

Abstract
Chronic granulomatous disease (CGD) is a rare immunodeficiency disease, which is characterized by the lack of a functional nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in phagocytes. The disease presents leukocytosis, anemia, hypergammaglobulinemia, and granuloma formation of the skin, lung, or lymph nodes. The mutation of the CYBB gene encoding gp91phox, located on chromosome Xp21.1 is one of the causes of CGD. We report a patient with X-linked CGD who carried a novel mutation, a c.1133A>G (paAsp378Gly) missense mutation, in the CYBB gene.
Keywords: Chronic granulomatous disease, immunodeficiency, CYBB gene, mutation
INTRODUCTION
Chronic granulomatous disease (CGD) is a rare genetic disease with a prevalence of 1/200,000-250,000 characterized by recurrent life-threatening bacterial or fungal infection and granuloma formation.1 The pathophysiology of CGD is the inactivation or a defect of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (phox) complex and a failure of production of superoxide anions. These genetic defects lead to the inability of phagocytes to destroy certain microbes, and present leukocytosis, anemia, hypergammaglobulinemia, and granuloma formation of the skin, lung, or lymph nodes.2
NADPH oxidase can produce hydrogen peroxide and superoxide which are needed in the bactericidal action of phagocytes.2 NADPH oxidase is composed of 2 plasma membrane subunits, gp91-phox and p22-phox, which comprise the cytochrome b558 complex.3,4,5,6 P47-phox, p67-phox, p40-phox and rac-2 are also important cytosolic oxidase components.3,4,5,6 NADPH oxidase has been identified with a variety of molecular mutations, and two thirds of these mutations are X-linked forms located in the CYBB gene (encoding gp91-phox) on chromosome Xp21.1. Some patients of CGD have an autosomal recessive trait, showing defects of other enzymes involved in the formation of peroxide compounds. The prevalence rates of CGD are almost similar across ethnic and racial groups, with about one-third of X-linked mutations occurring de novo.6
We describe here a patient with X-linked CGD who carried a novel mutation, a c.1133A>G (p.Asp378Gly) missense mutation in the CYBB gene.
CASE REPORT
A 3-year-old boy was transferred to our hospital due to recurrent and persistent pneumonia. He was born at 41 weeks of gestation without perinatal problems. His birth weight was 3.4 kg. His parents were not consanguineous, and did not have any medical history. He had been vaccinated as scheduled. He had a history of 3 hospitalizations because of viral pneumonia at 12, 18, and 27 months of age.
One month before admission, he showed symptoms including cough, sputum and fever. He was hospitalized with a diagnosis of pneumonia and was treated with antibiotics at another hospital. Although ceftriaxone and azithromycin were administrated for 15 days, he showed sustained fever and progression of pneumonic infiltration with pleural effusion. The antibiotics were changed to meropenem, amikacin and azithromycin, but he did not show response. He was then transferred to our hospital.
At admission, his body temperature was 39.5℃, pulse rate 129/min, respiration rate 28/min, and blood pressure 95/55 mmHg. On physical examination, his body weight was 15.3 kg (50-75 percentile) and height was 96.5 cm (50-75 percentile). Two ulcers of 3 mm in size were observed in the uvula. Fine crackles were heard on both lung fields. In his abdomen, the liver was palpable 1 finger-breadth below the right costal margin and the tip of the spleen was detected. Neurologic examination was normal.
Laboratory tests revealed a leukocyte count of 3,380/mm3 (neutrophil 70.1%, lymphocyte 22.2%, monocyte 7.4%) in peripheral blood. The total bilirubin was 1.2 mg/dL with an elevated level of AST (165 U/L) and ALT (435 U/L). The C-reactive protein level was 12.05 mg/dL. Chest radiography and computed tomography (CT) showed pneumonic consolidation and atelectasis in both the lower lobes and pleural effusion in the right lower lungs (Fig. 1). Respiratory syncytial virus was detected in the nasopharyngeal aspirates by multiplex polymerase chain reaction analysis. Candida parapsilosis was isolated from the blood culture. Granuloma formation in the liver parenchyma was found by liver biopsy. The patient's convoluted clinical course of illness and candidemia prompted us to evaluate the patient for immunological test. The results of immunological tests, including serum immunoglobulin level, lymphocyte subset analyses, and complement levels, were all normal. However, the neutrophil dihydrorhodamine (DHR) test revealed the near absence of fluorescence on granulocyte stimulation. The stimulation index (SI) was 1.5, which was compatible with X-linked CGD. The SI of his mother and that of a normal subject was 25.0 and 127.9, respectively (Fig. 2). Since CGD was strongly suspected further work-up was performed to evaluate the disease extent. Abdomen ultrasonography demonstrated hepatosplenomegaly, gallbladder edema, ascites, and enlargement of paraaortic and mesenteric lymph nodes. In addition, a bone scan showed a focal radiouptake in the proximal diaphysis of the right femur, suggesting osteomyelitis.Written informed consent was obtained for molecular genetic studies for CYBB gene mutations. We examined the genomic DNA sequence of the patient and his mother for the detection of CYBB gene mutations. Thirteen CYBB exons and exon/intron boundaries were sequenced. The sequence analysis of our patient revealed, in exon 9, a novel mutation A to G (base pairs 1133) (Fig. 3). When his CYBB gene was translated, aspartic acid at 378 was replaced by glycine, which is a novel mutation. The patient's mother was a carrier showing a normal CYBB gene in another allele.
