

Abstract
Provided here is a collective review of research on the extrarenal CYP27B1-hydroxylase that shapes our current and expanding vision of the role this enzyme plays in the intracrinonology and paracrinology, as opposed to the traditional endocrinology, of vitamin D to regulate the innate and adaptive immune response, particularly in human granuloma-forming diseases like tuberculosis. Special emphasis is placed on soluble factors (i.e., cytokines) in the local microenvironment of these human diseases that coordinate amplification and feedback inhibition of the macrophage CYP27B1-hydroxylase. Principal among these factors are Type I and Type II interferons (IFNs); the Type II IFN, IFN-γ, stimulates the production of 1,25-dihydroxyvitamin D (1,25(OH)2D) from 25-hydroxyvitamin D (25OHD) by the granuloma-forming disease-activated macrophage, while the Type I IFNs, IFN-α and IFN-ß, block the hydroxylation reaction. The type I IFN response is associated with more aggressive disease, while the Type II IFN response, the one that promotes 1,25(OH)2D production by the macrophage, is associated with more confined disease. Tilting the balance in the human immune response toward a type II IFN, confined disease phenotype in enabled by the presence of extracellular 25OHD levels that are sufficient to enable the type II IFNγ-promoted, substrate 25OHD-driven intracellular synthesis of 1,25(OH)2D.
Keywords: Vitamin D, Hydroxylase, Intracrine, Immunology, Innate immunity, Macrophage
I. Introduction
The first focus of this manuscript is to develop a retrospective view of the extrarenal CYP27B1-hydroxylase that shapes our current and expanding vision of the role this enzyme plays in the intracrinonology and paracrinology of vitamin D: that is the role of the CYP27B1 in converting extracellular substrate 25-hydroxyvitamin D (25OHD) to 1,25-dihydroxyvitamin D (1,25(OH)2D) and the synthesized hormone then activating the vitamin D receptor (VDR) and downstream gene expression in the cell in which it was produced or in a neighboring, VDR-expressing cell. The second aim of this review is to describe evidence from “human experiments” of disease that expression of the CYP27B1 in extrarenal tissues and cells, especially the macrophage, underpins the emerging field of the immunobiology of vitamin D in man. The third goal of what follows is to model and describe in some molecular detail the mechanism by which the activity of macrophage CYP27B1 modulates both the innate and adaptive immune response to microbial challenge in humans, especially when conditioned ex vivo in human serum obtained from the same host before and after 25OHD replacement in vivo.
II. Immunobiology of vitamin D
Many, if not most, of the significant biological consequences of dysregulated vitamin D balance in man are associated with changes in the extracellular concentration of substrate 25OHD, not the active hormone, 1,25(OH)2D [1]. Therefore, from the intracrine point of view the entrance of free 25OHD into the cell, its conversion to 1,25(OH)2D and the interaction of 1,25(OH)2D with the VDR to regulate gene expression all occur in the cell interior, away from events that can be easily detected and interpreted in the blood or urine (top panel, Figure 1). In fact, there are a host of cell types that have been shown now to harbor both the 1-hydroxylase and VDR (Table 1) and so have the potential to legislate the intracrine synthesis and actions of 1,25(OH)2D. This list also includes cells like the mammary and renal tubular epithelial cell that use megalin-mediated endosomal internalization of the serum vitamin D binding protein (DBP)-25OHD complex (bottom panel, Figure 1). In this case, it is still free 25D that must traverse the bits of plasma membrane that now make up the wall of the endosome in order to gain entry into the cell proper. Because it is what we have studied for the last thirty years and what we know best, this review will highlight on 25OHD-directed intracrine, autocrine and paracrine events in and around the human macrophage.
Figure 1.

