

Abstract
Epithelial-mesenchymal transition (EMT) plays a crucial role in cancer metastasis. In this study, we evaluated the effect of heat treatment on tumor growth factor-β1 (TGF-β1)-induced EMT in pancreatic cancer cells and tried to ascertain the mechanism related to any observed effects. Human pancreatic cancer cell lines (BxPC-3, PANC-1 and MIAPaCa-2) were stimulated by TGF-β1, and evaluated for morphological changes using immunofluorescence and EMT-related factors (i.e., E-cadherin, Vimentin, Snail or ZEB-1) using RT-PCR. To examine the effect of heat on EMT, the cancer cells were heat-treated at 43°C for 1 h then stimulated with TGF-β1. We then evaluated whether or not heat treatment changed the expression of EMT-related factors and cell migration and also whether Smad activation was inhibited in TGF-β signaling. After being treated with TGF-β1, pancreatic cancer cells resulted in EMT and cell migration was enhanced. Heat treatment inhibited TGF-β1-induced changes in morphology, inhibited the expression of EMT-related factors, and attenuated TGF-β1-induced migration in pancreatic cancer cells. Additionally, we observed that heat treatment blocked TGF-β1-induced phosphorylation of Smad2 in PANC-1 cells. Our results suggest that heat treatment can suppress TGF-β1-induced EMT and opens the possibility of a new therapeutic use of hyperthermia as a potential treatment for cancer metastasis.
Keywords: pancreatic cancer, epithelial-mesenchymal transition, heat treatment, hyperthermia, TGF-β1
Introduction
Pancreatic cancer is the fifth most common cause of cancer-related death in Japan. Factors responsible for high mortality rates are late diagnosis due to the lack of early symptoms, extreme difficulty in resecting the tumor, extensive metastasis, and high resistance to treatment.(1) Unfortunately, there remains no effective therapy available for this aggressive tumor. Gemcitabine is the clinical standard for most chemotherapy regimens for pancreatic cancer; however patients generally have limited response to this therapy. To date, randomized trials of two regimens—gemicitabine plus erlotinib and a combination of 5-FU (5-fluorouracil), leucovorin, irinotecan, and oxaliplatin (FOLFIRINOX) have demonstrated significant prolongation of overall survival (OS).(2,3) However, gemcitabine plus erlotinib resulted in a statistically significant but relatively small improvement (0.33 months) in median OS (6.24 vs 5.91 months),(2) while the FOLFIRINOX regimen proved quite toxic.(4) To this end, more effective, better tolerated regimens are required to improve the outcome of patients with advanced pancreatic cancer.
Hyperthermia has been shown to increase the cytotoxic effects of some anti-cancer agents by facilitating increased drug penetration into tissues and causing thermal destruction of cancer cells.(5) Numerous phase II and phase III studies have shown that hyperthermia is feasible and effective in a variety of solid tumors especially if combined with chemotherapy or radiotherapy.(6–9) Moreover, we have previously demonstrated the safety and efficacy of a combined regional hyperthermia and gemcitabine treatment in patients with advanced pancreatic cancer.(6) In this phase II study, patients with locally advanced pancreatic cancer had a better outcome than those with metastatic pancreatic cancer (median OS, 17.7 vs 5.2 months, respectively). The results in patients with locally advanced, non-metastatic pancreatic cancer compare favorably with previous studies of 5-FU or gemcitabine with radiation, which reported a median OS of 8–10 months.(10–12) Since it has been proven that regional hyperthermia combined with gemcitabine improve the prognosis of patients with locally advanced pancreatic cancer, we hypothesized that epithelial-mesenchymal transition (EMT) inhibition may be the principal mechanism by which hyperthermia deters the progression of pancreatic cancer.
