

Abstract
Interactions within the same cell population (homotypic) and between different cell types (heterotypic) are essential for tissue development, repair, and homeostasis. To elucidate the underlying mechanisms of these cellular interactions, co-culture models have been used extensively to investigate the role of cell–cell physical contact, autocrine and/or paracrine interactions on cell function, as well as stem cell differentiation. Specifically, the mixed co-culture model is often optimal for interpreting the effects of cell–cell contact on cellular behavior in vitro, while indirect co-culture can be used to study the effects of paracrine signaling on cell reactions. Additionally, cell–cell contact can be controlled by establishing physical barriers, which are used to regulate spatial and temporal cell distribution patterns in co-culture. In this chapter, we describe a method for forming a removable permeable divider for temporally and spatially controlling cellular interactions. This model can be used to study the impact of both cell–cell contact and paracrine signaling on the behavior of the mixed population as a whole and on the response of each subpopulation of cells in co-culture.
Keywords: Co-culture, Stem cells, Live cell tracking, Hydrogel divider, In vitro
1 Introduction
The maintenance, remodeling, and repair of biological tissue rely on the interaction of cells with other cell types and with their surrounding environment (1, 2). Specific to regenerative medicine, there is a growing interest in the nature and ramifications of the interactions between mesenchymal stem cells and resident cell populations in a given tissue or at the repair site, as stem cells represent a clinically feasible cell source for tissue engineering and regenerative medicine because of their trophic capabilities, as well as their potential to differentiate toward a number of mesenchymal lineages (3–5). The mechanisms behind the interactions between these cell types are, however, not well understood (6). For this reason, the development of in vitro model systems capable of modulating these interactions, which allow researchers to investigate the role of cellular communication in tissue formation and regeneration, is much needed.
To date, the majority of current co-culture models can be categorized into two types: those which examine the effects of direct cell–cell contact on co-cultured cell populations (7–12) and those which focus on paracrine signaling and response to soluble signaling factors (13–17). These studies can be conducted at relatively macro- (18, 20, 21) or microscales (22–25). The simplest model for examining the effects of cell–cell contact involves the mixing of two cell types and subsequently seeding a monolayer of this mixed population. This model allows for control over the extent of heterotypic and homotypic interactions through alteration of the seeding densities of each cell type and relative seeding ratio of the subpopulations (26). However, it is difficult to directly determine the relative contributions of each cell type to any observed effects of co-culture; thus, these studies are often accompanied in parallel by conditioned media experiments. In studies in which the effects of paracrine signaling are of interest, a segregated co-culture system can be utilized. This is done by first growing individual cell populations separately, followed by culturing both cell types in the same environment, in the absence of direct cell contact. This can be achieved either through the use of conditioned media studies or by forming a physical barrier or membrane between cell types that permits the exchange of soluble factors, while still preventing cell–cell contact. One advantage of this system is that the analysis of the individual response of each subpopulation of cells due to co-culture can be readily determined (26).
In the methods outlined below we describe a model which allows for the study of the effects of both cell–cell contact and paracrine signaling on subpopulations of cells in co-culture. Moreover, this model exerts spatial and temporal control over heterotypic cellular interactions through the development of a removable permeable hydrogel barrier. This in vitro model system permits the live tracking of populations throughout in vitro culture and allows for the study of individual cell subpopulation response to co-culture (20).
2 Materials
2.1 Hydrogel Divider Components
Phosphate-buffered saline (PBS, Sigma Chemical, St. Louis, MO, USA): One packet PBS powder into 1,000 mL deionized water (see Note 1).
Low gelling temperature agarose (Sigma Chemical, St. Louis, MO, USA).
Hydrogel solution: 4 % weight per volume (w/v) low gelling temperature agarose powder in 1× sterile PBS.
2.2 Live Cell Tracker Reagent Components
Fully supplemented (F/S) culture media: Dulbecco's modified essential medium (DMEM) with 10 % fetal bovine serum, 1 % nonessential amino acids, 1 % antibiotics (see Notes 2 and 3).
Serum-free culture media: DMEM with 1 % nonessential amino acids, 1 % antibiotics.
Vybrant™ DiO cell-labeling solution (Molecular Probes, Eugene, OR, USA) (see Note 4).
Vybrant™ DiD cell-labeling solution (Molecular Probes, Eugene, OR, USA).
3 Methods
Carry out all procedures at room temperature under sterile conditions unless otherwise specified.
3.1 Hydrogel Divider Formation and Placement
Sterile filter 1× PBS into a sterile container (see Notes 5 and 6).
Mix agarose powder into 1× PBS to achieve a final agarose concentration of 4 % w/v.
Autoclave for 25 min using a liquid cycle to sterilize.
Cast agarose gel by pipetting 2 mL of agarose solution into a 3.8 cm2 tissue culture plastic well and allow cooling (see Note 7).
