 10.1038/embor.2008.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu L, Cai C, Wang X, Liu M, Li X, Tang H. MicroRNA-142-3p, a new regulator of RAC1, suppresses the migration and invasion of hepatocellular carcinoma cells. FEBS Lett. 2011;585:1322–30. doi: 10.1016/j.febslet.2011.03.067. [DOI] [PubMed] [Google Scholar]
- 64.Sun Y, Varambally S, Maher CA, Cao Q, Chockley P, Toubai T, Malter C, Nieves E, Tawara I, Wang Y, et al. Targeting of microRNA-142-3p in dendritic cells regulates endotoxin-induced mortality. Blood. 2011;117:6172–83. doi: 10.1182/blood-2010-12-325647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan X, Zhang P, Zhou L, Yin B, Pan H, Peng X. Clock-controlled mir-142-3p can target its activator, Bmal1. BMC Mol Biol. 2012;13:27. doi: 10.1186/1471-2199-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olaru AV, Selaru FM, Mori Y, Vazquez C, David S, Paun B, Cheng Y, Jin Z, Yang J, Agarwal R, et al. Dynamic changes in the expression of MicroRNA-31 during inflammatory bowel disease-associated neoplastic transformation. Inflamm Bowel Dis. 2011;17:221–31. doi: 10.1002/ibd.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Necela BM, Carr JM, Asmann YW, Thompson EA. Differential expression of microRNAs in tumors from chronically inflamed or genetic (APC(Min/+)) models of colon cancer. PLoS One. 2011;6:e18501. doi: 10.1371/journal.pone.0018501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.