

Abstract
Objective
Evidence of increasing heritability of BMI over childhood can seem paradoxical given longer exposure to environmental influences. Genomic data were used to provide direct evidence of developmental increases in genetic influence.
Methods
BMI standard deviation scores (BMI-SDS) at ages 4 and 10 were calculated for 2,556 twin pairs in the Twins Early Development Study. Twin analyses estimated heritability of BMI-SDS at each age and the longitudinal genetic correlation. One randomly selected twin per pair was genotyped. Genome-wide complex trait analysis (GCTA) determined DNA-based heritability at each age and the longitudinal genomic correlation. Associations with a polygenic obesity risk score (PRS) using 28 obesity-related single nucleotide polymorphisms (SNPs) were assessed at each age, with bootstrapping to test the significance of the increase in variance explained.
Results
Twin-estimated heritability increased from age 4 (0.43; 95% CI: 0.35-0.53) to 10 (0.82; 0.74-0.88). GCTA-estimated heritability went from non-significant at 4 (0.20; −0.21 to 0.61) to significant at 10 (0.29; 0.01-0.57). Longitudinal genetic correlations derived from twins (0.58) and GCTA (0.66) were similar. The same PRS explained more variance at 10 than 4 years (R2 Δ:0.024; 0.002-0.078).
Conclusions
GCTA and PRS findings confirm twin-based results suggesting increasing genetic influence on adiposity during childhood despite substantial genetic stability.
Introduction
Body mass index (BMI) is a heritable trait; with moderate-to-high estimates (47–90%) reported from twin studies 1,2. A recent meta-regression showed that heritability was higher in children than in adults 1, and several studies have shown increasing heritability of BMI from early to late childhood 3–5. Using longitudinal data from the Twins Early Development Study (TEDS), we showed that the heritability of BMI increased substantially from 4 to 10 years (49–82%) 5, consistent with findings from the Danish Twin Registry (48–87%) 3. This may appear paradoxical given that increasing duration of children's exposure to environmental influences could be expected to increase the environmental effect. However, developmental increases in heritability have been demonstrated for other phenotypes 6, and it would be consistent with a model of gene expression depending on environmental exposure 7. Twin data also show high genetic correlations over time for BMI 5,8–10, suggesting that many of the same genes continue to influence BMI at different ages. However, because twin analyses are inferential, it is important to demonstrate the same pattern of results using genomic data.
Genome-wide association studies (GWAS) have now identified more than 32 common single nucleotide polymorphisms (SNPs) associated with BMI in adults and children 11,12. These can be combined into a polygenic obesity risk score (PRS) to quantify genomic influence on BMI. A PRS created from a subset of these SNPs explained increasing variance in BMI from early to late childhood in the Avon Longitudinal Study of Parents and Children (ALSPAC) 13, with a similar result in the 1946 British Birth Cohort Study 14. Although these studies did not directly compare quantitative and molecular genetic results, they support the view that increases in twin-estimated heritability can be explained by increasing phenotypic effects of the same genes, because the genetic correlation is constrained to 1 using the same PRS.
On the basis of obesity-related SNPs identified to date, the total variance explained is still well below the twin-estimated heritability 15. However, a novel quantitative method called Genome-wide Complex Trait Analysis (GCTA) 16 uses whole genome arrays to estimate the total additive genetic influence due to all common SNPs simultaneously. Like twin data, GCTA can be used to explore developmental increases in genetic association insofar as it provides age-specific estimates of the additive effects of all common SNPs tagged by the commercially available genetic chips, and an estimate of the longitudinal genetic correlation. SNP-derived heritability will be lower than twin-derived heritability if SNPs other than those tagged by the additive effects of common SNPs are important (such as rare variants), or if non-additive effects are important (gene–gene or gene–environment interactions).
In this study, we used GCTA, PRS, and twin analysis, in the same sample of children to test the hypotheses that twin-based evidence of increases in heritability of BMI from early to late childhood is supported by genomic results (GCTA and PRS), and that genomic and twin analyses both suggest that many of the same genes influence adiposity at both ages.
Methods
Sample
The sample for this study was from TEDS, a population-based cohort of >11,000 British twins 17. Informed consent for each part of the study was provided by the parents prior to data collection. Ethical approval was provided by King's College London's Ethics Committee.
