

Abstract
Proliferating ducts, termed “oval cells”, have long thought to be bipotential, i.e. produce both biliary ducts and hepatocytes during chronic liver injury. The precursor to oval cells is considered to be a facultative liver stem cell (LSC). Recent lineage tracing experiments indicated that the LSC is Sox9+ and can replace the bulk of hepatocyte mass in several settings. However, no clonal relationship between Sox9+ cells and the two epithelial liver lineages was established. We labeled Sox9+ mouse liver cells at low density with a multicolor fluorescent confetti reporter. Organoid formation validated the progenitor activity of the labeled population. Sox9+ cells were traced in multiple oval cell injury models using both histology and FACS. Surprisingly, only rare clones containing both hepatocytes and oval cells were found in any experiment. Quantitative analysis showed that Sox9+ cells contributed only minimally (<1%) to the hepatocyte pool, even in classic oval cell injury models. In contrast, clonally marked mature hepatocytes demonstrated the ability to self-renew in all classic mouse oval cell activation injuries. A hepatocyte chimera model to trace hepatocytes and non-parenchymal cells also demonstrated the prevalence of hepatocyte-driven regeneration in mouse oval cell injury models.
Conclusion
Sox9+ ductal progenitor cells give rise to clonal oval cell proliferation and bipotential organoids but rarely produce hepatocytes in vivo. Hepatocytes themselves are the predominant source of new parenchyma cells in prototypical mouse models of oval cell activation.
Keywords: liver regeneration, chronic liver injury, lineage tracing, hepatocyte, ductal proliferation
The proliferation of ductal liver progenitor cells, or “oval cells”, is a hallmark of chronic human and experimental liver injuries(1,2). In humans oval cells are prominently observed in common forms of chronic liver disease leading to cirrhosis(3). They are thought to provide an alternative path to parenchymal regeneration when hepatocyte replication is blocked(4). Oval cells are proposed to originate from a quiescent, facultative stem cell anatomically located at the interface between bile ducts and hepatocytes in the “Canal of Hering”. However, a debate regarding their precise origin dates back over 50 years(5-7). In rodent chronic liver injury models, oval cells can differentiate into hepatocytes upon recovery(8), transplantation (7,9) and in vitro differentiation (10,11). Interestingly, phenotypically defined duct-like cells isolated from normal liver do not demonstrate the same efficiency of hepatocyte differentiation, especially in transplantation assays(12,13). Defining the cell of origin responsible for the regeneration of hepatic parenchyma is key to devising pharmacologic strategies to modulate the oval cell response in chronic liver disease and for advancing cell-based liver therapy.
Recent lineage tracing experiments have yielded disparate results in well-studied mouse oval cell activation models (13-16). Furuyama et al. used a Sox9-IRES-CreERT2 lineage tracing approach and found the Sox9+ biliary compartment contributed the majority of new hepatocytes even during normal liver homeostasis(14). This was further accelerated by injury. Subsequent work by Malato et al. labeled all hepatocytes with Cre-recombinase delivered by adeno-associated virus(15). In contrast to Furuyama, they found that only a small percentage of hepatocytes were derived from non-parenchymal (NPC) precursors and only following certain injuries. A limitation of these and other prior studies (13,16,17) is that the biliary or non-parenchymal compartments were traced en masse, which precludes the identification of clonal relationships between hepatocytes and ductal progenitors. Evidence that tamoxifen can induce “ectopic” expression of ductal markers in hepatocytes (18) and that biliary transcription factors are expressed in normal hepatocytes (19) suggested that a clonal labeling strategy was needed to directly identify the origin of hepatocyte precursor cells in liver repair.
The aim of our study was to use in vivo clonal analysis to directly identify bipotential adult liver stem cells and understand their function in injury. We used low density clonal labeling in classic models of oval cell activation to separately track the progeny of adult biliary cells and hepatocytes. As a second approach we used hepatocyte-chimeras generated by transplantation to determine the contribution of NPC in models of oval cell activation. Our results indicate that bipotential hepatic progenitors of Sox9+ ductal origin do not contribute significantly to hepatocyte replacement even in traditional mouse oval cell injury models. Instead, hepatocytes themselves are the predominant source of new parenchymal cells.
Results
Clonal labeling of ductal progenitors
We hypothesized that clonally marking Sox9+ cells followed by oval cell injury would reveal bipotential clones containing both ducts (self-renewal) and hepatocytes (stem cell differentiation). Towards this end Sox9-CreERT2 R26R-Confetti multi-color stochastic reporter mouse was generated and used to establish the tamoxifen dose suitable for clonal labeling. Recombination of the confetti allele irreversibly turned on one of three mutually exclusive fluorescent proteins. Given that high doses of tamoxifen induces Sox9 expression in hepatocytes (18), we first sought to determine quantities that would avoid significant levels of hepatocyte marking (SFig. 1).
With limiting doses of tamoxifen, the Sox9-CreERT2 recombination rate was roughly a linear function of tamoxifen dose, as assessed by immunofluorescence and FACS-based analysis in phenotypically defined MIC1-1C3+ ductal progenitor cells (Fig. 1a)(12). Confetti-marked periportal hepatocytes were readily observed in uninjured animals treated with 250mg/kg tamoxifen and rarely after 125mg/kg tamoxifen. In contrast, hepatocyte marking was undetectable in animals treated with ≤62mg/kg tamoxifen.
Figure 1. Sox9 clonal lineage tracing identifies ductal progenitors but not hepatocytes in homeostasis.
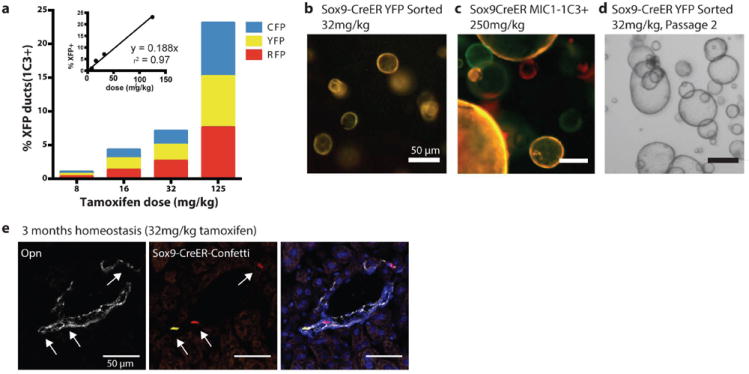
(a) Sox9-CreERT2 R26R-Confetti+/- mice dosed with tamoxifen at limiting dilutions produced recombination in ductal progenitor cells (CD45- CD31- MIC1-1C3+) (b) FACS sorted MIC1-1C3+, Sox9-CreERT2 Confetti+ marked single cells initiated self-renewing organoids (YFP sorted) after 10 days culture or (c) 14 days (YFP+, mCerulean+, or RFP+ sorter) (d) Clonally labeled organoids self-renewed after passage. (e) Sox9-CreERT2 clonally marked cells continued to express ductal marker Opn after 3 months of homeostasis (scale bars = 50μm).
In order to verify that low doses of tamoxifen indeed labeled ductal progenitors we applied the organoid formation assay(13). This progenitor assay identifies clonogenic cells with extensive self-renewal capacity that arebipotential in vitro and give rise to hepatocytes upon transplantation. Single FACS isolated Sox9-CreERT2 marked cells were seeded into organoid cultures. Organoid initiation was observed at all tamoxifen doses after 10 (Fig. 1b) or 14 days (Fig. 1c), demonstrating the stochastic marking of a functionally homogenous population. Organoids were of a single color, confirming they clonally initiated from single cells (Fig. 1c). The self-renewal potential of Sox9-CreERT2 marked organoids was shown by serial passage (Fig. 1d). This result establishes that low doses of tamoxifen marked the Sox9+ clonogenic progenitors in vivo.
Sox9+ tracing without injury
To determine whether Sox9+ cells are a continuous source of new hepatocytes in homeostasis, labeling was carried out in adult mice without hepatic injury. Adult Sox9-CreERT2 R26R-Confetti mice were injected with either 32 or 125 mg/kg tamoxifen (n=2). On a normal diet clonally labeled (32mg/kg) Sox9+ labeled cells continued to express only the biliary duct markers Sox9, A6, and Osteopontin but never the hepatocyte marker Hnf4a after 3 months (n=500 clones, Fig 1e) and 6 months (SFig. 2). When periportal hepatocytes were labeled with high levels of tamoxifen (>125mg/kg), they remained in the periportal location and did not stream or replace the hepatic parenchyma after 3 or 6 months (SFig. 2).
Sox9+ tracing in CDE induced oval cell injury
Steatotic injury induced by the CDE diet is one of the prototypical and most widely used oval cell induction regimens (13,14,16,17,20). To trace the fate of Sox9+ cells after CDE injury, cells were first marked by recombination followed by a two-week rest period for tamoxifen to dissipate (Fig. 2a). Mice were then treated according to previously described injury regimens associated with Sox9+/Opn+ progenitor-to-hepatocyte differentiation(16). After 3 weeks of CDE diet followed by a 2-week recovery period, sections were examined for evidence of duct-to-hepatocyte differentiation(16). Prominent ductal proliferation and branching was observed in animals when 2.5-4% of Sox9+ ducts were marked one of three colors each (mCerulean, YFP, or RFP; the total of MIC1-1C3+ cells was 8-11%, Fig. 2b). Unexpectedly, no clones containing both oval cells and hepatocytes were found. In fact, hepatocyte marking was extremely rare. Co-immunohistochemistry with Hnf4a, A6, Osteopontin, and Sox9 confirmed the progeny of Sox9-Cre marked cells were only biliary cells. 500 Confetti+Opn+ or A6+ clones were scored in multiple sections treated with CDE (Fig. 2c, n=4 mice). Only a single YFP+ hepatocyte clone was identified adjacent to a portal vein (Fig. 2d) but adjacent ductal cells were mCerulean+ or RFP+, indicating that the marked hepatocyte was not clonally related to the marked ductal cells.