Create a 5 mm hydrogel divider by cutting a 5 mm-wide strip along the center of the well using a scalpel. Slide the blade along the outer boundary of the well afterwards to separate the strip from the well.
Gently remove the 5 mm strip from the well and transfer to a new well plate.
Coat the bottom surface and sides of the divider with unsolidified agarose solution, using a sterile spatula or a pipette tip.
Place the coated divider into a new 3.8 cm2 well, and allow the agarose glue to dry and the divider to adhere to the sides and bottom of the tissue culture plastic surface (Fig. 1).
Parafilm seal the well plate containing the divider using parafilm and store at 37 °C until cell seeding to prevent gel dehydration.
Repeat steps 1–10 for each sample.
Fig. 1.

Hydrogel barrier formed following gel setting and removal of excess hydrogel
3.2 Cell Membrane Staining
All solutions used should be warmed to 37 °C before use.
Remove stem cell (Cell Type A) culture plates from cell culture incubator (see Note 8).
Establish a cell suspension via traditional trypsinization methods.
Remove cell suspension from the tissue culture plate and transfer the suspension into a sterile polypropylene centrifuge tube.
Centrifuge cells at 1,500 rpm for 12 min at room temperature to form a cell pellet.
Remove supernatant (see Note 9).
Resuspend cell pellet in a minimum volume of F/S culture media.
Count cells and assess viability (see Note 10).
Centrifuge cells at 1,500 rpm for 10 min at room temperature.
Resuspend cells in serum-free culture media at a density of 1 × 106/mL. Steps 15–20 should be performed in the dark, preferably (Fig. 2).
Add 5 μL DiD Vybrant™ Cell-Labeling Solution (1 mM in solvent) per mL of cell suspension. Mix well by gently pipetting up and down.
Incubate the cell suspension for 20 min in the dark at 37 °C (see Notes 11 and 12).
Centrifuge the cell suspension at 1,500 rpm for 5 min, preferably at 37 °C.
Remove supernatant, and gently resuspend cells in warm serum-free culture media.
Repeat steps 17 and 18 two more times.
Resuspend cells in F/S culture media at a density of 1 × 105/mL.
Incubate in the dark at 37 °C until ready for seeding (see Note 13).
Repeat steps 1–16 for second cell type (Cell Type B), using DiO Vybrant™ Cell-Labeling Solution (1 mM in solvent) instead of DiD Vybrant™ Cell-Labeling Solution in step 10.
Fig. 2.

Schematic of membrane staining using Vybrant™ Cell-Labeling Solutions (Molecular Probes, Eugene, OR, USA) to track both stem cells (red, DiD) and a second cell type (green, DiO). Cells are suspended at a concentration of 1 × 106/mL in serum-free media with 5 μL cell-labeling solution/mL
3.3 Cell Seeding
All steps should be performed in the dark, preferably.
- For hydrogel divider samples:
- Pipette 0.5 mL of Cell Type A cell suspension (5 × 104 total cells) directly onto the tissue culture plastic surface on the left side of the well, to the left of the divider (Fig. 3).
- Pipette 0.5 mL of Cell Type B cell suspension (5 × 104 total cells) onto the tissue culture plastic surface on the right side of the well (Fig. 3).
- Incubate for 30 min to allow for cell settling and attachment.
- Slowly add enough F/S culture media to completely cover the hydrogel divider.
- To allow for temporally controlled cell–cell contact, the hydrogel divider can later be removed by sliding a scalpel blade along the circumference of the well, severing the gel from the plastic well. Discard the hydrogel.
- For analysis of the behavior of individual subpopulations following culture:
- Pipette a 10 μL droplet of Cell Type A cell suspension onto a Thermanox coverslip.
- Pipette a 10 μL droplet of Cell Type B cell suspension onto a second cover slip.
- Incubate cover slips at 37 °C for 30 min to allow cells to attach.
- Place the cover slip with Cell Type A on the right side of the well, to the right of the hydrogel divider.
- Place the cover slip with Cell Type B onto the left side of the well, to the left of the hydrogel divider.
- Add enough F/S culture media to the well to cover the hydrogel divider.
- For monoculture control group samples:
- Pipette 1 mL of Cell Type A or Cell Type B (1 × 105 total cells) into each 3.8 cm2 tissue culture plastic well.
- Add 1 mL F/S culture media to each well.
- For direct co-culture control samples:
- In the dark, combine 50 % Cell Type A cell suspension and 50 % Cell Type B cell suspension in a sterile centrifuge tube.
- Pipette 1 mL of mixed suspension (1 × 105 total cells) into a 3.8 cm2 tissue culture plastic well.
- Add 1 mL F/S media to each well.
Fig. 3.