Genotyping
In 2010, genome-wide genotyping was carried out for one randomly selected member of each twin pair in 3,665 TEDS families as part of the Wellcome Trust Case Control Consortium 2 (WTCCC2) project 18. DNA was extracted from buccal cheek swabs. The Affymetrix 6.0 GeneChip was used to genotype about 1,000,000 SNPs using standard experimental protocols 19. IMPUTE version 2 software 20 was used to impute approximately 2.5 million additional SNPs. After stringent quality control was carried out as part of WTCCC2 20, the sample was reduced to about 1.7 million SNPs and 3,152 children.
Creating a polygenic obesity risk score
A PRS indexing genetic predisposition to obesity was calculated using 28 known obesity-related SNPs from a total of 34 identified in two published meta-analyses in adults 12 and children 11. The SNPs included are described in the Supporting Information (“Supplementary methods: creating a polygenic obesity risk score”). A PRS was created by calculating a mean score for each individual from the 28 SNPs, yielding a possible score ranging from 0 to 56 with higher scores indicating greater genetic predisposition to obesity. A weighted mean score was calculated to take account of relative effect sizes by multiplying each SNP by its beta coefficient in the published meta-analyses from which the SNPs were identified 11,12.
Measurement of BMI-SDS at age 4 and 10 years
Anthropometric data were collected in 1999 and 2005, when the children were 4 and 10 years old, respectively. Questionnaires and 2-meter tape measures were mailed to the families; parents were provided with detailed instructions regarding how to measure their children's height (to the nearest centimeter) and weight (to the nearest pound or tenth of a kilogram), and they were asked to record the date of each measurement. Correspondence between parent- and researcher-measured height and weight were 0.90 and 0.83 in a sample of 228 families 21.
BMI was calculated from height and weight (weight (kg)/height (m)2) and converted to Standard Deviation Scores (BMI-SDS) which take into account the child's age and sex, using 1990 UK growth reference data 22 with the program ImsGrowth 23. BMI-SDS were residualized for age- and sex-effects using a regression procedure, prior to analyses. To aid model optimization and cross-method comparison, we standardized BMI-SDS such that the variance was equal to one and the mean equal to zero.
Exclusions
About 2,556 children of the sample with genotyping data (n = 3,152) had height and weight data for at least one of the two ages, and their exact age at the time of measurement. As the GWAS sample was already screened for medical and other general exclusions, there were no further exclusions (for details on that sample, see ref. 19).
Statistical analyses
Twin analyses of the heritability of BMI-SDS at ages 4 and 10 years
To provide twin-based heritability of BMI-SDS at ages 4 and 10 for comparison, data from genotyped twins and their co-twins were modeled using a longitudinal Cholesky Decomposition Model in OpenMx 24. This method models variance and covariance for pairs of monozygotic (MZ) twins who are 100% genetically identical, and dizygotic (DZ) twins who share on average 50% of their segregating alleles. Differences between intraclass correlations for MZ and DZ pairs are used to partition the variance into genetic and environmental effects. Variance is attributed to: additive genetic influence (A), to the extent that MZ correlations are higher than those for DZ twins; shared or common (C) environmental influences, which is residual MZ twin resemblance not explained by genetics; and nonshared or unique environmental influences (E), which is the extent to which MZ twins differ, and includes measurement error. A longitudinal genetic correlation is derived by partitioning covariance between BMI-SDS at 4 and 10 years, for MZs and DZs. This estimates the extent to which common genetic effects underlie BMI-SDS at both ages, ranging from 0 (no common genetic effects) to 1 (all of the same genes are involved). Importantly, twin heritability captures genetic influence from the whole genome, and may include non-additive genetic influences. A detailed description of the twin method and related issues can be found elsewhere 25.
The fit of the model was not of primary interest in this study; however, to assure a “good fit,” we used the full model with all parameters, including A, C, and E for each age, and allowed longitudinal covariation among all of these parameters. Because previous analysis on the same sample did not indicate sex differences we modeled both sexes together.