Figure 2. Sox9+ ducts rarely give rise to hepatocytes in CDE injury.
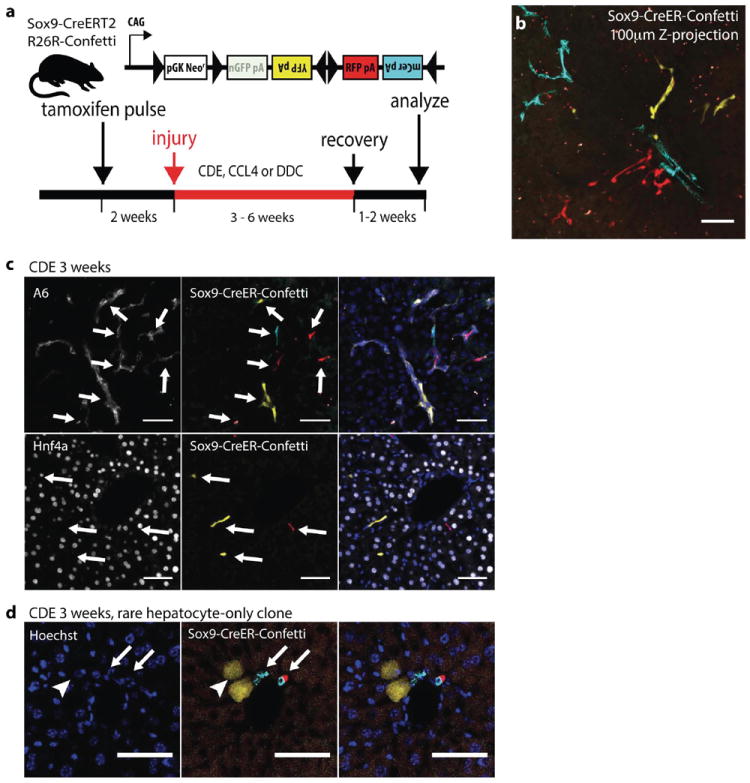
(a) Experimental scheme: 4-6 weeks old Sox9-CreERT2 R26R-Confetti+/- mice were given a single dose of tamoxifen followed by oval cell injury two weeks later (b) CDE diet produced ductal proliferation after low density Sox9-labeling. A Z-projection generated from confocal analysis of 100 μ m thick liver section (32mg/kg tamoxifen). Ductal proliferation was not associated with hepatocyte-differentiation (c) Immunostaining for ductal markers A6 (top), Hnf4a (bottom) confirmed Sox9-CreERT2 marked cells did not differentiate into hepatocytes after recovery from CDE injury. (d) 2 YFP+ cells with distinct hepatocyte morphology (arrowhead) were adjacent to clonally unrelated mCerulean+ and RFP+ cholangiocytes (arrows). Scale bars = 50μm.
FACS analysis of single cell dissociated livers was used as a second method to quantify and phenotype Sox9-marked cells. MIC-1C3 is a reliable surface marker for biliary duct cells, including progenitors(12). Conversely, 2F8 is a hepatocyte-specific surface marker(13). Fluorescently marked cells were mostly MIC-1C3 positive, indicating a ductal phenotype. In concordance with the histological results, labeled 2F8+ hepatocytes were rare, comprising less than 0.1% of the total (SFig. 3). Importantly, FACS sorted Sox9-CreER-Confetti+ cells from CDE injured animals retained their ability to form organoids in vitro at all labeling densities (SFig. 4).
Sox9+ tracing in CCl4 injury
We next assessed the fate of clonally labeled Sox9+ cells in chronic and acute CCL4 injury. After five-weeks of chronic CCL4 injury and a one-week recovery period, livers were examined for evidence of hepatocytic differentiation. In clonal tracing experiments, we found confetti-marked cells continued to co-localize with Opn+/Sox9+ ductal structures but never with Hnf4a+ hepatocytes (n=500 clones, n=3 animals; Fig. 3a). FACS-based hepatocyte quantification confirmed histologic findings that Sox9-CreERT2 marked hepatocytes were extremely rare after injury (SFig. 3). Acute CCl4 injury tracing produced the similar results (SFig. 5). Finally, Sox9-CreERT2-Confetti+ cells from CCl4 injured animals formed organoids in vitro at all labeling densities (SFig. 4).
Figure 3. Sox9+ ducts do not differentiate into hepatocytes in DDC or CCl4 injury.
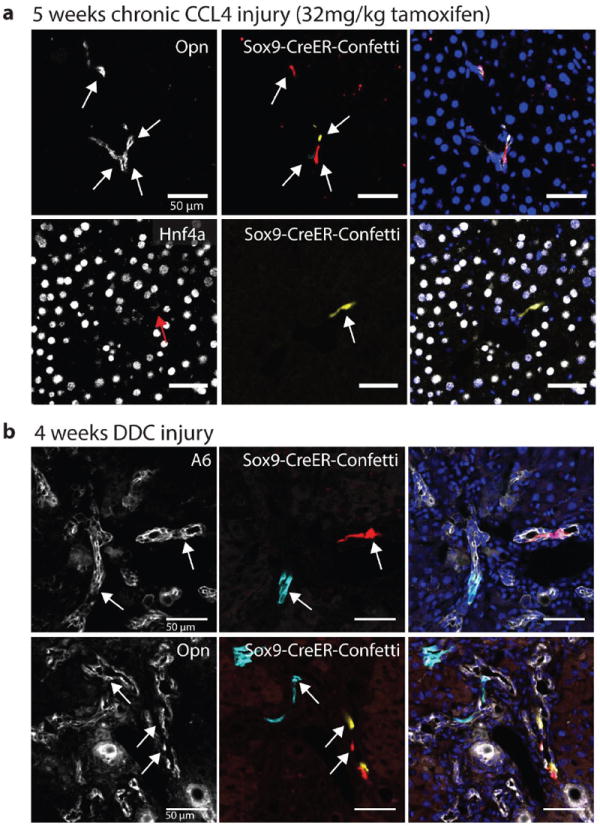
(a) Immunofluorescence for ductal marker Opn (top) and hepatocyte marker Hnf4a (bottom) showed Sox9-CreERT2 marked cells retained ductal fate after regeneration from 5 weeks CCl4 injury (arrows indicate a unique clone). (b) Immunofluorescence for ductal markers A6 (top) and Opn (bottom) continued to co-localize with Sox9-CreERT2 marked clones after 4 weeks of DDC injury. Scale bars = 50μm.
Sox9+ tracing in DDC induced oval cell proliferation
After Sox9-CreERT2 clonal labeling and a 2-week washout period, mice were given DDC (3,5-diethoxycarbonyl-1,4-dihydrocollidine) for 4 weeks(10,12). Similar to the CDE and CCl4 injury regimens we found that Confetti-marked cells exclusively localized with A6 or Opn+ ductal cells (500 clones, n=3 mice, Fig. 3b). This indicated that proliferating ducts do not differentiate into hepatocytes in this model.
Generating mice with chimeric livers
We next wished to validate our unexpected findings with a second approach independent of Cre-recombination. Chimeras generated by transplantation have previously been used to track the respective contributions of adult cell types in organ regeneration(21). Our overall approach was similar to the one used by Malato et al by generating animals with genetically marked hepatocytes, followed by oval cell injury(15). As in their study, the goal was to determine whether nonparenchymal precursors (not only ductal cells) would significantly contribute to hepatocyte regeneration following injury.
To obtain a high labeling efficiency of mature hepatocytes, we used the Fah-mutant mouse model which allows for the specific and reproducible replacement of >95% of hepatocytes with donor-marked hepatocytes through therapeutic liver repopulation(22). Fah-mutant hepatocytes accumulate toxic metabolites and are unable to proliferate(23). Importantly, the deleterious effects of Fah-deficiency and hepatocyte replication arrest are completely reversed by treatment with the small molecule nitisinone (NTBC) (SFig 10)(22).
We generated chimeric mice by transplanting syngeneic hepatocytes from ROSA-lacZ or ROSA-mTomato marker strains into recipient Fah-mutant mice (Fig. 4a). Liver repopulation was assessed 10-weeks after transplantation. 95-99.4% of hepatocytes were donor derived as measured by Fah-immunostaining or FACS (Fig. 5b,c). For histologic analysis, we focused on tissue away from the capsule where donor hepatocyte content consistently was >99% before injury (n=6).
Figure 4. Robust and specific replacement of adult hepatocytes but not other cells in Fah-chimeric mice.