Intracrine synthesis and action of 1,25-dihydroxyvitamin D (1,25-D). The upper panel of the cartoon demonstrates dependence on the serum vitamin D binding protein (DBP) to deliver substrate 25-hydroxyvitamin D (25-D) to its target cell and the movement of 25-D off the DBP and of free 25-D into the cell. Chaperone-mediated transport of 25-D (not depicted) to the inner mitochondrial membrane delivers substrate 25-D to the CYP27B1-hydroxylase for synthesis of 1,25-D. This event is followed by the binding of 1,25-D to VDR in an intracrine mode and dimerization with the retinoid X receptor to drive transactivation of genes bearing cis-acting vitamin D responsive elements. The lower panel shows an alternative mode of delivery of 25-D to the target cell via endocytic internalization of megalin-bound DBP and subsequent diffusion of 25-D across the endosomal membrane to the cell interior; the latter sequence of megalin-mediated events occurs in renal and mammary epithelial cells (see Table 1).
Table 1.
Human cells co-expressing the CYP27B1-hydroxylase and vitamin D receptor.
| • macrophage | • enterocyte |
| • dendritic cell | • decidual stromal cell |
| • parathyroid cell | • fetal trophoblast |
| • osteoblast | • prostate epithelial cell |
| • osteoclast | • vascular endothelial cell |
| • keratinocyte | • pancreatic ß cell |
| • mammary epithelial cell | • renal tubular cell |
The field of the immunobiology of vitamin D got its start in the early 1980s with three especially influential reports, all of which preceded the molecular cloning of the VDR and the CPY27B1. The first was from the Suda lab in 1981 [2]. These investigators showed for the first time that, when delivered to the extracellular culture medium, a physiologically relevant concentration of active 1,25(OH)2D metabolite induced maturation of white blood cells in the myeloid series; in other words, 1,25(OH)2D elicited a specific bioresponse in a cell not directly involved in intestinal calcium absorption and skeletal homeostasis. This was followed in the same year by a seminal case report appearing in the NEJM authored by Coburn and co-workers [3] of a hypercalcemic, anephric patient with the granuloma-forming disease sarcoidosis who harbored an inappropriately elevated serum concentration of 1,25(OH)2D. This was unequivocal evidence that the hormone was not being synthesized via the 1-hydroxylase in renal tissue. It is now well recognized that macrophages in granulomata from many human noninfectious, infectious and neoplastic diseases are capable of dysregualted synthesis of 1,25(OH)2D in quantities sufficient to have an endocrine, hypercalcemic action in the host [4]. The third piece of evidence originated from the Manolagas lab [5]; these investigators showed that activation of human lymphocytes with mitogen or specific antigen prompted expression of the VDR, making those cells potential paracrine targets for locally-produced and secreted 1,25(OH)2D.
III. Human macrophage CYP27B1-hydroxylase and granuloma-forming disease
We entered the field in 1983 with publication of the observations i) that macrophages harvested from the alveolar space of hypercalcemic patients with the granuloma-forming disease sarcoidosis were prolific synthesizers of 1,25(OH)2D ex vivo, ii), compared to the renal 1-hydroxylation reaction, the synthesis of 1,25(OH)2D in the macrophage was 25OHD substrate-dependent and iii) that a substantial portion of that endogenously synthesized hormone escaped the macrophage into the surrounding medium [6]. Shortly thereafter it was discovered that among the various cytokines concentrated in the alveolar lavage fluid of these patients it was the type II interferon (IFN), IFN-gamma (IFN-γ), that promoted 1,25(OH)2D synthesis most robustly ex vivo in these cells (Figure 2; [7]). In fact, a concentration-dependent increase in 25OHD-1-hydroxylating activity appeared to be fairly specific for IFN-γ and not the type I IFN, IFN-alpha (IFN-α). Going forward in this review attention will focus on the infectious human granuloma-forming disease tuberculosis (TB) as a model for the study of the extrarenal CYP27B1-hydroxylase in the human macrophage.
Figure 2.