Fig. 1.

(A) Chest X-ray showed patchy consolidation in the right middle lobe and increased peribronchial opacity in the bilateral lower lung fields. (B) Chest CT showed air bronchogram and consolidation in the left lower lobe and atelectasis with pleural effusion in the right lower lobe.
Fig. 2.
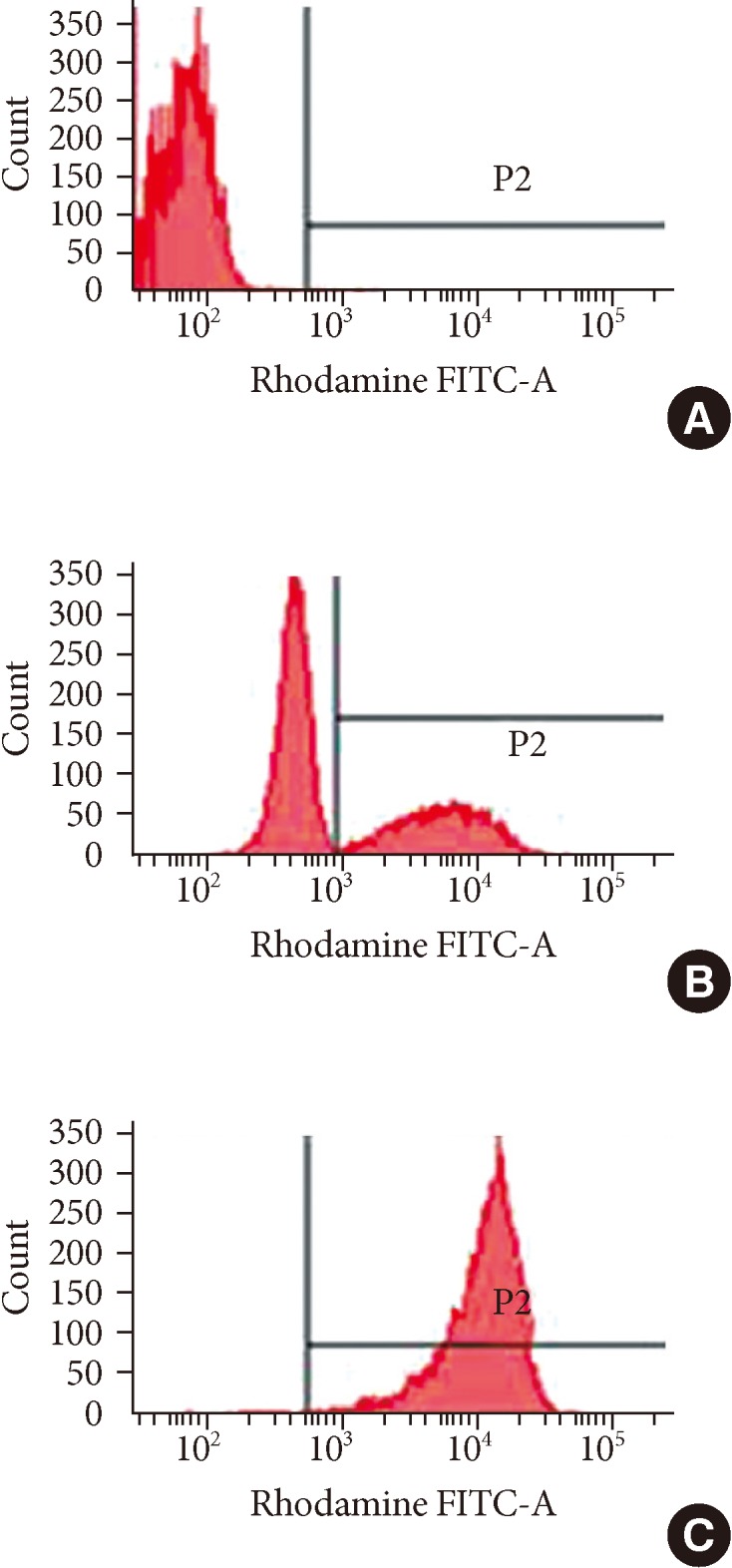
Histograms of the dihydrorhodamine (DHR) assay of granulocytes from the patient, his mother and a normal subject. DHR assay of the patient's granulocyte (A) revealed the near absence of fluorescence upon granulocyte stimulation. The stimulation index (SI) was 1.4 which was compatible with X-CGD. The DHR assay of granulocytes from the patient's mother (B) and a normal subject (C) showed a histogram with SI 25 and 127.9, respectively.