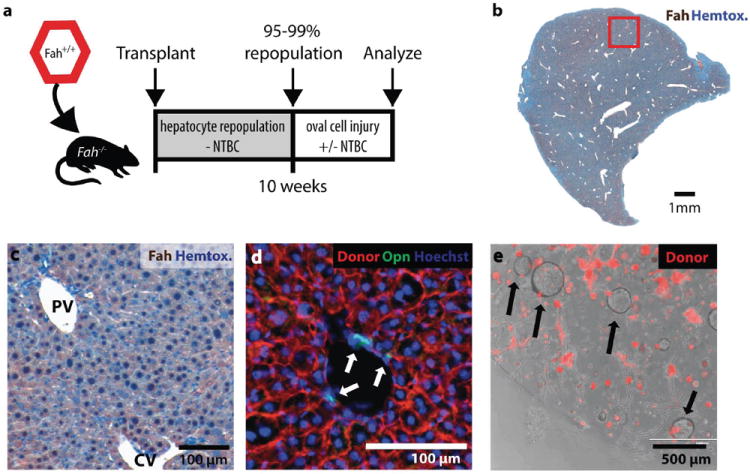
(a) Experimental scheme: gravity enriched wildtype or mTomatoFah+/+ hepatocytes were transplanted into the spleen of Fah-/- mice and allowed to repopulate (95-99%).
(b) Fah-immunostaining (brown) shows >99% of hepatocytes were donor derived after 10 weeks repopulation (bar = 1mm) (c) Complete central vein (CV) to portal vein (PV) repopulation with donor Fah+ hepatocytes (brown). (d) Donor cell marker (mTomato red) did not colocalize with Osteopontin+ (green) host duct cells. (e) Organoids (arrows) formed from dissociated chimeric liver were host derived (mTomato negative). mTomato+ hepatocytes did not form organoid spheres after 12 days culture.
Figure 5. Progenitor derived hepatocytes are not required for adaptation to oval cell injury in chimeric mice.
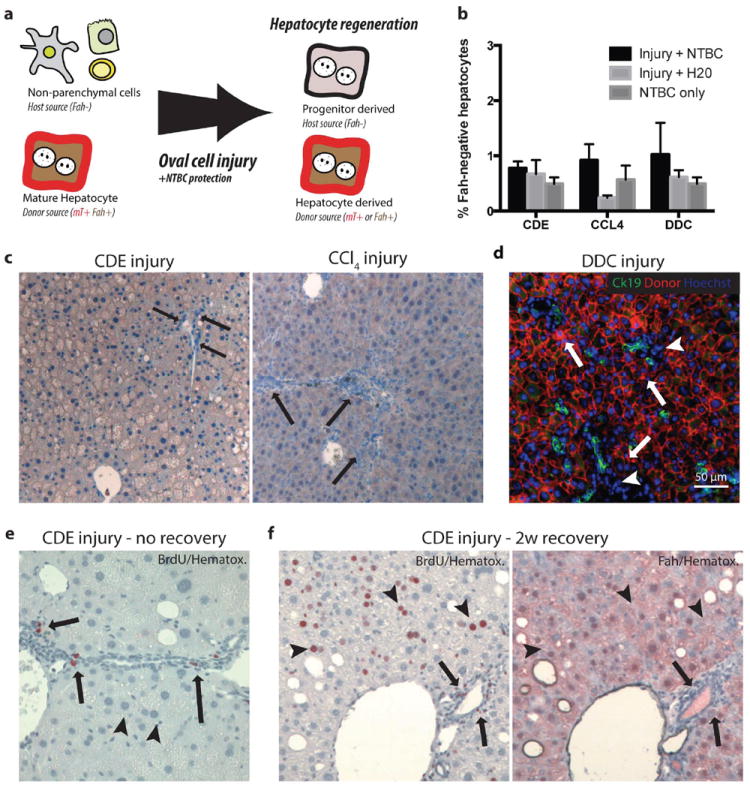
(a) Schematic: Hepatocyte transplantation into Fah-/- mice generated chimeric mice, host nonparenchymal cells were Fah- and unmarked. Donor hepatocytes were Fah+ and mTomato-marked. If nonparenchymal cells gave rise to hepatocytes after oval cell injury, we hypothesized that progenitor-derived Fah- hepatocytes would displace marked Fah+ hepatocytes. (b) Image quantification of Fah-negative hepatocytes demonstrates approximately 1% of hepatocytes remain host derived after injury. (c) No Fah-negative progenitor derived hepatocytes were observed at the majority portal regions (arrow = ductal proliferation) after 3 weeks CDE injury plus NTBC rescue or 10 weeks CCl4 injury plus NTBC (arrow = portal inflammation, ductal proliferation but no Fah- hepatocytes). (d) After 4 weeks DDC injury plus NTBC treatment CK19+ (green) ductal proliferations were adjacent to donor mTomato (red) hepatocytes (arrows) and inflammatory nonparenchmal cell (arrowhead, mTomato-negative) (bar=50μm). (e) BrdU uptake (2-hour pulse) was observed in proliferating ductal and nonparenchymal cells (arrow) but not hepatocytes at the peak of CDE injury. Hepatocytes show microvesicularsteatosis. (f) BrdU uptake (2 weeks BrdU water) during recovery from CDE injury was observed in hepatocytes (arrowheads), inflammatory cells, and cholangiocytes (arrows). Serial sectioning demonstrates that regenerating hepatocytes are lineage-marked mature hepatocytes (Fah+) and not derived from a nonparenchymal progenitor (Fah-).
No transplanted donor cells were found to express biliary progenitor markers (CK19/Osteopontin/MIC1-1C3) (>4,500 ducts surveyed, n=3 mice, Fig. 4) or other non-parenchymal cell markers (SFig. 6) by immunohistochemistry or FACS. In vitro organoid formation showed that all progenitors were host-derived (198/198, n=2 animals) (Fig. 4e). Because hepatocytes but not non-parenchymal cells were specifically replaced with marked transplanted cells, we proceeded to use chimeric mice to study the non-parenchymal cell compartment in oval cell injuries.
Lineage tracing in hepatocyte chimeras
Current models regarding oval cell response hold that the putative LSC is activated because the hepatocytes themselves cannot regenerate(4). This model predicts that the percentage of hepatocytes derived from non-hepatocyte precursors will significantly increase at the expense of the disabled donor hepatocytes during oval cell injury. In hepatocyte chimeric animals these cells can be readily identified, because they are of host origin and hence negative for the marker (either lacZ or mTomato). Newly born progenitor derived hepatocytes would be protected from the deleterious effects of Fah-deficiency if treated with NTBC (SFig. 7a). Conversely, new Fah-/- hepatocytes would be expected to be functionally impaired if the animal is off NTBC (SFig. 9). Thus, we hypothesized that only NTBC-treated chimeras would survive the oval cell injury, if progenitor derived new hepatocytes were necessary for survival.
After complete repopulation, chimeric mice were given the CDE diet for 3 weeks with or without NTBC. A control cohort received NTBC alone. After injury, mice recovered for two weeks to permit progenitor differentiation(16). Injury on NTBC therapy (newly born hepatocytes are functional) was associated with only a minimal increase in mean marker-negative host-derived hepatocytes (0.77% ± 0.12%) compared with injury alone (0.67% ± 0.25%, p=0.8) or NTBC alone (0.49% ± 0.11%, p=0.4) (n=3 per group, Figure 5b,c). Animal survival, body weight, or liver weight were also similar between groups, indicating the small differences between groups were not functionally significant (SFig. 6). A two-hour BrdU pulse given at the peak of CDE injury showed proliferation of NPC in the portal tract and microvesicularsteatosis in hepatocytes(Fig. 5f). Upon recovery, we detected robust hepatocyte regeneration and proliferation with 2-week continuous BrdU treatment (Fig. 5g). Serial section analysis indicated BrdU+ hepatocytes were derived from lineage-marked, mature hepatocytes (Fig. 5h).
Next, repeated toxic injury with twice-weekly CCL4 (0.5ul/kg) was given for 10 weeks with or without NTBC. Overall, only 0.24-0.92% of all hepatocytes were of host origin in animals after injury (n=3 per group, Fig. 5b). NTBC protection during injury produced no significant increase of host-derived hepatocytes (p = 0.18) or global measures of animal health compared with injury alone. BrdU uptake was observed in non-parenchymal cells 72 hours after injury and in donor-marked hepatocytes upon recovery from chronic injury (SFig. 7).
Finally, we tested whether chimeric Fah-/- mice required NTBC to adapt to 5 weeks of DDC injury. As in CCL4 and CDE injury, putative progenitor-derived mTomato-negative hepatocytes were only rarely found next to proliferating ducts(Fig. 5e). mTomato negative hepatocytes trended higher in the DDC-plus-NTBC group (1.03% versus 0.62%, p=0.4, Fig. 5i), but these differences did not effect survival or global measures of liver function (SFig. 7). FACS-based quantification confirmed that NTBC did not increase progenitor-derived hepatocytes compared with injury alone (p=0.88, n=3, SFig. 7).
Mature hepatocytes self-renew in oval cell injuries
To further test this self-renewal potential of hepatocytes, we examined the clonal dynamics of mature hepatocytes in oval cell injury. R26R-Confetti+/- micewere injected with AAV8-Ttr-Cre to establish sparse, specific hepatocyte labeling (SFig. 8). Each allele in a R26R-Confetti+/- mouse randomly recombined to mark cells in one mutually exclusive fluorescent color. This produced 6 observable color patterns due to the sometimes polyploid nature of adult hepatocytes. At baseline, over 95% of marked cells were identifiable as single cells (Fig. 6b). Two weeks later, partial hepatectomy was performed to eliminate residual rAAV by cell replication. Upon regeneration from partial hepatecomy, we observed either single marked cells or clonally related clusters of two cells, in agreement with others (24). Colonies of hepatocytes were stable as mostly one and two cell clones for up to 6 months (Fig. 6c).