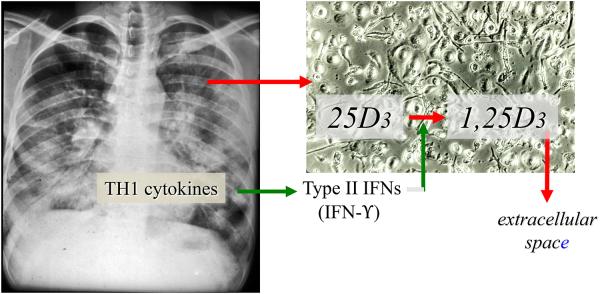
Human macrophages express the CYP27B1-hydroxylase. Macrophages harvested from the alveolar or pleural space of patients with active granuloma-forming disease of the lung (left panel; e.g., sarcoidosis and tuberculosis) and plated in monolayer at high density (right panel) synthesize and secrete 1,25-dihydroxyvitamin D3 (1,25D3) when incubated with physiologically relevant concentrations of substrate 25-hydroxyvitamin D3 (25D3). The specific activity of the 1-hydoxylation reaction is significantly increased if monolayer cultures of macrophages are preconditioned in medium containing rhIFN-γ or patient-derived pulmonary alveolar fluid enriched in IFN-γ.
The same kind of IFN-γ-directed promotion of macrophage 1-hydroxylating activity in sarcoidosis was also evidenced in the pleural effusion fluid from patients with active TB [8]. Compared to non-TB pleural fluid and serum from patients donating pleural effusion fluid, tuberculous pleural effusion fluid significantly stimulated sarcoid macrophage 1-hydroxylating activity ex vivo. Macrophage 1,25(OH)2D synthesis was correlated with the concentration of IFN-γ in the conditioning pleural effusion fluid. In these studies local production of 1,25(OH)2D resulted in a clear positive pleural effusion-to-serum gradient in “free “1,25(OH)2D levels from TB patients compared control subjects with non-TB pleural effusions [9]. This finding suggested that relatively more of the hormone that escaped the macrophage in a closed, compartmentalized inflammatory microenvironment, like the pleural space, existed in the “free state” (e.g., not bound to protein) and was thus less hindered in its ability to enter into and affect a neighboring, VDR-expressing inflammatory cell (i.e., T cell or B cell) in that same environment.
By the conclusion of the 1980s the above described work as well as that of other groups in the field clearly established that the macrophage was a source of 1,25(OH)2D in man. Further, 1,25(OH)2D could escape the disease-affected macrophage in sufficient quantities to exert a paracrine effect on neighboring VDR-expressing inflammatory cells, such as inhibition of proliferation of T and B cells with accompanying abrogation of lymphokine and immunoglobulin production, respectively [10]. In fact, in the face of wide spread granuloma-forming disease (e.g., sarcoidosis, TB, some lymphomas) the 1,25(OH)2D synthetic capacity of the macrophage rivaled that of the human kidney, exerting a hypercalciuric/hypercalcemic endocrine effect on calcium balance [4]. However, unlike the renal 1-hydroxylase, the activity of the macrophage 25OHD-1-hydroxylating reaction was regulated by other cytokines, particularly the type II IFN, IFN-γ, not by other calcium regulating hormones, such as parathyroid hormone, that have major effects on the activity of the renal enzyme.
Despite advances in our knowledge regarding the extrarenal 1-hydroxylase in the macrophage, there remained many crucial unanswered questions. For instance, what was the CYP gene product that was expressed in the macrophage? Was it the same gene product found in kidney cells? As described above, there is a relative lack of 25OHD-24-hydroxylating activity in the macrophage and this no doubt contributed to the relative efficiency of 1,25(OH)2D in this cell compared to renal tubular epithelial cell. Was there an alternative 24-hydroxylase expressed in the macrophage? Or, was it differentially regulated compared to its renal counterpart? Why was promotion of the macrophage 1-hydroxylase specific for type II IFNs in human granuloma-forming disease? How was it that synthetic 1-hydroxylating activity in macrophages harvested from humans with active disease persisted ex vivo in culture even in the absence of conditioning IFNs? Some of the questions have been answered and some are still coming to light. For example, it is now known that there exists a single gene that encodes the 1-hydroxylase, CYP27B1 [11] and the 24-hydroxylase, CYP24A1 [12]. Further, it is now clear that persistence of mTB in macrophages is an explanation for the continual expression of the CYP27B1 in human macrophages even after their removal from the host and conditioning cytokines present in the inflammatory microenvironment in the host.