Genome-wide Complex Trait Analysis at ages 4 and 10 years
GCTA was used to estimate DNA-based heritability at ages 4 and 10 years, and the longitudinal genomic correlation, for the 2,556 unrelated children with genotyping and anthropometric data. GCTA takes advantage of GWAS data to estimate the total amount of variance in a phenotype that can be explained by the additive effects of all common SNPs measured on commercial chips or in high linkage disequilibrium with them. The method adapts a linear mixed model (LMM) framework fitting genetic influence as a random polygenic effect. GCTA estimates the amount of genetic influence by associating mean genetic similarity calculated from all genetic loci to the phenotypic similarity between all pairs of unrelated individuals in the sample 26. Using a random effects model to estimate genetic influence means that this estimate is “unbiased” if all individuals in the sample are truly unrelated. For that reason, if any pairwise comparison returns genetic relatedness > 0.025, one of the pair is removed; on this basis five individuals were removed from the GCTA analysis. Given that GCTA is a genome-wide method, it is affected by population structure 27; therefore we used eight eigen vectors previously used on the same sample in our GWAS 19. To compare age-related differences in the amount of variance in BMI-SDS explained, and to estimate the longitudinal genetic correlation, we used a bivariate GCTA model 28; this differs from a univariate model in that it focuses on the covariance between two ages.
Associations between polygenic risk score and BMI-SDS at ages 4 and 10 years
Linear regression analyses were used to establish the association between the PRS with age- and sex-adjusted BMI-SDS at 4 and 10 years of age for the 2,556 unrelated children with genotyping and anthropometric data. Bootstrapping analyses were carried out to provide 95% confidence intervals for the R2 estimates derived from the linear regression analyses, and to test for differences between the R2 estimated at age 4 and age 10. To provide a more accurate test for bootstrapping the difference in R2 between the two ages, we sampled from individuals who had data points at both ages. The linear regression and bootstrapping analyses were performed in R version 2.15.0 29.
Results
Sample characteristics
The distribution of dizygotic (n = 1,535; 60%) and monozygotic (n = 1,021; 40%) twin pairs was approximately as expected; the sample included slightly more girls (n = 1,368; 54%) than boys (n = 1,188; 46%). The number of obesity risk-increasing alleles was normally distributed in the sample and ranged from 11 to 30 (mean = 12.48; SD = 2.85) (Figure 1). Anthropometric characteristics are shown in Table 1 for ages 4 and 10 years. At each age, the mean BMI-SDS was less than 0, indicating that the average relative body weight of the sample was slightly less than the UK reference values. In keeping with this, the sample had lower rates of overweight and obesity at each age than observed in national UK statistics. BMI-SDS at ages 4 and 10 were positively correlated (r = 0.40, P < 0.001).
Figure 1.
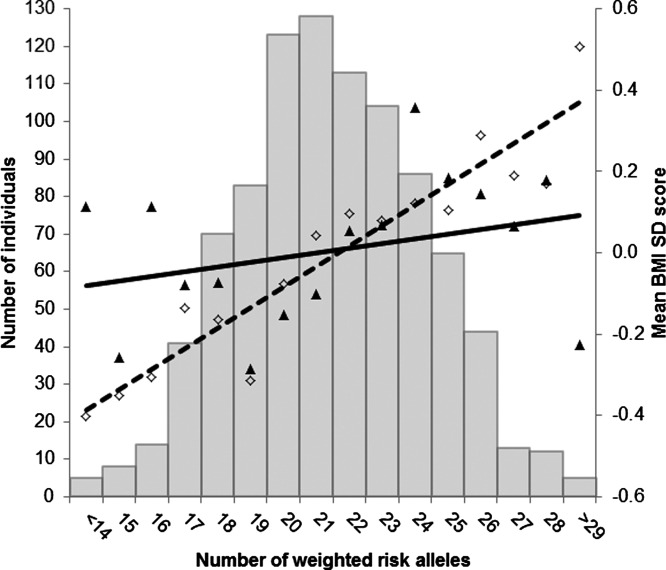
Regression of mean age- and sex-adjusted BMI-SDS values at ages 4 and 10 years across the risk-allele scores. The histogram shows that the number of weighted obesity risk alleles was normally distributed in the sample. The solid black triangles show the mean age- and sex-adjusted BMI-SDS values at age 4 across the weighted risk-allele scores; the black diamonds show the mean age- and sex-adjusted BMI-SDS values at age 10 across the weighted risk-allele scores. The solid black line shows the regression line for age- and sex-adjusted BMI-SDS at age 4 predicted from the PRS (R2 = 0.010; 95% CI: 4.3 e−09 to 0.042; P = 0.002). The dashed line shows the regression line for age- and sex-adjusted BMI-SDS at age 10 predicted from the PRS (R2 = 0.034; 95% CI: 0.009-0.093; P < 0.001).