Figure 6. Mature hepatocytes self-renew in oval cell injuries.
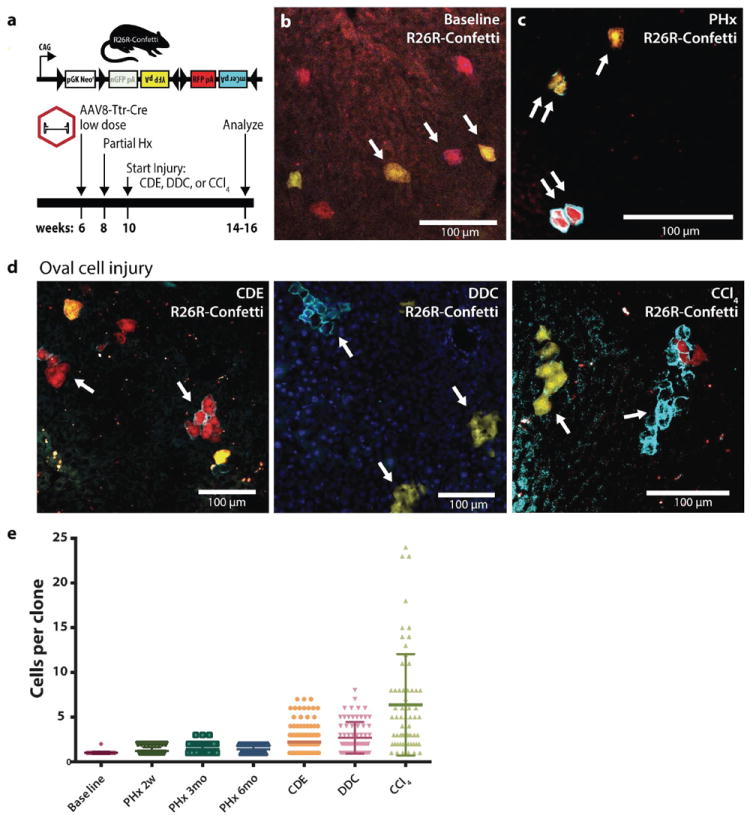
(a) Experimental scheme: Hepatocytes were marked at low density in heterozygous R26R-Confetti+/- mice using low dose rAAV8-Ttr-Cre. Partial hepatectomy was performed and injuries were initiated after complete regeneration. (b) Prior to hepatectomy single hepatocytes were marked. (c) Partial hepatectomy induced hepatocyte replication producing clones of 1 (single arrow) or two identically marked cells (double arrow). (d) Oval cell injury with CDE diet, DDC diet, or chronic carbon tetrachloride induced mature hepatocyte replication and self-renewal or clonal expansion. (e) Dot plot histogram of mature hepatocyte colony size at baseline, partial hepatectomy followed by 2 weeks to 6 months homeostasis, and after oval cell injury (line = mean ± SD). Scale bars = 100μm.
Next, we induced prototypical chronic liver injuries previously associated with oval cell activation two weeks after recovery from hepatectomy. Mean hepatocyte clone size increased considerably following injury (1.5-4.5 fold, Fig. 6g), indicating that mature hepatocytes marked prior to injury were an important source of new hepatocytes. In CDE and DDC injuries 20% of surviving hepatocytes were part of a cluster of 5 cells or more (SFig. 8). After chronic CCl4 injury, a majority of marked surviving hepatocytes were part of a clone of 10 or more cells.
Discussion
First, our results indicate that Sox9+ precursors do not continuously generate new hepatocytes in normal physiologic conditions, consistent with multiple other reports(15,16,18,25). Even when periportal hepatocytes were marked with Sox9-CreERT2 (high tamoxifen), marked hepatocytes did not continuously replace the parenchyma (14,17)(SFig. 2). rAAV clonal tracing (Fig. 6) similarly underscored the low turnover rate of hepatocytes in normal homeostasis and positional stability along the portal-central axis (SFig 8f).
Second, our data show that Sox9+ductal proliferation makes only a minor contribution to parenchymal regeneration in diverse liver injuries. Low-density clonal lineage tracing in oval cell activation injury, showed that Sox9+ cells gave rise to highly proliferative oval cells(18) but we found little evidence that oval cell activation was associated with efficient hepatocyte differentiation. Our results in DDC induced injury are similar to several previous studies (14-16,20) but in contrast to other reports (12,13,26). Our findings in CCl4-mediated regeneration are in closer agreement with older studies suggesting CCl4-mediated regeneration is primarily driven by hepatocyte self-renewal (4) and less consistent with a stem-cell based mechanism (13-15). We also find little evidence for hepatocytic differentiation of progenitors in the CDE oval cell activation regimen, in contrast to a recent study using Opn-lineage tracing(16).
Given the long half-life of active tamoxifen metabolites in mice (t1/2= 5-7 days)(27) and the sensitivity of CreERT2 at low tamoxifen at low levels (Fig. 1a), it is likely that cre-recombination continues for several half-lives after tamoxifen administration. In fact, we have directly shown that tamoxifen persists in the liver one week after injection (SFig 11), the time at which CDE injury was initiated in the Opn lineage tracing study(16). Consistent with our own result, others have found that tamoxifen can persist up to 4 weeks after injection(28). Thus the low hepatocyte labeling frequency observed by Español-Suñer et al. could be the result of CDE-induced Opn-expression in hepatocytes(29).
Formally, the results raise the possibility that our low-tamoxifen dosing strategy simply failed to mark a hypothetical biphenotypic Sox9low Krt19low resident stem cell. Two experiments argue against this. First, Sox9-CreERT2 marked cells behave functionally similar in an in vitro functional assay for progenitor activity regardless of tamoxifen dose, suggesting that high and low doses of tamoxifen label the same population of cells. Second, in our chimera lineage-tracing experiments, transplantation only replaced mature hepatocytes. Thus, this model system generally tests whether any NPC, such as a Sox9low biliary cell or even a Sox9-negative stellate cell (30), has the capacity to differentiate into hepatocytes. Therefore, it is unlikely that our strategy systematically overlooked a subset of biliary cells. Evidence that Sox9 and other biliary cell genes are expressed in mature hepatocytes (18,19) raises the possibility that the hypothetical Sox9low Krt19low Opnlow progenitors are simply injured periportal hepatocytes.
Our chimera experiments further confirmed the scarce contribution of NPC precursors to the hepatocyte pool after oval cell activation. NTBC treatment had no effect on animal survival or global measures of liver function in response to any of the oval cell injury regimens used (SFig. 7), providing functional evidence that rescue of a progenitor population was not required for adaptation to these injury models. This result strongly suggested Fah+/+ donor hepatocytes contribute significantly to liver function and that a newborn progenitor had only a minor contribution in oval cell injury.
Our study assessed liver function by survival, liver size, and animal weight (Fig. 5, SFig. 7) as previously described (26,31) but we could have missed other differences detectable by more sensitive synthetic liver function tests (31). Our results do not determine conclusively whether rare marked hepatocytes are a result of stem cell differentiation, transdifferentiation of a biliary progenitor, or direct Sox9-CreERT2-marking of hepatocytes themselves. Our experiments would have readily detected a hepatocytic contribution of 5% or more, the level we deem to have a functional impact in the short term. However, it is important to note that the cumulative effect of progenitor-to-hepatocyte differentiation at a constant low rate could hypothetically reach a functionally significant level over time in long-term chronic injury. Our experiments therefore do not rule out a functionally significant contribution of progenitors in long-term liver injury, such as chronic hepatitis in humans.
The ease of detecting self-renewing hepatocytes—in all injuries—starkly contrasted with the paucity of potential progenitor-derived hepatocytes in Sox9-CreERT2 clonal tracing experiments with the same R26R-Confetti reporter. The distribution AAV8-Ttr-Cre marked hepatocytes clone size indicates that a subset of hepatocytes divided many times while other hepatocytes did not expand in DDC and CDE injuries.
Continued hepatocyte replication raises the possibility that existing mouse injury regimens are not sufficient to evoke true progenitor activation (4). Due to the lack of lineage tracing tools in other organisms, it is currently unknown whether our results are generalizable to human liver injuries or rats, the organisms in which oval cell progenitors were first described(1,3).
While our study found no difference in the survival of chimeric animals with ducts that lacked hepatocyte potential, cholangiocyte and oval cell proliferation may still be important to overall regeneration. Two recent studies showed that blocking ductal proliferation in DDC injury decreased survival (26,31). Therefore, while ductal proliferation does not provide a significant source of new hepatocytes, expansion of the ductal compartment could instead act as a scaffold for periportal hepatocyte regeneration or by altering hepatocyte gene expression through juxtacrine or paracrine signaling.
Recent reports that mature hepatocytes retain the potential to morph into duct-like cells, raise the possibility that hepatocytes can become Sox9+ after injury and appear similar in morphology and gene expression to cholangiocytes (32-34). Our data provide an opening for the hypothesis that hepatocyte-regeneration in chronic injury models is entirely a result of hepatocyte-self renewal and differentiation of hepatocyte-derived oval cells—rather than activation of a resident facultative stem cell. Future work will be required to characterize the function of hepatocyte subsets in regeneration and develop better models of hepatocyte senescence to study putative progenitors.