Recently, it has been purported that EMT is crucial to cancer invasion and metastasis.(13) The EMT phenotype is characterized by (1) the loss of cell-to-cell adhesion with the disintegration of tight, adherens, and gap junctions, and (2) a phenotypic change where cells shift from an ”epithelial” morphology to an elongated fibroblast-like morphology which is associated with increased motility and tumor invasion.(14) The process of EMT involves the up-regulation of mesenchymal markers such as vimentin, N-cadherin and fibronectin, and the down-regulation of epithelial adhesion molecules such as E-cadherin and cytokeratins.(15,16) EMT is triggered by the interplay of extracellular signals (such as collagen) and many secrete soluble factors such as Wnt, transforming growth factor-β (TGF-β), fibroblast growth factor, epidermal growth factor, hepatocyte growth factor, and platelet-derived growth factors. Among these signaling pathways, the Wnt, TGF-β, Hedgehog, Notch, and nuclear factor-κB (NF-κB) signaling pathways are critical for EMT induction.(15,17)
Coupled with its involvement in cancer metastasis, emerging lines of evidence also suggest that there is a molecular link between EMT phenotype and chemo-resistance or radio-resistance.(18–21) Reports have suggested that EMT is linked to drug resistance in pancreatic cancer cell.(19,21) Thus, it is important to control EMT to improve the clinical outcome of cancer patients, because doing this could inhibit cancer metastasis and assist patients to overcome chemo- or radio-resistance. With this in mind, the objective of this study was to evaluate the effect of hyperthermia on TGF-β1-induced EMT in pancreatic cancer cell lines to assess if it is a promising alternative to current treatments. Additionally, we aim to shed light on the mechanism of any significant effects caused by hyperthermia.
Materials and Methods
Cell lines and culture
The human pancreatic cell lines PANC-1 and MIAPaCa-2 were obtained from RIKEN Bioresource Center Cell Bank (Tsukuba, Japan) and BxPC-3 from DS Phama Biomedical Co (Osaka, Japan). PANC-1 and BxPC-3 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% fetal bovine serum (FBS), L-glutamine, and penicillin (100 U/ml)/streptomycin (100 µg/ml). MIAPaCa-2 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) low glucose supplemented with 10% FBS, L-glutamine, and penicillin (100 U/ml)/streptomycin (2.5 µg/ml). Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Induction of EMT and heat treatment
EMT induction was undertaken using TGF-β1 (R&D Systems, Minneapolis, MN). PANC-1, MIAPaCa-2 and BxPC-3 cells were incubated in a serum-free medium supplemented with 10 ng/ml TGF-β1 in a humidified 5% CO2 atmosphere at 37°C for 48 h. Before exposing cancer cells to TGF-β1, cells were incubated in a temperature-controlled CO2 incubator at 43°C for 60 min. After the cells were pretreated with heat, they were exposed to passive cooling in a temperature controlled CO2 incubator at 37°C before further examination.
Evaluation of cell migration using wound healing assay
Wound-healing assay was performed as previously described with minor modifications.(22) Briefly, the cells were seeded in P60 culture dishes and cultured until they reached confluence. The cells were then incubated with serum-free culture medium for 24 h, and scraped with a 10 µl extra-long micro-pipette tip, denuding a strip of the monolayer approximately 500 µl in diameter. Cultures were washed twice with PBS to remove cell debris and incubated with serum-free culture medium with and without TGF-β1. After incubation, the cells were photographed with a digital camera, and the migrated area was measured using NIH Image (ver. 1.63) software. To ensure that the same wounds were compared, we used a permanent marker to make positioning marks at the bottom of the culture dishes. The migration area in the wound was calculated according to the following formula: cell free area at 0 h subtracted from the cell free area at 12, 18, and 24 h. At least 8 fields were analyzed and the migrated area was expressed as a percentage of the 0 h cell.