Table 1.
Summary statistics of anthropometrics for the analysis sample at ages 4 and 10 years (n = 2,556 childrena)
| Mean (sd) or n (%) | ||
|---|---|---|
| Age 4 | Age 10 | |
| Age at measurement (years) | 4.00 (0.11) | 9.90 (0.85) |
| Weight (kg) | 16.53 (2.40) | 33.46 (7.78) |
| Height (m) | 1.02 (0.05) | 1.39 (0.08) |
| BMIb (kg/m2) | 15.79 (2.10) | 17.03 (2.57) |
| BMI-SDSc | −0.12 (1.59) | −0.02 (1.12) |
| Weight statusd | ||
| Healthy weight | 1241 (87.3) | 1995 (87.8) |
| Overweight | 94 (6.6) | 197 (8.7) |
| Obese | 87 (6.1) | 81 (3.5) |
The sample characteristics presented are for the 2,556 unrelated children with genotyping and BMI-SDS data for at least one age point; 1,422 unrelated children had genotyping and BMI-SDS data at age 4; 2,273 children had genotyping and BMI-SDS data at age 10.
BMI, body mass index.
BMI-SDS, BMI standard deviation score: BMI adjusted for age and sex using UK 1990 reference data 22.
Weight status calculated from BMI-SDS using UK 1990 reference data: healthy weight, BMI-SDS <91st centile; overweight, BMI-SDS ≥ 91st centile, and <98th centile; obese, BMI-SDS ≥ 98th centile 22.
Twin analyses of the heritability of BMI-SDS at ages 4 and 10 years
As reported previously 5, heritability increased significantly from 0.43 (95% confidence interval (CI): 0.35-0.53) at age 4 to 0.82 (95% CI: 0.74-0.88) at age 10 (Table 2, Figure 2). The genetic correlation between BMI-SDS at the two ages was 0.58 (95% CI: 0.48-0.68). The shared environment effect showed the opposite trend—it was considerable and significant at age 4 (0.41; 95% CI: 0.32-0.49), but very small and not significant by age 10 (0.06; 95% CI: 0.00-0.14). Full results from the twin analysis are available in Supporting Information Table S1.
Table 2.
Comparison of twin-, genome-wide complex trait analysis (GCTA)-, and polygenic risk score (PRS)-estimates of heritability at 4 and 10 years
| Twin study estimate of heritability (95% CI)a | GCTA estimate of genetic influence (95% CI) | Association (R2) between PRS and BMI-SDS (95% CI)bc | |
|---|---|---|---|
| BMI-SDS age 4 | 0.43 (0.35-0.53) | 0.20 (−0.21 to 0.61) | 0.010 (4.3 e−09 to 0.042) |
| BMI-SDS age 10 | 0.82 (0.74-0.88) | 0.29 (0.01 to 0.57) | 0.034 (0.009-0.093) |
Twin-estimated heritability was significantly higher at age 10 compared to age 4.
Bootstrapping was used to test the difference in the association (R2Δ) between the PRS and age- and sex-adjusted BMI-SDS at 4 and 10 years.
R2 estimate at age 10 was significantly greater than at age 4 (R2Δ=0.024, 95% CI = 0.002–0.078).
Figure 2.
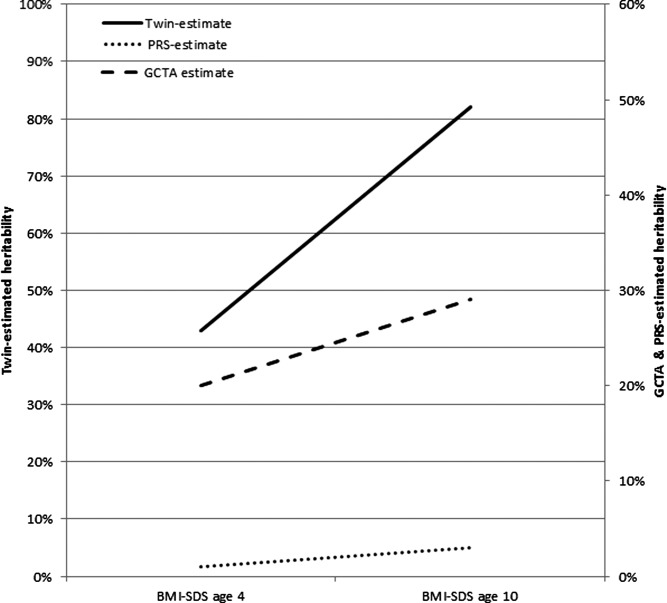
Comparison of twin- (solid black line), GCTA- (dashed line), and PRS-estimated (dotted line) heritability of BMI-SDS at age 4 and age 10. The variance explained by genetic effects increased from age 4 to age 10, using all three methods. The age-related increase in variance explained was significant for twin-estimated heritability and for the PRS; the GCTA-estimated heritability went from non-significant at age 4 to significant at age 10.