Materials and methods
Mice husbandry and Cre-recombination
Sox9-CreERT2 mice(35) were backcrossed 3 generations with C57bl/6 mice and then bredwith R26R-Confetti mice(36). Tamoxifen (Sigma) was dissolved in sesame oil (20 mg/ml) and administered intraperitoneally to 4-6 week old mice. Dosing ranged from 8-250mg/kg. As a negative control for recombination, mice were given sesame oil. A two-week washout period was allowed for tamoxifen levels to dissipate prior to starting injuries. For AAV8-Ttr-Cre lineage tracing, heterozygous R26R-Confetti mice were given 2 × 1010 viral genomes by retro-orbital injection. Two weeks later, 50-66% partial hepatectomy was performed. Two weeks were allowed for recovery/regeneration prior to injury initiation. The Oregon Health and Science University IACUC approved all animal experiments described.
Hepatocyte transplantation and chimeric Fah-/- mice
C57bl/6 Fah-/- mice were maintained on 8mg/L NTBC (2-(2- nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione) (Yecuris Corp.) drinking water until 1 day prior to transplant. Primary hepatocytes were freshly isolated by a 2-step collagenase perfusion from Fah-wildtype mice carrying either ROSA26-lacZ or ROSA26-mTomato/mGFP genes. Hepatocytes were enriched by a series of low speed (1’ × 50g) centrifugation steps(22). Then 500,000 live hepatocytes resuspended in 150uL in DMEM/10% FBS/0.5% green food coloring were injected percutaneously into the spleen of Fah-/- mice 3-6 weeks of age. Fah-/- mice that showed evidence of weight stabilization by 4 weeks were allowed to repopulate for 6 additional weeks before injuries were initiated.
Diets and injury regimens
Mice were maintained on Purina 5015 diet with or without supplementation with 0.1% wt/wt DDC (3,5-diethoxycarbonyl-1,4-dihydrocollidine)(12) (Harlan Laboratories). To induce steatosis, mice were fed a diet deficient in choline (MP Biomedicals)(13,16) supplemented with 0.1% (wt/vol) ethioninewater (Sigma) for 3 weeks and allowed to recover for 2 weeks on Purina 5015 before harvest. CCL4 injury was induced with i.p.injection of 1uL/kg CCL4 (acute injury; Sigma) or 0.5uL/kg body weight 2 times/week for 5-10 weeks (chronic) and examined 1 week after the last injection CCL4 (13,15). Control mice were given an equal volume of corn oil vehicle. BrdU was injected 2 hours prior to sacrifice (150mg/kg) or given continuously in drinking water (0.5mg/mL) supplemented with 1% dextrose where indicated.
Immunohistochemistry and imaging
Livers were perfused with cold 4% paraformaldehyde/PBS and post-fixed for 2-6 hours. Tissues were then either cut on a vibrating microtome in 50-100μm sections or cryopreserved in 25% sucrose/PBS for cryosectioning in 8-10μm sections. Immunohistochemistry was performed on fixed/frozen cryosections with all steps conducted at 4°C. mCerulean, eYFP, tdimer(RFP), and mTomato were directly detected on a Zeiss LSM780 with similar laser and filter settings as previously described(36). High-level Cre-expression is required to produce nGFP recombination, and was not observed with the doses of tamoxifen used. Primary antibodies (Supplementary Table 1) were detected with AF647-conjugated secondary antibodies (Jackson ImmunoResearch), counterstained with Hoechst 33258, and mounted with Prolong Gold (Invitrogen).
Cell culture
Biliary ducts were isolated by collagenase-based perfusion(12). Isolated ducts were either FACS sorted into single cell suspensions or directly mixed with matrigel, as previously described(13). Organoid cultures were analyzed or passaged after 10-14 days.
Supplementary Material
Supporting Information Figure 1: Sox9-CreERT2 marks hepatocytes with high tamoxifen doses
Recombination was induced in Sox9-CreERT2 R26R-lacZ mice with 250mg/kg tamoxifen. Mice were examined (a) after 24 hours normal chow or (b) after prototypical steatosis injury that included a 1-week rest period, 3 weeks choline-deficient, ethionine supplemented diet, and a 2 week recovery period. (c) Quantification of Sox9-CreERT2 marked cells by epithelial cell type after normal chow or CDE injury recreates the increase in marked hepatocytes after injury previously reported in the literature. (d) 250mg/kg tamoxifen induced recombination Sox9-CreERT2 R26R-confetti recombination in both hepatocytes and ducts. Hepatocytes and ducts were identified in each of three Confetti colors (mCerulean, RFP, and YFP). (e) High dose tamoxifen tracing in Sox9-CreERT2 R26R-Confetti mice injured with CDE for 3 weeks and a 1-week recovery results in marked hepatocytes (arrow head) and Opn+ ductal cells (arrow). (f) High dose tamoxifen tracing in Sox9-CreERT2 R26R-Confetti in mice injured with repeated CCl4 administration results in marked HNF4a+ hepatocytes (arrowhead) and ductal cells (arrow).
Supporting Information Figure 2: Sox9-CreERT2 marked ducts and periportal hepatocytes do not continuously “stream” in homeostasis
Sox9-CreERT2 R26R-lacZ mice were treated with (a) 32mg/kg tamoxifen or (b) 125mg/kg tamoxifen and maintained on normal chow for 6 months. Marked ducts (arrows) or periportal hepatocytes (arrowheads) did not progressively replace the bulk of the hepatocyte mass (bar = 200 μm, inset bar = 50μm). (c) Recombination in Sox9-CreERT2 R26R-Confetti mice treated with 125mg/kg tamoxifen showed recombination primarily in Sox9+ cells that did not give rise to hepatocytes after 3 months homeostasis. (d) Rare perioportal hepatocyte (arrowhead) maintained a periportal position and did not proliferate or replace the bulk of hepatocytes after 3 months homeostasis.
Supporting Information Figure 3: FACS-based analysis of Cre-marked Confetti cells
(a) Single-cell suspensions of liver nonparenchymal cells were FACS sorted with the following gating strategy to identify biliary progenitors. Gates were FSC/SSC, low trigger pulse width (not shown), propridium iodide negative (live cells, not shown), followed by MIC1-1C3+ Cd31- Cd45- Cd11b-. (b) MIC1-1C3 cells were then scored based on eYFP, mCerulean, and tdimer RFP status in a mouse treated with CDE and 125mg/kg tamoxifen (c) or CDE and 16mg/kg tamoxifen. (d) MIC1-1C3 ductal cells were unmarked in AAV-Ttr-Cre treated Confetti mice (and sesame oil treated Sox9-CreERT2 R26R-Confetti mice). (e) Gravity enriched hepatocytes were identified based on FSC/SSC and were PI-, OC2-2F8+, Cd31-, Cd45-. (f) Hepatocytes were interrogated for eYFP and tdimer RFP in injured Sox9-CreERT2 R26R-Confetti animals and (g) virally marked AAV8-Ttr-Cre marked hepatocytes (1-3% marked). (h) FACS-based quantification of Sox9-CreERT2 R26R-Confetti marked hepatocytes confirmed they were rare. (i) FACS-based analysis was used to confirm image based scoring of chimeric Fah-/- mice mTomato hepatocyte chimeras. The percentage of host mT-negative hepatocytes were plotted for each of three groups (mean± SEM, n=3 per group).
Supporting Information Figure 4: Sox9-CreERT2 marks phenotypically defined MIC1-1C3+ cells that form liver organoids
Non-parenchymal liver cells from Sox9-CreERT2 R26R-Confetti FACS were FACS sorted (YFP+ MIC1-1C3+ CD31- CD45- CD11b-) and seeded into organoid culture conditions. Organoids formed from YFP+ cells at equivalent rates in mice treated with (a) 32mg/kg or (b) 250mg/kg tamoxifen after 12 days culture. (c) Albumin mRNA expression in confetti+ MIC1-1C3+ CD31- CD45- CD11b- marked with high (250mg/kg) or low tamoxifen (32mg/kg) and differentiated towards a hepatic fate. (d) Clonally labeled ducts formed organoids retained the ability to form organoids in vitro after injury with CDE diet or (e) chronic CCl4 injury.
Supporting information 5: Sox9+ ducts rarely give rise to hepatocytes in acute CCl4 injury
(a) Experimental scheme for acute CCl4 tracing: Sox9-CreERT2 R26R-Confetti+/- mice were given a single acute toxic injury (1ul/kg CCl4) (b) 21 days recovery after injury, most Sox9-CreERT2 marked cells co-localized with ductal marker Opn (arrow = unique clone). (c) A single RFP+ Hnf4a+ cluster of Sox9-CreERT2 marked hepatocytes (arrow head) adjacent to a cell with a ductal Hnf4a- ductal cell is suggestive of a clonal relationship. (d) FACS quantification of Sox9-CreERT2 marked cells after acute CCl4 regeneration in phenotypically defined biliary cells (MIC1-1C3+) where approximately 4% of ducts are marked RFP or YFP+ with 32mg/kg tamoxifen. (e) OC2-2F8+ hepatocyte fractions show regeneration following CCl4 injury is associated with a 2.5-fold increase marked hepatocytes compared with in corn oil only. Less than 0.01% of hepatocytes were Confetti marked.
Supporting Information Figure 6: Hepatocyte transplantation into Fah-/- mice specifically replaced hepatocytes but not other cell types.