Fig. 3.
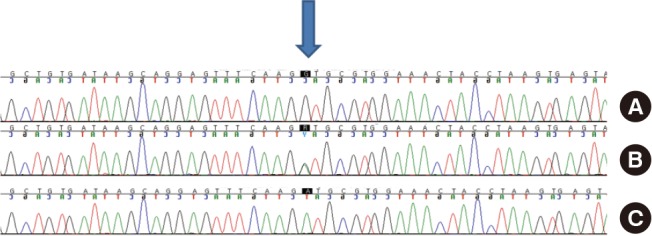
Analysis of a CYBB gene exon of the patient (A), his mother (B) and a normal subject (C). Chromatogram of a CYBB exon of the patient showed a novel complex mutation, c.1133A>G (p.Asp378Gly).
Finally, he was diagnosed with CGD accompanying Candida parapsilosis septicemia and pneumonia. He received amphotericin B dexoycholate for 4 weeks followed by oral itraconazole to treat pneumonia and suspected fungal osteomyelitis. He was also placed on trimethoprim/sulfamethoxazole prophylaxis. After this infection issues were stabilized, he received allogeneic hematopoetic stem cell transplantation by unrelated peripheral blood stem cell, 1 year later. After the transplantation, the SI in the DHR test rose to 99.9. Although grade 1 skin rash from acute graft-versus-host-disease occurred, it was well controlled by immunosuppressant medications without infection. His general condition is stable as of post-transplant day 120.
DISCUSSION
CGD is a primary phagocytic disorder involving defective superoxide formation and intracellular microbe killing as a result of the reduced NADPH oxidase complex.7 CGD patients have recurrent life-threatening bacterial and fungal infections, formation of chronic granulomas, and poor wound healing.8 Aspergillus fumigatus is one of the most common fungal infections in patients with CGD, which can be locally invasive or disseminated.9 In addition, these patients are susceptible to infections with catalase-positive bacteria, such as Staphylococcus aureus, Serratia marcescens, Salmonella species, Klebsiella species, Pseudomonas cepacia, and Norcadia.10,11,12 The incidence rate of CGD is reported as 1 case per 200,000-250,000 live births without apparent racial or ethnic predilection in the Unites States.13 The life expectancy of patients with the X-linked form of CGD is known to be from 20 to 40 years and that of AR-CGD patients ranges from 38 to 50 years.14
A previous European report showed that X-linked CGD (gp91-phox deficient) accounted for 67% of patients and autosomal recessive inheritance for 33% of patients.15 In the genetic analysis of Danish patients with CGD, 59% of patients had autosomal recessive mutations located in either NCF1 or CYBA, and 40% of patients demonstrated an X-linked mutation in the CYBB gene.16 In a Korean study, 12 patients from 10 unrelated families in Jeju-do underwent genetic analysis, and showed an identical homozygous single-base substitution of C to T in exon 1 of the CYBA gene.17 In a genetic subtype analysis of 158 Japanese patients with CGD, 118 patients (75%) had gp91-phox deficiency, 17 (11%) had p22-phox deficiency, 11 (7%) had p47-phox deficiency, and 12 (7.5%) had p67-phox deficiency.18 The most common mutations found in the CYBB gene were single nucleotide substitutions, followed by insertions, deletions and combinations of small deletions and insertions.19 In a recent study, however, nonsense or missense mutations represented 35% of mutations, while splice site mutations, and deletions or insertions comprised 18% and 43% of mutations, respectively.20 An international X-CGD database including over 300 CYBB mutations19,21 showed that most mutations were found throughout the 13 exons or at exon/intron boundaries, and that almost 200 of these mutations were unique.22 However, no clear correlation was found between genotype and phenotype for CGD with mild symptoms.22,23 The clinical severity of the disease is likely to be related to the defects in specific NADPH oxidase proteins and the extent of residual activity, but not with the type of mutations.24
We reported a case of a patient with CGD who had fungal septicemia due to Candida parapsilosis. He had an unusual clinical course of pneumonia, which was unresponsive to treatment with conventional empirical antibiotics. Therefore, we suspected immunodeficiency based on his clinical history and laboratory features, and performed immunologic tests including DHR flow cytometric assay, which is a rapid and sensitive screening test for CGD.25 In this test, nonfluorescent DHR 123 when phagocytosed by normal activated neutrophils is oxidized by hydrogen peroxide to fluorescent rhodamine 123. The SI is calculated by dividing the mean fluorescence intensity of stimulated neutrophils by that of the unstimulated neutrophils.25,26 In addition, he showed hepatosplenomegaly, and liver biopsy demonstrated granuloma formation. In our patient, the diagnosis of CGD was confirmed by direct sequencing of the CYBB gene, which showed a c.1133A>G (p.Asp378Gly) missense mutation. It is a novel mutation which has not been reported in the literature, although there were 2 previous Korean reports about the mutations of the CYBB gene.27,28
In summary, we identified a novel CYBB gene mutation in a boy with CGD. Further studies with more patients are needed to examine the genotype of CGD and to elucidate correlations between genotype and phenotype in patients with CGD.