GCTA estimates of the heritability of BMI-SDS at ages 4 and 10 years
GCTA estimated heritability increased from a non-significant 0.20 (95% CI: −0.21 to 0.61) at age 4 to a significant 0.29 (95% CI: 0.01-0.57) at age 10 (Table 2, Figure 2). However, the change in point estimates was not significant, indicated by the overlapping 95% confidence intervals. The point-estimate for the GCTA-derived genetic correlation was high, although because of the large standard error, was not significant (r = 0.66; 95% CI: −0.28 to 1.60). Full GCTA results are available in Supporting Information Table S2.
Associations between the obesity-related PRS and BMI-SDS at ages 4 and 10 years
In multiple linear regression analyses, the PRS was significantly associated with BMI-SDS at both ages, with the size of the association increasing from age 4 (R2 = 0.010; 95% CI: 4.3 e−09 to 0.042; P = 0.002) to age 10 (R2 = 0.034; 95% CI: 0.009-0.093; P < 0.001) (Table 2, Figures 1 and 2). Bootstrapping analyses confirmed that the association was significantly higher at age 10 than at age 4 (R2Δ = 0.024; bootstrapped 95% CI = 0.002-0.078) (Supporting Information Figure S1).
Discussion
In this study, we used genome-wide genotyping data to create an obesity-related polygenic risk score (PRS) and for a GCTA analysis, to test the hypotheses that developmental increases in the twin estimates of the heritability of BMI-SDS are supported by genomic data, and that GCTA and twin analyses confirm that many of the same genes are influencing BMI-SDS at both ages. In line with previous estimates from TEDS, twin-estimated heritability of BMI-SDS increased significantly from 4 to 10 years (h2Δ = 35%) 5; in keeping with other studies that have explored developmental increases in heritability of BMI in this age group 3,4. Although the trend of increasing heritability in the GCTA analysis was in the same direction as the twin results, the analyses lacked power to detect whether this change was significant. The point estimate of the GCTA bivariate longitudinal genetic correlation was very similar to the bivariate longitudinal twin correlation (0.58 and 0.66, respectively); providing DNA-based support for the inferential twin-based statistic, and indicating that the increasing heritability is driven to a large extent by many of the same weight-related genes exerting progressively greater effects on weight.
The twin-estimated genetic correlation was virtually the same as that reported in the Netherlands Twin Registry for BMI between age 4 and age 10 (0.52) 10. There was also a significant increase in the association between the PRS and BMI-SDS from age 4 to 10 (R2Δ = 0.024), which suggests that many of the same BMI-related genes exert a greater effect as children get older. However, it is also possible that the influence of some loci in the PRS is age-dependent, such that different or additional loci come into effect at age 10. This would explain why the genetic correlations are not 1.
Previous findings using variants in the FTO and MC4R genes support the idea that the same genes have increasing effect as children get older. A meta-analysis of eight cohorts of European ancestry showed that the primary obesity-related common genetic variant in the FTO gene increased its effect on BMI progressively from 0.7% at 5–7 years, to 1.0% at 7–9 years, and 1.3% at 9–11 years 30. Analyses of an obesity-associated variant in the MC4R gene showed a similar pattern in the 1946 British Birth Cohort; associations with age- and sex-adjusted weight increased during childhood and adolescence by 0.005 units per year.