Immunocytochemistry
PANC-1, MIAPaCA-2 and BxPC-3 cells cultured in 35-mm µ-dishes (iBidi, Munich, Germany) were washed with PBS and fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were then incubated with anti-E-cadherin (R&D Systems) and anti-vimentin (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 h. Subsequently, cells were incubated with fluorescence-labeled secondary antibodies (Alexa Fluor 594, Life Technologies, Tokyo, Japan) for 1 h at room temperature and staining was observed using epi-illumination on a laser scanning confocal microscope (Olympus, Tokyo, Japan).
Reverse transcription-polymerase chain reaction (RT-PCR)
The expression levels of E-cadherin, Vimentin, Snail and ZEB-1 mRNA were determined using real-time PCR. The samples used for mRNA isolation were removed from the pancreatic cancer cells (PANC-1, MIAPaCa-2 and BxPC-3). Total RNA was isolated using the acid guanidinium phenol chloroform method with Isogen (Nippon Gene Co. Ltd., Tokyo, Japan). The isolated RNA was stored at –80°C until use in real-time PCR. Subsequently, 1 µg of extracted RNA was reverse-transcribed into first-strand complementary DNA (cDNA) using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster, CA). Real-time PCR for E-cadherin, Vimentin, Snail, ZEB-1 and GAPDH was performed with the 7300 Real-Time PCR system (Applied Biosystems) using the DNA-binding dye SYBR Green to detect the PCR products. The primers had the following sequences: for E-cadherin, sense 5'-GTCAGTTCAGACTCCAGCCC-3' and antisense 5'-AAATTCACTCTGCCCAGGACG-3'; for Vimentin, sense 5'-TCTACGAGGAGGAGATGCGG-3' and antisense 5'-GGTCAAGACGTGCCAGAGAC-3'; for Snail, sense 5'-ACCACTATGCCGCGCTCTT-3' and antisense 5'-GGTCGTAGGGCTGCTGGAA-3'; for ZEB-1, sense 5'-TGGGATCAACCACCAATGG-3' and antisense 5'-AAGTAACCCTGTGTATTTCTGGATGA-3'; for GAPDH 5'-ACCACAGTCCATGCCATCACT-3' and antisense 5'-CCATCACGCCACAGTTTCC-3'.
Western blotting
Cells were washed twice with ice-cold PBS. After removing the upper PBS, the cell pellets were lysed in Lysis Buffer (CelLytic M; Sigma-Aldrich Co., St. Louis, MO), retrieved with a cell scraper and stirred and incubated on ice for 15 min. The supernatants were collected and stored at –80°C, and total proteins were mixed with an SDS sample buffer. The samples were then subjected to 10% SDS-PAGE and blotted onto a polyvinylidene fluoride membrane (Atto Corporation, Tokyo, Japan). The membrane was then incubated with 10% EzBlock (Atto Corporation) in TBS-T [10 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.1% Tween-20 V/V] for 30 min at room temperature and washed with TBS-T three times. The membrane was incubated for 1 h at room temperature with anti-E-cadherin (R&D Systems), anti-phospho-Smad2 (UPSTATE, Lake Placid, NY) and anti-Smad2 (Cell Signaling Technology, Beverly, MA) in TBS-T (diluted 1:500). Following this the membrane was then incubated with the secondary anti-rabbit and mouse IgG antibodies (GE Healthcare, Tokyo, Japan) in TBS-T (diluted 1:1000) for 1 h at room temperature. Immuno-complexes were detected using Western blotting (ECL plus; GE Healthcare Bio-Sciences K.K., Tokyo, Japan).
Statistical Analysis
All analyses were performed using the GraphPad Prism 5 program (GraphPad Software Inc., San Diego, CA). The results are presented as mean ± SEM An analysis of variance (ANOVA) and Tukey’s Multiple Comparison Test were used to compare the mean values. The criterion for statistical significance was taken as p<0.05.
Results
Heat treatment inhibits morphplogic changes consistent with EMT in the presence of TGF-β1
After being treated with TGF-β1, the morphology in pancreatic cancer cells changed from a typical epithelium to a mesenchymal spindle shape. Heat treatment for 1 h prior to TGF-β1 exposure inhibited morphologic changes consistent with EMT. Especially in MIAPaCa-2 which is originally circular, the morphology was quite obvious (Fig. 1).