Footnotes
There are no financial or other issues that might lead to conflict of interest.
References
- 1.Malech HL, Hickstein DD. Genetics, biology and clinical management of myeloid cell primary immune deficiencies: chronic granulomatous disease and leukocyte adhesion deficiency. Curr Opin Hematol. 2007;14:29–36. doi: 10.1097/00062752-200701000-00007. [DOI] [PubMed] [Google Scholar]
- 2.Gallin JI, Buescher ES, Seligmann BE, Nath J, Gaither T, Katz P. NIH conference. Recent advances in chronic granulomatous disease. Ann Intern Med. 1983;99:657–674. doi: 10.7326/0003-4819-99-5-657. [DOI] [PubMed] [Google Scholar]
- 3.Dinauer MC, Pierce EA, Bruns GA, Curnutte JT, Orkin SH. Human neutrophil cytochrome b light chain (p22-phox). Gene structure, chromosomal location, and mutations in cytochrome-negative autosomal recessive chronic granulomatous disease. J Clin Invest. 1990;86:1729–1737. doi: 10.1172/JCI114898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkos CA, Allen RA, Cochrane CG, Jesaitis AJ. Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relative molecular weights of 91,000 and 22,000. J Clin Invest. 1987;80:732–742. doi: 10.1172/JCI113128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quinn MT, Parkos CA, Jesaitis AJ. Purification of human neutrophil NADPH oxidase cytochrome b-558 and association with Rap 1A. Methods Enzymol. 1995;255:476–487. doi: 10.1016/s0076-6879(95)55050-x. [DOI] [PubMed] [Google Scholar]
- 6.Volpp BD, Nauseef WM, Donelson JE, Moser DR, Clark RA. Cloning of the cDNA and functional expression of the 47-kilodalton cytosolic component of human neutrophil respiratory burst oxidase. Proc Natl Acad Sci U S A. 1989;86:7195–7199. doi: 10.1073/pnas.86.18.7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang EM, Marciano BE, DeRavin S, Zarember KA, Holland SM, Malech HL. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2011;127:1319–1326. doi: 10.1016/j.jaci.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Desjardins A, Coignard-Biehler H, Mahlaoui N, Frange P, Bougnoux ME, Blanche S, Fischer A, Blumental S, Lortholary O. Chronic granulomatous disease: pathogenesis and therapy of associated fungal infections. Med Sci (Paris) 2012;28:963–969. doi: 10.1051/medsci/20122811015. [DOI] [PubMed] [Google Scholar]
- 10.Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, Goldblatt D, Parker L, Cant AJ. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. 2008;152:211–218. doi: 10.1111/j.1365-2249.2008.03644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, Rossi P, Gattorno M, Rabusin M, Azzari C, Dellepiane RM, Pietrogrande MC, Trizzino A, Di Bartolomeo P, Martino S, Carpino L, Cossu F, Locatelli F, Maccario R, Pierani P, Putti MC, Stabile A, Notarangelo LD, Ugazio AG, Plebani A, De Mattia D IPINET. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol. 2008;126:155–164. doi: 10.1016/j.clim.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 12.Soler-Palacín P, Margareto C, Llobet P, Asensio O, Hernández M, Caragol I, Español T. Chronic granulomatous disease in pediatric patients: 25 years of experience. Allergol Immunopathol (Madr) 2007;35:83–89. doi: 10.1157/13106774. [DOI] [PubMed] [Google Scholar]
- 13.Winkelstein JA, Marino MC, Johnston RB, Jr, Boyle J, Curnutte J, Gallin JI, Malech HL, Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 2000;79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Ben-Ari J, Wolach O, Gavrieli R, Wolach B. Infections associated with chronic granulomatous disease: linking genetics to phenotypic expression. Expert Rev Anti Infect Ther. 2012;10:881–894. doi: 10.1586/eri.12.77. [DOI] [PubMed] [Google Scholar]