IV. Human macrophage and tuberculosis
The human pulmonary macrophage serves as a reserve for viable mTB in infected humans. The mycobacteria are taken up by phagosomal vacuoles and reside there unless killed intracellularly. The fact is that roughly a third of the worlds’ population or 2.3 billion people carries live bacteria around in these phagosomal vacuoles without disease [13]. However, each year mycobacterium in only about a tenth of a percent of those infected (1.75 M individuals) escape containment in a great enough load to cause perforation of the macrophage plasma membrane, reinfection and death of the human host. At the same time mTB is trying to survive in these vacuoles, the host cell is mounting an intracellular antimicrobial campaign. It does so by fusion of the mTB-containing phagosomes with lysosomes and then deploying a repertoire of endogenous antibiotic molecules, such as LL-37 or cathelicidin; these antimicrobial molecules are incorporated into phagolysomal vacuoles and kill ingested mTB [14].
A real breakthrough in the field occurred with the discovery of Toll-like receptors and their role in mediating the innate immune response to invading microbes [15]. On the basis of cellular location there are two populations of TLRs in the human macrophage [16], those in the plasma membrane and internalized endolysomal membrane-bound TLRs. The plasma membrane-anchored TLRs 1, 2, 4, 5 and 6 are activated by lipopeptides, glycopeptides as well as native proteins (pathogen associated membrane patterns or PAMPs). TLRs 3, 7, 8 and 9 (and sometimes TLR4) project their ligand binding domains to the interior of the outside-in-endosome; the activating ligands for the endosomal-localized TLRs consists largely of single strand and double strand nucleic acids. Regardless of cellular positioning TLRs employ intracellular coupling proteins, either MyD88 or TRAM and TRIF to engage a series of downstream kinases promoting the nuclear translocation of the transacting factors. These transactivators include NFκB and interferon regulatory factors (IRFs) to promote the innate immune response and direct the adaptive immune response, respectively. In the case of infection with mTB the PAMP is a 19kD protein of the mycobacterial membrane and the PRR is the Toll-like receptor 2/1 dimer pair in the plasma membrane [17].
A decade ago now we undertook a series of straight forward experiments with primary cultures of human monocytes harvested from the blood of normal donors and the 19kD mTB PAMP to activate the TLR 2/1 [18]. The initial plan was to determine the mechanism of TLR2/1-induced antimicrobial activity by comparing the responses of human monocytes and GM-CSF-derived dendritic cell since TLR-activated monocytes and macrophages, but not dendritic cells, killed internalized mTB. When unbiased microarray analysis of both TLR-activated, monocytes and dendritic cells was performed, the VDR gene came up quickly and robustly in mTB-killing macrophages but not in dendritic cells. In a series of experiments we showed that if one can get enough free 1,25(OH)2D to the VDR in an mTB-infected, TLR-activated macrophage, as might occur in and around the lung in patients with active TB (see section II above), then in an intracrine mode the liganded VDR had the potential to i) initiate transcription of the VDR-responsive cathelicidin gene [19, 18] and ii) promote production of the antimicrobial cathelicidin gene product LL37, phagolysosome development and bacterial killing (Figure 3A). The act of blocking the VDR with a competitive non-activating analogue of 1,25(OH)2D or blocking 1,25(OH)2D-VDR-directed cathelicidin gene expression by siRNA blocked mTB antimicrobial activity [20].
Figure 3.