Fig. 1.

Morphological changes after TGF-β1 and heat treatment in MIAPaCa-2 cells. Control: untreated cells. TGF-β1: Cells were treated with TGF-β1 (10 ng/ml) for 48 h. TGF-β1 + HT (heat treatment): Cells were heat-treated at 43°C for 1 h then stimulated with TGF-β1.
Effects of heat treatment on molecular marker of EMT
Immuno-fluorescence staining for E-cadherin (epithelial marker) was done on PANC-1, MIAPaCa-2, and BxPC-3 cells. The expression of E-cadherin was observed in only BxPC-3 cells, however, it was not observed even in the absence of TGF-β1 in the other two cell lines (i.e., PANC-1, MIAPaCa-2) (data not shown). After exposure to TGF-β1 for 48 h, E-cadherin expression of BxPC-3 cells was weaker, but it was reversed when pre-treated with heat (Fig. 2A). To further confirm our observation, we assessed the E-cadherin protein expression levels by Western blot (Fig. 2B). The expression of E-cadherin slightly decreased after exposure to TGF-β1, and post heat treatment its expression distinctly increased. As shown in Fig. 3A, in three pancreatic cancer cell lines that underwent EMT after being treated with TGF-β1, the expression of vimentin, which is one of the mesenchymal markers, was observed in cells that acquired a mesenchymal spindle-shape. Heat treatment reduced the expression of vimentin induced by TGF-β1 (Fig. 3A). We assessed the levels of vimentin and transcriptional suppressors of E-cadherin (i.e., Snail or ZEB-1) by quantitative RT-PCR. In BxPC-3 and MIAPaCa-2 cells, the expression of vimentin was significantly up-regulated by TGF-β1 treatment, however post heat treatment vimentin expression was blocked. In PANC-1 cells, vimentin expression showed a similar albeit insignificant tendency (Fig. 3B). In regards to the transcriptional suppressors of E-cadherin, the expression of Snail was up-regulated by TGF-β1 treatment in PANC-1 and MIAPaCa-2 cells; subsequently this up-regulation was significantly blocked by heat treatment in MIAPaCa-2 cells. In PANC-1 cells, heat treatment tended to attenuate TGF-β1-induced Snail expression (Fig. 3B). In BxPC-3 cells, the expression of Snail did not change significantly after exposure to both TGF-β1 and heat treatment (data not shown); this lead us to take a look at ZEB-1 expression in BxPC-3 cells. We observed that ZEB-1 expression was up-regulated by TGF-β1 treatment in BxPC-3 cells, and this up-regulation was then significantly blocked by heat treatment (Fig. 3B).
Fig. 2.

Immunofluorescence staining and Western blot analysis of E-cadherin in BxPC-3 cells. After exposure to TGF-β1 (10 ng/ml) for 48 h, E-cadherin expression in BxPC-3 cells weakened, but it was reversed by 1 h of heat treatment. HT: heat treatment.
Fig. 3.
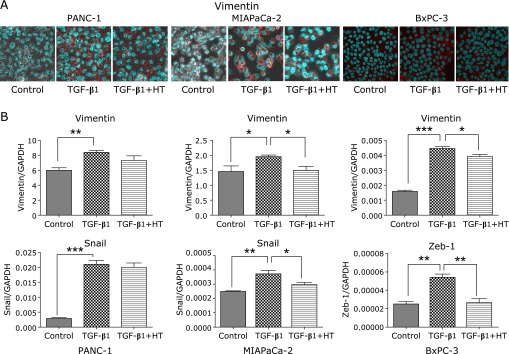
Immunofluorescence staining for Vimentin (A) and reverse transcription-polymerase chain reaction analysis of Vimentin and Snail or ZEB-1 expressions (B) in three pancreatic cell lines. (A) Immunofluorescence staining for Vimentin (red) and nucleus (green). (B) The bars depict the relative expression levels of Vimentin and Snail or ZEB-1 after normalization to GAPDH. *p<0.05, **p<0.01, ***p<0.001. HT: heat treatment.