(a) 6 weeks after transplant with mTomato (red) marked mature donor hepatocytes, donor cells express hepatocyte marker Fah (green), but not duct marker A6 (white), demonstrating the detection of Fah-negative mTomato-negative host hepatocytes near the capsule (arrowhead), (bar = 50 μm). (b) mTomato donor marker (blue) surrounds Hnf4a+ hepatocytes (green) but not nonparenchymal cells nuclei (Hoescht only = red) c) CD31+ endothelium large vessels and sinusoids (d) CD11b+ mononuclear cells, and (e) GFAP+ stellate cells, and do not localize with mTomato, indicating these nonparenchymal cells are of host origin. (f) Fah immunostaining (green) shows donor hepatocyte repopulation was near complete but some Fah-/- hepatocytes (arrow) were retained in the periportal region adjacent to Opn+ cells (g) FACS-based analysis mTomato repopulated chimeric animals indicated no ductal cells were donor derived (0/4308 MIC1-1C3+).
Supporting Information Figure 7: Chimeric Fah mice treated with DDC and CDE do not require NTBC
(a) Schematic for testing the function of new progenitor derived hepatocytes in chimeric mice. Biliary and nonparenchymal cells are Fah-deficient. Fah-mutant hepatocyte require treatment with compound NTBC for normal survival. The liver stem cell model predicted NTBC rescue of progenitor-derived hepatocytes would be necessary for regeneration in when hepatocyte replication was disabled in oval cell/chronic injury. (b) Liver weights as a percentage of body weight were measured in chimeric animals treated with 10 weeks CCl4 injury with 1 week recovery or 3 weeks CDE. NTBC rescue of putative progenitor-derived hepatocytes was not required for chimeric animal survival or to maintain global measures of liver function such as body weight. (c) Body weight changes in chimeras treated with CDE injury (±NTBC) or NTBC only (d) chronic CCl4 injury and (e) DDC injury. (f) Quantification of body weight changes in cohorts of chimeric animals at the peak of oval cell injury. (g) FACS-based quantification of mTomato-negative hepatocytes in chimeras after DDC injury (n=3 per group). (h) Chimeric mice recovering from chronic CCl4 injury (2 hour BrdU pulse) showed BrdU incorporation in both host portal non-parenchymal cells (arrow) and hepatocytes (arrowhead). (i) Fah-immunohistochemistry in serial sections showed replicating hepatocytes (arrowhead) to be donor derived mature hepatocytes. (j) BrdU+ hepatocytes marked after 1 week BrdU administration in the recovery period following 10 weeks CCl4. (k) Hepatocyte BrdU incorporation (2 hour pulse) in uninjured chimeras was observed in ~1 in 25 high-powered fields.
Supporting Information Figure 8: AAV8-Ttr-Cre specificity and hepatocyte clonal dynamics
AAV8-Ttr-Cre induced recombination in R26R-Confetti+/- mice specifically in (a) Hnf4a+ hepatocytes but not (b) Osteopontin+ ducts. (c) Confetti marked clones after acute CCl4 (1ul/kg) mediated pericentral hepatocyte injury and 21 days regeneration (d) The cumulative percentage of clonally marked hepatocytes that are part of a large cluster increases following injury, demonstrating that mature hepatocytes are a common source of new hepatocytes. (e) Histogram representation of hepatocyte colony size following regeneration. (f) Distance of each Confetti marked clone to the nearest portal vein was measured at baseline (n=4) and after 3 or more months of homeostasis (n=2).
Supplementary Information Figure 9: NTBC rescues Fah-/- hepatocyte replication in regeneration from partial hepatectomy
(a) Ten days prior to 2/3 partial hepatectomy Fah-/- animals were assigned to continue receiving NTBC (8mg/L water) or taken off the drug. Partially repopulated Fah-/- hepatocyte chimeric mice were put back on NTBC at this time. Upon 2/3 partial heptectomy, BrdU was continously administered in the drinking water. Replication was assessed after 4 days partial hepatectomy. (b) Fah-/- animals on NTBC showed robust BrdU incorporation in parenchymal and non-parenchymal cells following regeneration from partial hepatectomy. (c) Fah-/- off NTBC showed BrdU incorporation primarily in non-prenchymal cells following partial hepatectomy due to hepatocyte replication blockade. (d) Chimeric Fah-/- animal put back on NTBC showed BrdU incorporation in parenchymal cells that (e) were both Fah-/- and Fah+/+ based on Fah-immunostaining in a serial section.
Supporting Information Figure 10: Extended recovery does not induce hepatic differentiation in Sox9-derived oval cells
(a) 32mg/kg tamoxifen was administered i.p. to Sox9CreERT2 R26R-Confetti mice at least two weeks prior to initiate of oval cell activation injury, as previously described (Fig. 2). CDE injury was given for 3 weeks, chronic CCl4 and DDC injury were given for 5 weeks. For recovery, animals were given normal chow (5LOD diet) for 4-15 weeks, as indicated. (b) Following CDE injury and 4 weeks recovery A6+ ductal proliferations persist. Confetti+ cells continue to colocalize with A6+ cells (200/200 clones, n=1 animal). (c) Following CCl4 injury and 4 weeks recovery limited ductal proliferation is observed. Confetti+ cells continue to colocalize with A6+ cells (75/75 clones, n=1 animal). (d) Following DDC injury and 4 or 15 weeks recovery, mild ductal proliferation still persists. Confetti+ cells were arranged in distinctly ductal morphology (200/200 clones, n=1 animal).
Supporting Information Figure 11: CreERT2 recombination continues a week after tamoxifen administration
(a) ROSACreERT2 were crossed with ROSAmT/mG mice to generate a mouse line that irreversibly switches from red to green fluorescence upon tamoxifen-mediated recombination. (b) In vitro tamoxifen administration to ROSACreERT2/mTmG primary hepatocytes switched cells from red to green fluorescence (bar =20 um). Fluorescence was monitored 2 and 6 days after tamoxifen treatment. (c) Transplantation experiment schematic: Fah-/- mice were given 100mg/kg i.p. tamoxifen (or vehicle) 1 or 7 days before intrasplenic transplant with ROSACreERT2/mTmG hepatocytes. NTBC water was withdrawn 1 day before transplant and 7 days were allowed for graft selection and fluorescent protein turnover. (d) The recombination rate in donor ROSACreERT2/mTmG hepatocytes was assessed by immunofluorescence in the livers of transplanted mice and (e) was quantified as the percentage of donor cells that were mGFP+.
Supporting Materials and Methods
Additional methods are provided in supplemental materials including:
Supporting information Table 1: Antibodies for immunohistochemistry and FACS.
Supporting information Table 2: Scientific names of transgenic mice
Acknowledgments
We acknowledge Leslie Wakefield, Feorillo Galivo, Annelise Haft, Angela Major, Stefanie Kaech Petrie, and Mandy Boyd for their excellent technical assistance. The Sox9-CreERT2 mouse was kindly provided by Dr. Maike Sander of UCSD. Dr. Holger Willenbring of UCSF provided the AAV-Ttr-Cre construct. We thank the Oregon Health & Science University for use of the Advanced Light Microscopy Core, Flow Cytometry Core, and Molecular Virology Support Core and the Texas Medical Center Digestive Disease Center Molecular Morphology Core, DK 56338 for its services.
Financial Support
This study was supported by the NIH/NIDDK (F30-DK095514 to BT, R01 DK051592 to MG, P30 DK56338 to MF) and the Knight Cancer Institute.
List of Abbreviations
- LSC
liver stem cell
- BrdU
Bromo-deoxyuridine
- CDE
choline-deficient ethionine-supplemented
- CCl4
carbon tetrachloride
- DDC
3,5-diethoxycarbonyl-1,4-dihydrocollidine
- NTBC
2-(2- nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione
- HNF4α
hepatocyte nuclear factor 4 alpha
- OPN
osteopontin
- SOX9
SRY-related HMG box transcription factor 9
- YFP
yellow fluorescent protein
- RFP
red fluorescent protein
- PV
portal vein
- CV
central vein
- NPC
non-parenchymal cell
- FACS
fluorescence activated cell sorting
- PBS
Phosphate-buffered saline
- i.p.
intraperitoneal
- IACUC
Institutional Animal Care and Use Committees
Footnotes
Additional methods are provided in the supplementary online information.