Regulation of the vitamin D-dependent antimycobacterial response in human monocytederived macrophages. Panel A shows that Mycobacterium tuberculosis (mTB) stimulation of the Toll-like receptor (TLR) 2/1 leads to induction of co-expression of the CYP27B1-hydroxylase and vitamin D receptor (VDR), intracrine induction of the cathelicidin gene product LL-37 expression and mycobacterial killing inside phagolysosomes. The key element for the occurrence of these serial events is the provision of adequate free, non-serum vitamin D binding protein ((DBP)bound) 25-hydroxyvitamin D (25D) substrate to the mitochondrial CYP27B1. Panel B shows that TLR activation leads to MYD88-coupled, NFκB-directed IL-15 and IL-1ß expression. IL-15 drives expression of the CYP27B1, VDR and 1,25DVDR-directed cathelicidin gene. The circuit is amplified by IL-15 and IL-1ß-induced TH1 cell proliferation with subsequent IFN-γ production to drive STAT-1-mediated increases interferon regulatory factor (IRF) and NF-kB signaling. The net result is i) more IL-15-powered CYP27B1 and VDR expression, ii) increased cathelicidin and ß defensin-4 expression and iii) recruitment of the cell’s autophagy and inflammasome capacity to support microbial killing; inflammasome activation increases the production and release of mature IL-1ß. Feedback inhibition of this amplification loop occurs through i) IL-1ß stimulated TH2 cell proliferation, ii) IFN-ß-driven IL-4 and IL-10 synthesis, iii) blockade endogenous 1,25D synthesis and iv) release of 1,25D into the extracellular environment in quantities sufficient to preferentially inhibit the proliferation of TH1 over TH2 cells, promoting suppression of the adaptive immune response.
During the course of these experiments there was another relevant vitamin D-associated gene, in addition to the VDR, discovered to be markedly upregulated in TLR2/1-activated, mTB-killing macrophages, CYP27B1, the sole structural gene product capable of metabolizing 25OHD to 1,25(OH)2D with high efficiency. Inhibition of CYP27B1-hydroxlating activity, blocked i) intracrine 1,25(OH)2D synthesis, ii) activation of the VDR in that cell, iii) upregulation of expression of cathelicidin gene products and iv) mTB killing in human macrophages. In other words, the TLR2/1-stimulated human macrophage had all of the equipment to make and respond to 1,25(OH)2D in terms of killing mTB. However, TLR2/1 activation repeatedly failed to elicit a transcriptional response through the VDR with endogenously synthesized 1,25(OH)2D until it was discovered that i) the fetal calf serum (FCS) used for incubation of cultured cells in these experiments was woefully 25OHD-deficient and ii) adequate endogenous synthesis of 1,25(OH)2D was possible by conditioning cells in 25OHD-sufficient human serum. So, in the presence of adequate substrate 25OHD in the extracellular space enough 1,25(OH)2D is generated inside the cell to activate the cathelicidin killing response to invading bacteria (Figure 3A).
V. Human macrophage CYP27B1-hydroxylase: Dependence on the extracellular 25D level
The above observations beg the question as to what serum levels of the 25OHD metabolite in living humans are sufficient to mount a cathelicidin response to a TLR challenge. We have now collected cathelicidin gene expression data (unpublished) in 19kD-TLR2/1-stimulated human macrophages conditioned in more than 100 human sera obtained before and after treatment in vivo with 500,000 IU vitamin D. Those sera contained increasing concentrations of total 25OHD, ranging from 5 to 95 ng/ml and total 1,25(OH)2D from 10 to 90 pg/ml. Among these samples, there was a significant (p=0.02) direct correlation between TLR-elicited cathelicidin gene expression and the conditioning serum 25OHD level, but not the 1,25(OH)2D level or expression of the CYP27B1 in macrophages. Further, when individual subjects harbouring 25OHD <30ng/ml are treated with 500,000 IU vitamin D, it is possible to reostatically upregulate macrophage cathelicidin gene expression in step with the increase in the total 25OHD level achieved in the serum after vitamin D replacement [21].