Our findings are also consistent with previous studies that have explored developmental increases in associations between adiposity and an obesity-related PRS. In ALSPAC, a PRS comprising eight obesity-related SNPs showed a weak association with BMI-SDS up to 3.5 years, but a rapid increase in the size of the association from 3.5 to 11 years 13. Lifecourse analyses for a 29-SNP PRS in the Dunedin Study 31, and an 11-SNP PRS in the 1946 Birth Cohort 14; both showed that the association with BMI increased year on year. Using comparable age data, the present findings in TEDS were almost identical to the Dunedin cohort which used a comparable PRS; and slightly higher than ALSPAC which utilized fewer SNPs (TEDS at 4 years: 0.8%; Dunedin at 3 years: 0.6%; ALSPAC at 3.5 years: 0.2%; TEDS at 10 years: 3.4%; Dunedin at 9 years: 3.2%; ALSPAC at 10 years: 1.6%). This study adds to the evidence base that genetic influences on weight increase from early to late childhood.
One previous study of cognitive abilities directly compared longitudinal GCTA correlations with twin-based estimates, and found increasing heritability in both types of analysis, and similarly high genetic correlations with each method 32. We had a small sample size relative to the point estimate for our BMI analyses at age 4 (n = 1,419), rendering the current study underpowered to detect a significant genetic correlation. The findings therefore need to be replicated using a larger sample.
One explanation put forward for age-related increases in heritability for a variety of phenotypes (e.g., externalizing behaviors, anxiety symptoms, and depressive symptoms) is that active gene–environment correlations increase as children get older and gain independence 6. Early childhood environments are largely controlled by parents, but as children grow older they can select out environments that “indulge” their genetic propensities. We have hypothesized that obesity-related genes influence weight via appetite 33; FTO's effects on childhood BMI have been shown to be mediated via satiety sensitivity 34,35. FTO is unlikely to be expressed fully unless the individual has the freedom to consume to satiety—a privilege that comes with age, when children are able to make decisions about when and how much to eat. Other obesity-related SNPs are also hypothesized to influence weight via appetite 36; making gene–environment correlation a plausible explanation for the rising genetic influence.
Increasing genetic effect on BMI from early to late childhood may also reflect processes that began even earlier in life. The Dunedin Study showed that early life growth from birth to 3 years mediated some of the genetic risk of obesity in adolescence 31. It is also possible that new loci come into effect at age 10; however, the twin- and GCTA-derived genetic correlations indicate that many of the same genes are involved at both ages.
Although analyzing heritability as the relative influence of genetics is standard in the literature, we also analyzed the absolute rather than relative variance explained by genetics at the two ages in an attempt to find out more about the increase in heritability. Heritability increased from 4 to 10 years not because the absolute amount of genetic variance increased but because the absolute amount of environmental variance (and thus total variance) decreased from 4 to 10 years. Nonetheless, it remains the case that of the phenotypic variance at each age, the proportion due to genetic variance (i.e., heritability) increases, as does the variance explained by GCTA and PRS.
These findings have public health implications. If genetic predisposition to obesity is expressed increasingly from infancy, the early school years provide an important intervention window for initiatives aimed at reducing or preventing the development of childhood obesity. Recent data from the National Childhood Obesity Center in Sweden showed that the efficacy of long-term behavioral treatment for severely obese children was considerably higher in 6–9 year olds than older children (10–13 years) or adolescents (14–16 years) 37.
The findings from the current study also have methodological implications; they show that the genetic architecture underlying developmental increases captured by twin data can be replicated using DNA alone, providing strong support for the reliability of inferential statistics derived from the twin method. Given the time and expense incurred collecting DNA in large population-based cohorts, twins offer a convenient and affordable alternative to describing the genetic architecture of complex traits.
This study has several limitations. The sample used in this analysis only included children with genotyping and BMI data at 4 or 10 years (n = 2,556). This excluded a substantial proportion of the total TEDS sample. It is possible that parents of overweight and obese children were less willing to report weights, limiting the generalizability of the results. In relation to this, the sample was leaner than the 1990 reference value at age 4 (BMI-SDS, −0.12) but approximated the reference value at age 10 (BMI-SDS, −0.02), which may reflect the fact that although twins are born smaller than singletons, singleton-twin differences in body size decrease as twins get older and catch up 38. This may render the age 10 findings more generalizable than the findings at age 4. In addition, rates of overweight and obesity were relatively low at both ages, indicating that the sample was somewhat leaner than current UK children of the same age. Finally, the sample size limits the conclusions that can be drawn from the longitudinal GCTA analysis.