References
- 1.Farber E. Similarities in the sequence of early histological changes induced in the liver of the rat by ethionine, 2-acetylamino-fluorene, and 3’-methyl-4-dimethylaminoazobenzene. Cancer Res. 1956;16:142–148. [PubMed] [Google Scholar]
- 2.Sell S. Comparison of liver progenitor cells in human atypical ductular reactions with those seen in experimental models of liver injury. Hepatology. 1998;27:317–331. doi: 10.1002/hep.510270202. [DOI] [PubMed] [Google Scholar]
- 3.Roskams T, De Vos R, Van Eyken P, Myazaki H, Van Damme B, Desmet V. Hepatic OV-6 expression in human liver disease and rat experiments: evidence for hepatic progenitor cells in man. Journal of Hepatology. 1998;29:455–463. doi: 10.1016/s0168-8278(98)80065-2. [DOI] [PubMed] [Google Scholar]
- 4.Itoh T, Miyajima A. Liver regeneration by stem/progenitor cells. Hepatology. 2013 [Google Scholar]
- 5.Grisham JW, Porta E. Origin and fate of proliferated hepatic ductal cells in the rate: electron microscopic and autoradiographic studies. Exp Mol Pathol. 1964;86:242–261. doi: 10.1016/0014-4800(64)90057-7. [DOI] [PubMed] [Google Scholar]
- 6.Sell S. Is there a liver stem cell? Cancer Res. 1990;50:3811–3815. [PubMed] [Google Scholar]
- 7.Wang X, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Grompe M. The origin and liver repopulating capacity of murine oval cells. Proc Natl Acad Sci USA. 2003;100(Suppl 1):11881–11888. doi: 10.1073/pnas.1734199100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evarts RP, Nagy P, Nakatsukasa H, Marsden E, Thorgeirsson SS. In vivo differentiation of rat liver oval cells into hepatocytes. Cancer Res. 1989;49:1541–1547. [PubMed] [Google Scholar]
- 9.Suzuki A, Sekiya S, Onishi M, Oshima N, Kiyonari H, et al. Flow cytometric isolation and clonal identification of self-renewing bipotent hepatic progenitor cells in adult mouse liver. Hepatology. 2008;48:1964–1978. doi: 10.1002/hep.22558. [DOI] [PubMed] [Google Scholar]
- 10.Sackett SD, Li Z, Hurtt R, Gao Y, Wells RG, et al. Foxl1 is a marker of bipotential hepatic progenitor cells in mice. Hepatology. 2009;49:920–929. doi: 10.1002/hep.22705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rountree CB, Barsky L, Ge S, Zhu J, Senadheera S, Crooks GM. A CD133-expressing murine liver oval cell population with bilineage potential. Stem Cells. 2007;25:2419–2429. doi: 10.1634/stemcells.2007-0176. [DOI] [PubMed] [Google Scholar]
- 12.Dorrell C, Erker L, Schug J, Kopp JL, Canaday PS, et al. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes Dev. 2011;25:1193–1203. doi: 10.1101/gad.2029411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huch M, Dorrell C, Boj SF, van Es JH, Li VSW, et al. In vitro expansion of single Lgr5(+) liver stem cells induced by Wnt-driven regeneration. Nature. 2013 doi: 10.1038/nature11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2010;43:34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- 15.Malato Y, Naqvi S, Schürmann N, Ng R, Wang B, et al. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J Clin Invest. 2011;121:4850–4860. doi: 10.1172/JCI59261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Español-Suñer R, Carpentier R, Van Hul N, Legry V, Achouri Y, et al. Liver Progenitor Cells Yield Functional Hepatocytes in Response to Chronic Liver Injury in Mice. Gastroenterology. 2012;143:1564–1575.e7. doi: 10.1053/j.gastro.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 17.Iverson SV, Comstock KM, Kundert JA, Schmidt EE. Contributions of new hepatocyte lineages to liver growth, maintenance, and regeneration in mice. Hepatology. 2011;54:655–663. doi: 10.1002/hep.24398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carpentier R, Suñer RE, Van Hul N, Kopp JL, Beaudry J-B, et al. Embryonic ductal plate cells give rise to cholangiocytes, periportal hepatocytes, and adult liver progenitor cells. Gastroenterology. 2011;141:1432–8. 1438.e1–4. doi: 10.1053/j.gastro.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isse K, Lesniak A, Grama K, Maier J, Specht S, et al. Preexisting epithelial diversity in normal human livers: a tissue-tethered cytometric analysis in portal/periportal epithelial cells. Hepatology. 2013;57:1632–1643. doi: 10.1002/hep.26131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18:572–579. doi: 10.1038/nm.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kragl M, Knapp D, Nacu E, Khattak S, Maden M, et al. Cells keep a memory of their tissue origin during axolotl limb regeneration. Nature. 2009;460:60–65. doi: 10.1038/nature08152. [DOI] [PubMed] [Google Scholar]
- 22.Overturf K, Al-Dhalimy M, Tanguay R, Brantly M, Ou CN, et al. Hepatocytes corrected by gene therapy are selected in vivo in a murine model of hereditary tyrosinaemia type I. Nat Genet. 1996;12:266–273. doi: 10.1038/ng0396-266. [DOI] [PubMed] [Google Scholar]
- 23.Willenbring H, Sharma AD, Vogel A, Lee AY, Rothfuss A, et al. Loss of p21 Permits Carcinogenesis from Chronically Damaged Liver and Kidney Epithelial Cells despite Unchecked Apoptosis. Cancer Cell. 2008;14:59–67. doi: 10.1016/j.ccr.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miyaoka Y, Ebato K, Kato H, Arakawa S, Shimizu S, Miyajima A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol. 2012;22:1166–1175. doi: 10.1016/j.cub.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 25.Bralet MP, Branchereau S, Brechot C, Ferry N. Cell lineage study in the liver using retroviral mediated gene transfer. Evidence against the streaming of hepatocytes in normal liver. Am J Pathol. 1994;144:896–905. [PMC free article] [PubMed] [Google Scholar]
- 26.Takase HM, Itoh T, Ino S, Wang T, Koji T, et al. FGF7 is a functional niche signal required for stimulation of adult liver progenitor cells that support liver regeneration. Genes Dev. 2013;27:169–181. doi: 10.1101/gad.204776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeGregorio MW, Coronado E, Osborne CK. Tumor and serum tamoxifen concentrations in the athymic nude mouse. Cancer Chemother Pharmacol. 1989;23:68–70. doi: 10.1007/BF00273519. [DOI] [PubMed] [Google Scholar]
- 28.Reinert RB, Kantz J, Misfeldt AA, Poffenberger G, Gannon M, et al. Tamoxifen-Induced Cre-loxP Recombination Is Prolonged in Pancreatic Islets of Adult Mice. PLoS ONE. 2012;7:e33529. doi: 10.1371/journal.pone.0033529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Español-Suñer R, Lemaigre FP, Leclercq IA. Reply. Gastroenterology. 2013;145:255–256. doi: 10.1053/j.gastro.2013.05.037. [DOI] [PubMed] [Google Scholar]
- 30.Yang L, Jung Y, Omenetti A, Witek RP, Choi S, et al. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells. 2008;26:2104–2113. doi: 10.1634/stemcells.2008-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishikawa T, Factor VM, Marquardt JU, Raggi C, Seo D, et al. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology. 2012;55:1215–1226. doi: 10.1002/hep.24796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27:719–724. doi: 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeliazkova P, Jörs S, Lee M, Zimber-Strobl U, Ferrer J, et al. Canonical Notch2 signaling determines biliary cell fates of embryonic hepatoblasts and adult hepatocytes independent of Hes1. Hepatology. 2013;57:2469–2479. doi: 10.1002/hep.26254. [DOI] [PubMed] [Google Scholar]
- 34.Michalopoulos GK, Barua L, Bowen WC. Transdifferentiation of rat hepatocytes into biliary cells after bile duct ligation and toxic biliary injury. Hepatology. 2005;41:535–544. doi: 10.1002/hep.20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, et al. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138:653–665. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell. 2010;143:134–144. doi: 10.1016/j.cell.2010.09.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1: Sox9-CreERT2 marks hepatocytes with high tamoxifen doses
Recombination was induced in Sox9-CreERT2 R26R-lacZ mice with 250mg/kg tamoxifen. Mice were examined (a) after 24 hours normal chow or (b) after prototypical steatosis injury that included a 1-week rest period, 3 weeks choline-deficient, ethionine supplemented diet, and a 2 week recovery period. (c) Quantification of Sox9-CreERT2 marked cells by epithelial cell type after normal chow or CDE injury recreates the increase in marked hepatocytes after injury previously reported in the literature. (d) 250mg/kg tamoxifen induced recombination Sox9-CreERT2 R26R-confetti recombination in both hepatocytes and ducts. Hepatocytes and ducts were identified in each of three Confetti colors (mCerulean, RFP, and YFP). (e) High dose tamoxifen tracing in Sox9-CreERT2 R26R-Confetti mice injured with CDE for 3 weeks and a 1-week recovery results in marked hepatocytes (arrow head) and Opn+ ductal cells (arrow). (f) High dose tamoxifen tracing in Sox9-CreERT2 R26R-Confetti in mice injured with repeated CCl4 administration results in marked HNF4a+ hepatocytes (arrowhead) and ductal cells (arrow).
Supporting Information Figure 2: Sox9-CreERT2 marked ducts and periportal hepatocytes do not continuously “stream” in homeostasis
Sox9-CreERT2 R26R-lacZ mice were treated with (a) 32mg/kg tamoxifen or (b) 125mg/kg tamoxifen and maintained on normal chow for 6 months. Marked ducts (arrows) or periportal hepatocytes (arrowheads) did not progressively replace the bulk of the hepatocyte mass (bar = 200 μm, inset bar = 50μm). (c) Recombination in Sox9-CreERT2 R26R-Confetti mice treated with 125mg/kg tamoxifen showed recombination primarily in Sox9+ cells that did not give rise to hepatocytes after 3 months homeostasis. (d) Rare perioportal hepatocyte (arrowhead) maintained a periportal position and did not proliferate or replace the bulk of hepatocytes after 3 months homeostasis.