- 15.van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, Español T, Fischer A, Kurenko-Deptuch M, Mouy R, Petropoulou T, Roesler J, Seger R, Stasia MJ, Valerius NH, Weening RS, Wolach B, Roos D, Kuijpers TW. Chronic granulomatous disease: the European experience. PLoS One. 2009;4:e5234. doi: 10.1371/journal.pone.0005234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakobsen MA, Katzenstein TL, Valerius NH, Roos D, Fisker N, Mogensen TH, Jensen PØ, Barington T. Genetical analysis of all Danish patients diagnosed with chronic granulomatous disease. Scand J Immunol. 2012;76:505–511. doi: 10.1111/j.1365-3083.2012.02771.x. [DOI] [PubMed] [Google Scholar]
- 17.Kim YM, Park JE, Kim JY, Lim HK, Nam JK, Cho M, Shin KS. Genetic analysis of 10 unrelated Korean families with p22-phox-deficient chronic granulomatous disease: an unusually identical mutation of the CYBA gene on Jeju Island, Korea. J Korean Med Sci. 2009;24:1045–1050. doi: 10.3346/jkms.2009.24.6.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nunoi H, Ishibashi F, Mizukami T, Hidaka F. Clinical evaluation of interferon-gamma treatment to chronic granulomatous disease patients with splice site mutations. Jpn J Infect Dis. 2004;57:S25–S26. [PubMed] [Google Scholar]
- 19.Roos D. X-CGDbase: a database of X-CGD-causing mutations. Immunol Today. 1996;17:517–521. doi: 10.1016/0167-5699(96)30060-1. [DOI] [PubMed] [Google Scholar]
- 20.Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, Avcin T, de Boer M, Bustamante J, Condino-Neto A, Di Matteo G, He J, Hill HR, Holland SM, Kannengiesser C, Köker MY, Kondratenko I, van Leeuwen K, Malech HL, Marodi L, Nunoi H, Stasia MJ, Ventura AM, Witwer CT, Wolach B, Gallin JI. Hematologically important mutations: X-linked chronic granulomatous disease (third update) Blood Cells Mol Dis. 2010;45:246–265. doi: 10.1016/j.bcmd.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heyworth PG, Curnutte JT, Rae J, Noack D, Roos D, van Koppen E, Cross AR. Hematologically important mutations: X-linked chronic granulomatous disease (second update) Blood Cells Mol Dis. 2001;27:16–26. doi: 10.1006/bcmd.2000.0347. [DOI] [PubMed] [Google Scholar]
- 22.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 23.Cooper DN, Krawczak M. The mutational spectrum of single base-pair substitutions causing human genetic disease: patterns and predictions. Hum Genet. 1990;85:55–74. doi: 10.1007/BF00276326. [DOI] [PubMed] [Google Scholar]
- 24.Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, Zarember KA, Malech HL, Holland SM, Gallin JI. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600–2610. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vowells SJ, Sekhsaria S, Malech HL, Shalit M, Fleisher TA. Flow cytometric analysis of the granulocyte respiratory burst: a comparison study of fluorescent probes. J Immunol Methods. 1995;178:89–97. doi: 10.1016/0022-1759(94)00247-t. [DOI] [PubMed] [Google Scholar]
- 26.Jirapongsananuruk O, Malech HL, Kuhns DB, Niemela JE, Brown MR, Anderson-Cohen M, Fleisher TA. Diagnostic paradigm for evaluation of male patients with chronic granulomatous disease, based on the dihydrorhodamine 123 assay. J Allergy Clin Immunol. 2003;111:374–379. doi: 10.1067/mai.2003.58. [DOI] [PubMed] [Google Scholar]
- 27.Oh HB, Park JS, Lee W, Yoo SJ, Yang JH, Oh SY. Molecular analysis of X-linked chronic granulomatous disease in five unrelated Korean patients. J Korean Med Sci. 2004;19:218–222. doi: 10.3346/jkms.2004.19.2.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee SY, Choi EY, Go SH, Rhim JW, Lee SD, Kim JG. A case of X-linked chronic granulomatous disease diagnosed in identical twin. Infect Chemother. 2007;39:332–337. [Google Scholar]