In conclusion, in this study we used GCTA and PRS analyses to show that twin-based evidence of increases in the heritability of BMI-SDS from early to later childhood are supported by genomic data; and twin and GCTA analyses suggest that many of the same genes are influencing adiposity at both ages. These findings underline the importance of intervening early in life for the prevention of childhood obesity.
Supporting Information
Additional Supporting Information may be found in the online version of this article.
References
- 1.Elks CE, den HM, Zhao JH, Sharp SJ, Wareham NJ, Loos RJ. Variability in the heritability of body mass index: a systematic review and meta-regression. Front Endocrinol (Lausanne) 2012;3:29. doi: 10.3389/fendo.2012.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maes HH, Neale MC, Eaves LJ. Genetic and environmental factors in relative body weight and human adiposity. Behav Genet. 1997;27:325–351. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- 3.Dubois L, Ohm KK, Girard M, et al. Genetic and environmental contributions to weight, height, and BMI from birth to 19 years of age: an international study of over 12,000 twin pairs. PLoS One. 2012;7:e30153. doi: 10.1371/journal.pone.0030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silventoinen K, Rokholm B, Kaprio J, Sorensen TI. The genetic and environmental influences on childhood obesity: a systematic review of twin and adoption studies. Int J Obes (Lond) 2010;34:29–40. doi: 10.1038/ijo.2009.177. [DOI] [PubMed] [Google Scholar]
- 5.Haworth CM, Carnell S, Meaburn EL, Davis OS, Plomin R, Wardle J. Increasing heritability of BMI and stronger associations with the FTO gene over childhood. Obesity (Silver Spring) 2008;16:2663–2668. doi: 10.1038/oby.2008.434. [DOI] [PubMed] [Google Scholar]
- 6.Bergen SE, Gardner CO, Kendler KS. Age-related changes in heritability of behavioral phenotypes over adolescence and young adulthood: a meta-analysis. Twin Res Hum Genet. 2007;10:423–433. doi: 10.1375/twin.10.3.423. [DOI] [PubMed] [Google Scholar]
- 7.Carnell S, Wardle J. Appetite and adiposity in children: evidence for a behavioral susceptibility theory of obesity. Am J Clin Nutr. 2008;88:222–9. doi: 10.1093/ajcn/88.1.22. [DOI] [PubMed] [Google Scholar]
- 8.Lajunen HR, Kaprio J, Keski-Rahkonen A, et al. Genetic and environmental effects on body mass index during adolescence: a prospective study among Finnish twins. Int J Obes (Lond) 2009;33(5):559–567. doi: 10.1038/ijo.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silventoinen K, Pietilainen KH, Tynelius P, Sorensen TIA, Kaprio J, Rasmussen F. Genetic and environmental factors in relative weight from birth to age 18: The Swedish Young Male Twins Study. Int J Obes. 2007;31:615–621. doi: 10.1038/sj.ijo.0803577. [DOI] [PubMed] [Google Scholar]
- 10.Silventoinen K, Bartels M, Posthuma D, et al. Genetic regulation of growth in height and weight from 3 to 12 years of age: a longitudinal study of Dutch twin children. Twin Res Hum Genet. 2007;10:354–363. doi: 10.1375/twin.10.2.354. [DOI] [PubMed] [Google Scholar]
- 11.Bradfield JP, Taal HR, Timpson NJ, et al. A genome-wide association meta-analysis identifies new childhood obesity loci. Nat Genet. 2012;44:526–531. doi: 10.1038/ng.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elks CE, Loos RJ, Sharp SJ, et al. Genetic markers of adult obesity risk are associated with greater early infancy weight gain and growth. PLoS Med. 2010;7:e1000284. doi: 10.1371/journal.pmed.1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elks CE, Loos RJ, Hardy R, et al. Adult obesity susceptibility variants are associated with greater childhood weight gain and a faster tempo of growth: the 1946 British Birth Cohort Study. Am J Clin Nutr. 2012;95:1150–1156. doi: 10.3945/ajcn.111.027870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hebebrand J, Volckmar AL, Knoll N, Hinney A. Chipping away the ‘missing heritability’: GIANT steps forward in the molecular elucidation of obesity—but still lots to go. Obes Facts. 2010;3:294–303. doi: 10.1159/000321537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haworth CM, Davis OS, Plomin R. Twins Early Development Study (TEDS): a genetically sensitive investigation of cognitive and behavioral development from childhood to young adulthood. Twin Res Hum Genet. 2012;16:117–125. doi: 10.1017/thg.2012.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barrett JC, Lee JC, Lees CW, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trzaskowski M, Eley TC, Davis OS, et al. First genome-wide association study on anxiety-related behaviours in childhood. PLoS One. 2013;8:e58676. doi: 10.1371/journal.pone.0058676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wardle J, Carnell S, Haworth CM, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87:398–404. doi: 10.1093/ajcn/87.2.398. [DOI] [PubMed] [Google Scholar]
- 22.Cole TJ, Freeman JV, Preece MA. Body-mass index reference curves for the UK, 1990. Arch Dis Childhood. 1995;73:25–29. doi: 10.1136/adc.73.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cole TJ. Software for LMS method: LMS Growth Program [computer program: MS Excel add-ins]. Version 2.64. London, England: Child Growth Foundation; August 21, 2008.