Supporting Information Figure 3: FACS-based analysis of Cre-marked Confetti cells
(a) Single-cell suspensions of liver nonparenchymal cells were FACS sorted with the following gating strategy to identify biliary progenitors. Gates were FSC/SSC, low trigger pulse width (not shown), propridium iodide negative (live cells, not shown), followed by MIC1-1C3+ Cd31- Cd45- Cd11b-. (b) MIC1-1C3 cells were then scored based on eYFP, mCerulean, and tdimer RFP status in a mouse treated with CDE and 125mg/kg tamoxifen (c) or CDE and 16mg/kg tamoxifen. (d) MIC1-1C3 ductal cells were unmarked in AAV-Ttr-Cre treated Confetti mice (and sesame oil treated Sox9-CreERT2 R26R-Confetti mice). (e) Gravity enriched hepatocytes were identified based on FSC/SSC and were PI-, OC2-2F8+, Cd31-, Cd45-. (f) Hepatocytes were interrogated for eYFP and tdimer RFP in injured Sox9-CreERT2 R26R-Confetti animals and (g) virally marked AAV8-Ttr-Cre marked hepatocytes (1-3% marked). (h) FACS-based quantification of Sox9-CreERT2 R26R-Confetti marked hepatocytes confirmed they were rare. (i) FACS-based analysis was used to confirm image based scoring of chimeric Fah-/- mice mTomato hepatocyte chimeras. The percentage of host mT-negative hepatocytes were plotted for each of three groups (mean± SEM, n=3 per group).
Supporting Information Figure 4: Sox9-CreERT2 marks phenotypically defined MIC1-1C3+ cells that form liver organoids
Non-parenchymal liver cells from Sox9-CreERT2 R26R-Confetti FACS were FACS sorted (YFP+ MIC1-1C3+ CD31- CD45- CD11b-) and seeded into organoid culture conditions. Organoids formed from YFP+ cells at equivalent rates in mice treated with (a) 32mg/kg or (b) 250mg/kg tamoxifen after 12 days culture. (c) Albumin mRNA expression in confetti+ MIC1-1C3+ CD31- CD45- CD11b- marked with high (250mg/kg) or low tamoxifen (32mg/kg) and differentiated towards a hepatic fate. (d) Clonally labeled ducts formed organoids retained the ability to form organoids in vitro after injury with CDE diet or (e) chronic CCl4 injury.
Supporting information 5: Sox9+ ducts rarely give rise to hepatocytes in acute CCl4 injury
(a) Experimental scheme for acute CCl4 tracing: Sox9-CreERT2 R26R-Confetti+/- mice were given a single acute toxic injury (1ul/kg CCl4) (b) 21 days recovery after injury, most Sox9-CreERT2 marked cells co-localized with ductal marker Opn (arrow = unique clone). (c) A single RFP+ Hnf4a+ cluster of Sox9-CreERT2 marked hepatocytes (arrow head) adjacent to a cell with a ductal Hnf4a- ductal cell is suggestive of a clonal relationship. (d) FACS quantification of Sox9-CreERT2 marked cells after acute CCl4 regeneration in phenotypically defined biliary cells (MIC1-1C3+) where approximately 4% of ducts are marked RFP or YFP+ with 32mg/kg tamoxifen. (e) OC2-2F8+ hepatocyte fractions show regeneration following CCl4 injury is associated with a 2.5-fold increase marked hepatocytes compared with in corn oil only. Less than 0.01% of hepatocytes were Confetti marked.
Supporting Information Figure 6: Hepatocyte transplantation into Fah-/- mice specifically replaced hepatocytes but not other cell types.
(a) 6 weeks after transplant with mTomato (red) marked mature donor hepatocytes, donor cells express hepatocyte marker Fah (green), but not duct marker A6 (white), demonstrating the detection of Fah-negative mTomato-negative host hepatocytes near the capsule (arrowhead), (bar = 50 μm). (b) mTomato donor marker (blue) surrounds Hnf4a+ hepatocytes (green) but not nonparenchymal cells nuclei (Hoescht only = red) c) CD31+ endothelium large vessels and sinusoids (d) CD11b+ mononuclear cells, and (e) GFAP+ stellate cells, and do not localize with mTomato, indicating these nonparenchymal cells are of host origin. (f) Fah immunostaining (green) shows donor hepatocyte repopulation was near complete but some Fah-/- hepatocytes (arrow) were retained in the periportal region adjacent to Opn+ cells (g) FACS-based analysis mTomato repopulated chimeric animals indicated no ductal cells were donor derived (0/4308 MIC1-1C3+).
Supporting Information Figure 7: Chimeric Fah mice treated with DDC and CDE do not require NTBC
(a) Schematic for testing the function of new progenitor derived hepatocytes in chimeric mice. Biliary and nonparenchymal cells are Fah-deficient. Fah-mutant hepatocyte require treatment with compound NTBC for normal survival. The liver stem cell model predicted NTBC rescue of progenitor-derived hepatocytes would be necessary for regeneration in when hepatocyte replication was disabled in oval cell/chronic injury. (b) Liver weights as a percentage of body weight were measured in chimeric animals treated with 10 weeks CCl4 injury with 1 week recovery or 3 weeks CDE. NTBC rescue of putative progenitor-derived hepatocytes was not required for chimeric animal survival or to maintain global measures of liver function such as body weight. (c) Body weight changes in chimeras treated with CDE injury (±NTBC) or NTBC only (d) chronic CCl4 injury and (e) DDC injury. (f) Quantification of body weight changes in cohorts of chimeric animals at the peak of oval cell injury. (g) FACS-based quantification of mTomato-negative hepatocytes in chimeras after DDC injury (n=3 per group). (h) Chimeric mice recovering from chronic CCl4 injury (2 hour BrdU pulse) showed BrdU incorporation in both host portal non-parenchymal cells (arrow) and hepatocytes (arrowhead). (i) Fah-immunohistochemistry in serial sections showed replicating hepatocytes (arrowhead) to be donor derived mature hepatocytes. (j) BrdU+ hepatocytes marked after 1 week BrdU administration in the recovery period following 10 weeks CCl4. (k) Hepatocyte BrdU incorporation (2 hour pulse) in uninjured chimeras was observed in ~1 in 25 high-powered fields.
Supporting Information Figure 8: AAV8-Ttr-Cre specificity and hepatocyte clonal dynamics
AAV8-Ttr-Cre induced recombination in R26R-Confetti+/- mice specifically in (a) Hnf4a+ hepatocytes but not (b) Osteopontin+ ducts. (c) Confetti marked clones after acute CCl4 (1ul/kg) mediated pericentral hepatocyte injury and 21 days regeneration (d) The cumulative percentage of clonally marked hepatocytes that are part of a large cluster increases following injury, demonstrating that mature hepatocytes are a common source of new hepatocytes. (e) Histogram representation of hepatocyte colony size following regeneration. (f) Distance of each Confetti marked clone to the nearest portal vein was measured at baseline (n=4) and after 3 or more months of homeostasis (n=2).
Supplementary Information Figure 9: NTBC rescues Fah-/- hepatocyte replication in regeneration from partial hepatectomy
(a) Ten days prior to 2/3 partial hepatectomy Fah-/- animals were assigned to continue receiving NTBC (8mg/L water) or taken off the drug. Partially repopulated Fah-/- hepatocyte chimeric mice were put back on NTBC at this time. Upon 2/3 partial heptectomy, BrdU was continously administered in the drinking water. Replication was assessed after 4 days partial hepatectomy. (b) Fah-/- animals on NTBC showed robust BrdU incorporation in parenchymal and non-parenchymal cells following regeneration from partial hepatectomy. (c) Fah-/- off NTBC showed BrdU incorporation primarily in non-prenchymal cells following partial hepatectomy due to hepatocyte replication blockade. (d) Chimeric Fah-/- animal put back on NTBC showed BrdU incorporation in parenchymal cells that (e) were both Fah-/- and Fah+/+ based on Fah-immunostaining in a serial section.
Supporting Information Figure 10: Extended recovery does not induce hepatic differentiation in Sox9-derived oval cells
(a) 32mg/kg tamoxifen was administered i.p. to Sox9CreERT2 R26R-Confetti mice at least two weeks prior to initiate of oval cell activation injury, as previously described (Fig. 2). CDE injury was given for 3 weeks, chronic CCl4 and DDC injury were given for 5 weeks. For recovery, animals were given normal chow (5LOD diet) for 4-15 weeks, as indicated. (b) Following CDE injury and 4 weeks recovery A6+ ductal proliferations persist. Confetti+ cells continue to colocalize with A6+ cells (200/200 clones, n=1 animal). (c) Following CCl4 injury and 4 weeks recovery limited ductal proliferation is observed. Confetti+ cells continue to colocalize with A6+ cells (75/75 clones, n=1 animal). (d) Following DDC injury and 4 or 15 weeks recovery, mild ductal proliferation still persists. Confetti+ cells were arranged in distinctly ductal morphology (200/200 clones, n=1 animal).
Supporting Information Figure 11: CreERT2 recombination continues a week after tamoxifen administration
(a) ROSACreERT2 were crossed with ROSAmT/mG mice to generate a mouse line that irreversibly switches from red to green fluorescence upon tamoxifen-mediated recombination. (b) In vitro tamoxifen administration to ROSACreERT2/mTmG primary hepatocytes switched cells from red to green fluorescence (bar =20 um). Fluorescence was monitored 2 and 6 days after tamoxifen treatment. (c) Transplantation experiment schematic: Fah-/- mice were given 100mg/kg i.p. tamoxifen (or vehicle) 1 or 7 days before intrasplenic transplant with ROSACreERT2/mTmG hepatocytes. NTBC water was withdrawn 1 day before transplant and 7 days were allowed for graft selection and fluorescent protein turnover. (d) The recombination rate in donor ROSACreERT2/mTmG hepatocytes was assessed by immunofluorescence in the livers of transplanted mice and (e) was quantified as the percentage of donor cells that were mGFP+.
Supporting Materials and Methods
Additional methods are provided in supplemental materials including:
Supporting information Table 1: Antibodies for immunohistochemistry and FACS.
Supporting information Table 2: Scientific names of transgenic mice