VI. Human macrophage CYP27B1-hydroxylase: Concerted regulation ex vivo
What is the mechanism(s) by which TLR activation regulates CYP27B1 expression in the macrophage? Is the TLR2/1 signaling pathway the sole regulator of the CYP27B1 and VDR in human macrophages or is there concerted regulation of the vitamin D metabolic and gene regulatory pathway in macrophages? Krutzik and colleagues [22] discovered that TLR2/1-stimulated IL-15. Compared to other common cytokines that drive monocyte differentiate (IL-4 and GM-CSF), IL-15 had the greatest stimulatory effect on CYP27B1 gene expression albeit with much more modest stimulatory effect on the VDR. As shown in Figure 3B, this places autocrine-acting IL-15 downstream of TLR-driven NFκB and upstream of the CYP27B1, VDR and 1,25D-VDR-directed cathelicidin gene expression. However, as predicted (see Figure 1), it was local production and action of the TH1 cytokine and type II IFN, IFN-γ that was the most potent stimulator of 1,25(OH)2D in macrophages cultured from the lungs of patients with sarcoidosis and TB. Figure 3B summarizes many years of work investigating the mechanism by which IFN-γ activates the macrophage vitamin D metabolic and gene regulatory system and promotes microbial killing [23, 24]. TLR stimulation promotes the generation and action of NFκB to upregulate IL-15 and IL-1ß. The circuit is amplified by IL-15 and IL-1ß-induced TH1 cell proliferation and TH1 cell-directed IFN-γ production to drive STAT-1-mediated increases IRF and NFκB signaling. The net result is more IL-15-powered CYP27B1 and VDR expression, increased cathelicidin and ß defensin-4 expression with recruitment of the cell’s autophagy and inflammasome capacity to support microbial killing. Inflammasome activation increases the production and release of mature IL-1ß. IL-1ß acts to recruit more TH1 cell participation in the IFN-γ response, completing a very aggressive feed-forward immune response that is shared between the innate and adaptive systems.
While this feed-forward, IFN-γ-driven amplification loop is very effective at ridding the host cell of mTB, persistent activation of this loop would eventually drive the adaptive immune response to be detrimental to the host. There appear to be three distinct means by which feedback inhibition of this feed-forward loop occurs (Figure 3B). One is through IL-1ß-stimulated TH2 cell proliferation and generation of IL-4; IL-4 blocks endogenous 1,25(OH)2D synthesis [25]. These events are a natural counterbalance to IL-1ß-stimulated TH1 proliferation and IFN-γ promotion of CYP27B1, VDR and cathelicidin expression. When assessed at the level of 25OHD metabolism in human macrophages, IFN-γ stimulates and IL-4 suppresses 1,25(OH)2D synthesis, while IFN-γ inhibits and IL-4 stimulates production of 24,25-dihydroxyvitamin D (24,25(OH)2D; [25]). The net effect of exposure of macrophages to IL-4 is diversion of substrate 25OHD away from the VDR-interacting metabolite, 1,25(OH)2D, and toward the non-biologically active 24,25(OH)2D metabolite. The second means by which feedback inhibition of the feed-forward mTB killing loop is achieved is by generation of a second TH2 product, the type II cytokine IFN-ß [26]. IFN-ß interacting with its receptor on macrophages in a paracrine mode increases IL-10 production by that cell. In turn, IL-10 antagonizes IFN-γ-driven 1,25(OH)2D synthesis principally through inhibition of expression of the CYP27B1 gene product with little or no effect of the CYP24A1. A third mechanism for feedback inhibition of an overzealous macrophage response to mTB is the release of macrophage-synthesized 1,25(OH)2D into the extracellular environment as is known to occur clinically (see section II). In the paracellular inflammatory microenvironment containing activated and VDR-expressing T cells, 1,25(OH)2D is known to preferentially exhibit inhibition of the proliferation of TH1 over that of TH2 cells [10]; the net result is a relative increase of TH2 over TH1 cells and relative accumulation of TH2 cell cytokine products, IFN-ß and IL-4, that inhibit the CYP27B1-hydroxylating activity of 25OHD to 1,25(OH)2D (Figure 3B).
VI. Conclusions
In summary, while the Type II IFN, IFN-γ, stimulates the production of 1,25(OH)2D by the granuloma-forming disease-activated macrophage, the Type I IFNs -α and -ß, use IL-10 to block the hydroxylation reaction. At least in the granuloma-forming disease, leprosy, the type I IFN response is associated with more aggressive disease, while the Type II IFN, IFN-γ, promotes 1,25(OH)2D production by the cell and is associated with more confined disease [26]. If tilting the balance in the human immune response toward the type II IFNs and away from the type I IFNs extends to other granuloma-forming diseases, like TB and sarcoidosis, then it is desirable that extracellular 25OHD levels are sufficient to enable the type II IFNγ-driven intracellular synthesis of 1,25(OH)2D and its intracrine mode of action.