- 24.Boker S, Neale M, Maes H, Wilde M, Spiegel M, Brick T. OpenMx: an open source extended structural equation modeling framework. Psychometrika. 2011;76:306–317. doi: 10.1007/s11336-010-9200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plomin R, DeFries JC, Knopik VS, Neiderhiser JM, editors. Behavioral Genetics. 6th ed. New York: Worth Publishers; 2012. [Google Scholar]
- 26.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Weedon MN, Purcell S, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–812. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28:2540–2542. doi: 10.1093/bioinformatics/bts474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- 30.Sovio U, Mook-Kanamori DO, Warrington NM, et al. Association between common variation at the FTO locus and changes in body mass index from infancy to late childhood: the complex nature of genetic association through growth and development. PLoS Genet. 2011;7:e1001307. doi: 10.1371/journal.pgen.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belsky DW, Moffitt TE, Houts R, et al. Polygenic risk, rapid childhood growth, and the development of obesity: evidence from a 4-decade longitudinal study. Arch Pediatr Adolesc Med. 2012;166:515–521. doi: 10.1001/archpediatrics.2012.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trzaskowski M, Davis OS, DeFries JC, Yang J, Visscher PM, Plomin R. DNA evidence for strong genome-wide pleiotropy of cognitive and learning abilities. Behav Genet. 2013;43:267–273. doi: 10.1007/s10519-013-9594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Llewellyn CH, van Jaarsveld CH, Plomin R, Fisher A, Wardle J. Inherited behavioral susceptibility to adiposity in infancy: a multivariate genetic analysis of appetite and weight in the Gemini birth cohort. Am J Clin Nutr. 2012;95:633–639. doi: 10.3945/ajcn.111.023671. [DOI] [PubMed] [Google Scholar]
- 34.Wardle J, Carnell S, Haworth CM, Farooqi IS, O'Rahilly S, Plomin R. Obesity associated genetic variation in FTO is associated with diminished satiety. J Clin Endocrinol Metab. 2008;93:3640–3643. doi: 10.1210/jc.2008-0472. [DOI] [PubMed] [Google Scholar]
- 35.Wardle J, Llewellyn C, Sanderson S, Plomin R. The FTO gene and measured food intake in children. Int J Obes (Lond) 2009;33:42–45. doi: 10.1038/ijo.2008.174. [DOI] [PubMed] [Google Scholar]
- 36.Llewellyn CH, Trzaskowski M, van Jaarsveld CHM, Plomin R, Wardle J. Satiety mechanisms in genetic risk of obesity. JAMA Pediatrics. 2013. doi: 10.1001/jamapediatrics.2013.4944. [DOI] [PMC free article] [PubMed]
- 37.Danielsson P, Kowalski J, Ekblom O, Marcus C. Response of severely obese children and adolescents to behavioral treatment. Arch Pediatr Adolesc Med. 2012;166:1103–1108. doi: 10.1001/2013.jamapediatrics.319. [DOI] [PubMed] [Google Scholar]
- 38.Buckler JM, Green M. A comparison of the early growth of twins and singletons. Ann Hum Biol. 2004;31:311–332. doi: 10.1080/03014460410001670120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.