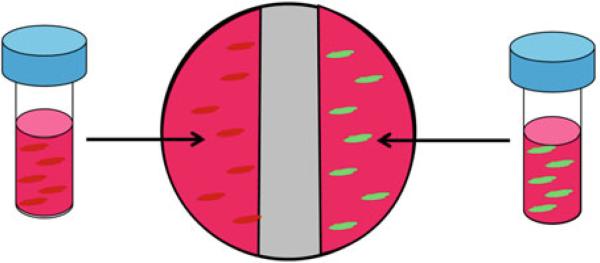
Seeding of Cell Types A and B into a 3.2 cm2 tissue culture plastic well with hydrogel divider in place
Acknowledgements
This work was supported by NIH grant R01-AR055280 (H.H.L.) and the NSF GRFP (D.R.B.).
Footnotes
PBS should be sterile filtered on the same day it is going to be used.
Antibiotics used should include, at minimum, penicillin– streptomycin. The addition of other antibiotics is optional.
Culture media should be stored at 4 °C when not in use. It can be stored for up to 1 month prior to use.
Vybrant™ Cell-Labeling Solutions should be stored at 4 °C upon arrival and should be stored in the dark.
Filter PBS using a sterile filter with 0.22 μm pores.
Sterile-filtered PBS can be stored at room temperature in a glass container following filtering for up to 6 months.
Allow agarose solution to cool/gel for 10–20 min at room temperature. Be sure to examine gel every 5 min, and begin step 5 as soon as the gel is formed, in order to avoid gel dehydration.
For co-culture, the primary cells used are typically between passage 2 and passage 5.
Tilt cell pellet upwards and aspirate slowly from the bottom side of the conical tube to avoid disturbing the pellet.
For accuracy, count cells at least three times, and take the average cell number over the three counts.
Molecular Probes has published a short list of optimized cell staining incubation times for specific cell lines that are less than 20 min. Check this list before proceeding with a 20-min incubation period.
Start by incubating cells in labeling solution for 20 min and subsequently optimize if uniform labeling is not achieved after 20 min of incubation.
Be sure to resuspend cells in solution periodically to prevent cell clumping.
References
- 1.Bhatia SN, Balis UJ, Yarmush ML, Toner M. Effect of cell–cell interactions in preservation of cellular phenotype: cocultivation of hepatocytes and nonparenchymal cells. FASEB J. 1999;13:1883–1900. doi: 10.1096/fasebj.13.14.1883. [DOI] [PubMed] [Google Scholar]
- 2.Lu HH, Jiang J. Interface tissue engineering and the formulation of multiple-tissue systems. Adv Biochem Eng Biotechnol. 2006;102:91–111. [PubMed] [Google Scholar]
- 3.Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276:71–74. doi: 10.1126/science.276.5309.71. [DOI] [PubMed] [Google Scholar]
- 4.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 5.Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem. 2006;98:1076–1084. doi: 10.1002/jcb.20886. [DOI] [PubMed] [Google Scholar]
- 6.Fan H, Liu H, Toh SL, Goh JC. Enhanced differentiation of mesenchymal stem cells co-cultured with ligament fibroblasts on gelatin/silk fibroin hybrid scaffold. Biomaterials. 2008;29:1017–1027. doi: 10.1016/j.biomaterials.2007.10.048. [DOI] [PubMed] [Google Scholar]
- 7.Richardson SM, Walker RV, Parker S, Rhodes NP, Hunt JA, Freemont AJ, Hoyland JA. Intervertebral disc cell-mediated mesenchymal stem cell differentiation. Stem Cells. 2006;24:707–716. doi: 10.1634/stemcells.2005-0205. [DOI] [PubMed] [Google Scholar]
- 8.Plotnikov EY, Khryapenkova TG, Vasileva AK, Marey MV, Galkina SI, Isaev NK, Sheval EV, Polyakov VY, Sukhikh GT, Zorov DB. Cell-to-cell cross-talk between mesenchymal stem cells and cardiomyocytes in co-culture. J Cell Mol Med. 2008;12:1622–1631. doi: 10.1111/j.1582-4934.2007.00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sheng H, et al. A critical role of IFN-gamma in priming MSC-mediated suppression of T cell proliferation through up-regulation of B7-H1. Cell Res. 2008;18:846–857. doi: 10.1038/cr.2008.80. [DOI] [PubMed] [Google Scholar]
- 10.Csaki C, Matis U, Mobasheri A, Shakibaei M. Co-culture of canine mesenchymal stem cells with primary bone-derived osteoblasts promotes osteogenic differentiation. Histochem Cell Biol. 2009;131:251–266. doi: 10.1007/s00418-008-0524-6. [DOI] [PubMed] [Google Scholar]