Highlights.
We highlight translational research on the extrarenal CYP27B1-hydroxylase
CYP27B1- orchestrates the intracrinonology and paracrinology of vitamin D
Macrophage CYP27B1 activity is a key to the immunobiology of vitamin D in man
Principal factors controlling the CYP27B1 are 25OHD and Type I and II IFNs
Acknowledgements
Research reported in this publication was supported by the NIH under awards number TR000124, AI073539, AR059033 and AR063020. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Adams JS, Hewison M. Update in vitamin D. J Clin Endocrinol Metab. 2010;95:471–478. doi: 10.1210/jc.2009-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abe E, Miyaura C, Sakagami H, Takeda M, Konno K, Yamazaki T, Yoshiki S, Suda T. Differentiation of mouse myeloid leukemia cells induced by 1 alpha,25-dihydroxyvitamin D3. Proc Natl Acad Sci U S A. 1981;78:4990–4994. doi: 10.1073/pnas.78.8.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbour GL, Coburn JW, Slatopolsky E, Norman AW, Horst RL. Hypercalcemia in an anephric patient with sarcoidosis: evidence for extrarenal generation of 1,25-dihydroxyvitamin D. N Engl J Med. 1981;305:440–443. doi: 10.1056/NEJM198108203050807. [DOI] [PubMed] [Google Scholar]
- 4.Adams JS, Hewison M. Extrarenal expression of the 25-hydroxyvitamin D-1-hydroxylase. Arch Biochem Biophys. 2012;523:95–102. doi: 10.1016/j.abb.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Provvedini DM, Tsoukas CD, Deftos LJ, Manolagas SC. 1,25-dihydroxyvitamin D3 receptors in human leukocytes. Science. 1983;221:1181–1183. doi: 10.1126/science.6310748. [DOI] [PubMed] [Google Scholar]
- 6.Adams JS, Sharma OP, Gacad MA, Singer FR. Metabolism of 25-hydroxyvitamin D3 by cultured pulmonary alveolar macrophages in sarcoidosis. J Clin Invest. 1983;72:1856–1860. doi: 10.1172/JCI111147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adams JS, Gacad MA. Characterization of 1 alpha-hydroxylation of vitamin D3 sterols by cultured alveolar macrophages from patients with sarcoidosis. J Exp Med. 1985;161:755–765. doi: 10.1084/jem.161.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams JS, Modlin RL, Diz MM, Barnes PF. Potentiation of the macrophage 25-hydroxyvitamin D-1-hydroxylation reaction by human tuberculous pleural effusion fluid. J Clin Endocrinol Metab. 1989;69:457–460. doi: 10.1210/jcem-69-2-457. [DOI] [PubMed] [Google Scholar]
- 9.Barnes PF, Modlin RL, Bikle DD, Adams JS. Transpleural gradient of 1,25-dihydroxyvitamin D in tuberculous pleuritis. J Clin Invest. 1989;83:1527–1532. doi: 10.1172/JCI114048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams JS, Hewison M. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab. 2008;4:80–90. doi: 10.1038/ncpendmet0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeyama K, Kitanaka S, Sato T, Kobori M, Yanagisawa J, Kato S. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science. 1997;277:1827–1830. doi: 10.1126/science.277.5333.1827. [DOI] [PubMed] [Google Scholar]
- 12.Ohyama Y, Noshiro M, Eggertsen G, Gotoh O, Kato Y, Bjorkhem I, Okuda K. Structural characterization of the gene encoding rat 25-hydroxyvitamin D3 24-hydroxylase. Biochemistry. 1993;32:76–82. doi: 10.1021/bi00052a011. [DOI] [PubMed] [Google Scholar]
- 13.Mandavilli A. A clash of cultures. Nat Med. 2007;13:268–270. doi: 10.1038/nm0307-268. [DOI] [PubMed] [Google Scholar]