- 11.Aguirre A, Planell JA, Engel E. Dynamics of bone marrow-derived endothelial progenitor cell/mesenchymal stem cell interaction in co-culture and its implications in angiogenesis. Biochem Biophys Res Commun. 2010;400:284–291. doi: 10.1016/j.bbrc.2010.08.073. [DOI] [PubMed] [Google Scholar]
- 12.Proffen BL, Haslauer CM, Harris CE, Murray MM. Mesenchymal stem cells from the retropatellar fat pad and peripheral blood stimulate ACL fibroblast migration, proliferation, and collagen gene expression. Connect Tissue Res. 2013;54:14–21. doi: 10.3109/03008207.2012.715701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang T, Xu Z, Jiang W, Ma A. Cell-to-cell contact induces mesenchymal stem cell to differentiate into cardiomyocyte and smooth muscle cell. Int J Cardiol. 2006;109:74–81. doi: 10.1016/j.ijcard.2005.05.072. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Chai C, Jiang XS, Teoh SH, Leong KW. Co-culture of umbilical cord blood CD34+ cells with human mesenchymal stem cells. Tissue Eng. 2006;12:2161–2170. doi: 10.1089/ten.2006.12.2161. [DOI] [PubMed] [Google Scholar]
- 15.Lu HH, Wang IE. Multi-scale co-culture models for orthopaedic interface tissue engineering. In: Gonsalves KE, et al., editors. Biomedical nanostructures. Wiley; New York: 2007. [Google Scholar]
- 16.Lee IC, Wang JH, Lee YT, Young TH. The differentiation of mesenchymal stem cells by mechanical stress or/and co-culture system. Biochem Biophys Res Commun. 2007;352:147–152. doi: 10.1016/j.bbrc.2006.10.170. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, Tran N, Chen HQ, Kahn CJ, Marchal S, Groubatch F, Wang X. Time-related changes in expression of collagen types I and III and of tenascin-C in rat bone mesenchymal stem cells under co-culture with ligament fibroblasts or uniaxial stretching. Cell Tissue Res. 2008;332:101–109. doi: 10.1007/s00441-007-0564-6. [DOI] [PubMed] [Google Scholar]
- 18.Jiang J, Nicoll SB, Lu HH. Co-culture of osteoblasts and chondrocytes modulates cellular differentiation in vitro. Biochem Biophys Res Commun. 2005;338:762–770. doi: 10.1016/j.bbrc.2005.10.025. [DOI] [PubMed] [Google Scholar]
- 19.Wang IE, Lu HH. Role of cell-cell interactions on the regeneration of soft tissue-to-bone interface. Conf Proc IEEE Eng Med Biol Soc. 2006;1:783–786. doi: 10.1109/IEMBS.2006.259456. [DOI] [PubMed] [Google Scholar]
- 20.Wang IE, Shan J, Choi R, Oh S, Kepler CK, Chen FH, Lu HH. Role of osteoblastfibroblast interactions in the formation of the ligament-to-bone interface. J Orthop Res. 2007;25:1609–1620. doi: 10.1002/jor.20475. [DOI] [PubMed] [Google Scholar]
- 21.Hamilton SK, Bloodworth NC, Massad CS, Hammoudi TM, Suri S, Yang PJ, Lu H, Temenoff JS. Development of 3D hydrogel culture systems with on-demand cell separation. Biotechnol J. 2013;8:485–495. doi: 10.1002/biot.201200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhatia SN, Yarmush ML, Toner M. Controlling cell interactions by micropatterning in co-cultures: hepatocytes and 3T3 fibro-blasts. J Biomed Mater Res. 1997;34:189–199. doi: 10.1002/(sici)1097-4636(199702)34:2<189::aid-jbm8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 23.Takayama S, McDonald JC, Ostuni E, Liang MN, Kenis PJ, Ismagilov RF, Whitesides GM. Patterning cells and their environments using multiple laminar fluid flows in capillary networks. Proc Natl Acad Sci U S A. 1999;96:5545–5548. doi: 10.1073/pnas.96.10.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma SH, Lepak LA, Hussain RJ, Shain W, Shuler ML. An endothelial and astrocyte co-culture model of the blood-brain barrier utilizing an ultra-thin, nanofabricated silicon nitride membrane. Lab Chip. 2005;5:74–85. doi: 10.1039/b405713a. [DOI] [PubMed] [Google Scholar]
- 25.Kane BJ, Zinner MJ, Yarmush ML, Toner M. Liver-specific functional studies in a microfluidic array of primary mammalian hepatocytes. Anal Chem. 2006;78:4291–4298. doi: 10.1021/ac051856v. [DOI] [PubMed] [Google Scholar]
- 26.Bogdanowicz DR, Lu HH. Studying cell–cell communication in co-culture. Biotechnol J. 2013;8:395–396. doi: 10.1002/biot.201300054. [DOI] [PMC free article] [PubMed] [Google Scholar]