- 14.Vandamme D, Landuyt B, Luyten W, Schoofs L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol. 2012;280:22–35. doi: 10.1016/j.cellimm.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Thoma-Uszynski S, Stenger S, Takeuchi O, Ochoa MT, Engele M, Sieling PA, Barnes PF, Rollinghoff M, Bolcskei PL, Wagner M, Akira S, Norgard MV, Belisle JT, Godowski PJ, Bloom BR, Modlin RL. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–1547. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 16.Netea MG, Wijmenga C, O’Neill LA. Genetic variation in Toll-like receptors and disease susceptibility. Nat Immunol. 2012;13:535–542. doi: 10.1038/ni.2284. [DOI] [PubMed] [Google Scholar]
- 17.Lopez M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol. 2003;170:2409–2416. doi: 10.4049/jimmunol.170.5.2409. [DOI] [PubMed] [Google Scholar]
- 18.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, Kamen DL, Wagner M, Bals R, Steinmeyer A, Zugel U, Gallo RL, Eisenberg D, Hewison M, Hollis BW, Adams JS, Bloom BR, Modlin RL. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–1773. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 19.Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol. 2004;173:2909–2912. doi: 10.4049/jimmunol.173.5.2909. [DOI] [PubMed] [Google Scholar]
- 20.Liu PT, Stenger S, Tang DH, Modlin RL. Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J Immunol. 2007;179:2060–2063. doi: 10.4049/jimmunol.179.4.2060. [DOI] [PubMed] [Google Scholar]
- 21.Adams JS, Ren S, Liu PT, Chun RF, Lagishetty V, Gombart AF, Borregaard N, Modlin RL, Hewison M. Vitamin D-directed rheostatic regulation of monocyte antibacterial responses. J Immunol. 2009;182:4289–4295. doi: 10.4049/jimmunol.0803736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krutzik SR, Hewison M, Liu PT, Robles JA, Stenger S, Adams JS, Modlin RL. IL-15 links TLR2/1-induced macrophage differentiation to the vitamin D-dependent antimicrobial pathway. J Immunol. 2008;181:7115–7120. doi: 10.4049/jimmunol.181.10.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu PT, Schenk M, Walker VP, Dempsey PW, Kanchanapoomi M, Wheelwright M, Vazirnia A, Zhang X, Steinmeyer A, Zugel U, Hollis BW, Cheng G, Modlin RL. Convergence of IL-1beta and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PLoS One. 2009;4:e5810. doi: 10.1371/journal.pone.0005810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee HM, Krutzik SR, Schenk M, Sieling PA, Teles R, Montoya D, Iyer SS, Bruns H, Lewinsohn DM, Hollis BW, Hewison M, Adams JS, Steinmeyer A, Zugel U, Cheng G, Jo EK, Bloom BR, Modlin RL. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med. 2011;3:104ra102. doi: 10.1126/scitranslmed.3003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edfeldt K, Liu PT, Chun R, Fabri M, Schenk M, Wheelwright M, Keegan C, Krutzik SR, Adams JS, Hewison M, Modlin RL. T-cell cytokines differentially control human monocyte antimicrobial responses by regulating vitamin D metabolism. Proc Natl Acad Sci U S A. 2010;107:22593–22598. doi: 10.1073/pnas.1011624108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, Modlin RL. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339:1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bacchetta J, Sea JL, Chun RF, Lisse TS, Wesseling-Perry K, Gales B, Adams JS, Salusky IB, Hewison M. Fibroblast growth factor 23 inhibits extrarenal synthesis of 1,25-dihydroxyvitamin D in human monocytes. J Bone Miner Res. 2013;28:46–55. doi: 10.1002/jbmr.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu PT, Wheelwright M, Teles R, Komisopoulou E, Edfeldt K, Ferguson B, Mehta MD, Vazirnia A, Rea TH, Sarno EN, Graeber TG, Modlin RL. MicroRNA-21 targets the vitamin D-dependent antimicrobial pathway in leprosy. Nat Med. 2012;18:267–273. doi: 10.1038/nm.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]