Heat treatment inhibits the phosphorylation of Smad2
To elucidate the mechanism by which heat treatment inhibits TGF-β1-induced EMT, the role of heat in regulating of Smad2 expression and phosphorylation, which is involved in the signaling pathway of TGF-β, was investigated in PANC-1 cells by Western blot. Exposure of cells to TGF-β1 resulted in the phosphorylation of Smad2 and heat treatment blocked the TGF-β1-induced phosphorylation of Smad2. Although exposure to TGF-β1 decreased Smad2, heat treatment did not affect total Smad2 levels in cells exposed to TGF-β1 (Fig. 4).
Fig. 4.

Western blot analysis of Smad2 and p-Smad2 in PANC-1 cells. HT: heat treatment.
Heat treatment suppresses migration potential of PANC-1 cells
Next, using wound healing assays, we examined the effect of heat treatment on the migratory capability of PANC-1 cells. Twenty four hours after the induction of the scratch wound, cell migration into the wound was captured by microscope. Significant wound healing was seen after 24 h in cells treated with TGF-β1 compared to the control (absence of TGF-β1). Heat treatment inhibited wound closure in cells treated with TGF-β1 (Fig. 5), therefore it is safe to conclude that heat treatment can inhibit TGF-β1-induced migration in PANC-1 cells.
Fig. 5.

Migration assay of PANC-1 cells exposed to TGF-β1 with or without heat treatment. Twenty four hours after wound scratch, cell migration into the wound was captured by microscope. Cell migration was quantified in eight experiments: each line represents the ratio of time 0. *p<0.05, ***p<0.001. HT: heat treatment.
Discussion
Our focus in this study was to assess if heat treatment could inhibit EMT in pancreatic cancer cells and to further understand the mechanism involved. The results obtained so far indicate that heat treatment suppressed TGF-β1-induced EMT in pancreatic cancer cells. Moreover, our results strongly suggest that heat treatment inhibits TGF-β1-induced Smad2 activation in PANC-1 cells. This inhibition of Smad2 is considered to be one of the principal mechanisms for how heat treatment prevents TGF-β1-induced EMT. While there is one other research group that supports our result that heat treatment inhibits EMT in hepatocellular carcinoma cells,(23,24) the precise mechanisms of this inhibitory effect was not uncovered in their published reports and is still unknown. This is the first report to our knowledge that has described the mechanisms of the inhibitory effect of heat on EMT, involving the TGF-β/Smad signaling pathway.
Our results so far indicate that hyperthermia holds promise as a treatment for cancer metastasis. This is not surprising as several reports exist that suggest that hyperthermia has the potential to attenuate malignancy metastasis. Nagashima et al.(25) reported that hyperthermia, consisting of two 40-min sessions of radio-frequency capacitive local heating at 43°C, significantly decreased cervical lymph node metastasis from oral squamous cell carcinomas in hamsters. Moreover, clinical trial has demonstrated that local hyperthermia could be effective at treating cervical lymph node metastasis of oral cancer.(26) Although the exact mechanism of how hyperthermia inhibits metastases remains unclear, the expressions of several genes related to cancer metastasis such as membrane type 1-matrix metalloproteinase (MT1-MMP),(27) vascular endothelial growth factors (VEGFs),(28,29) and urokinase type plasminogen activator receptor (uPAR)(30) were shown to be down-regulated by heating. This supports the notion that hyperthermia alters the metastatic capacities of tumors by regulating metastasis related genes. While some progress is being made in this regard, there are few reports about the effects of hyperthermia on EMT, which is crucial in cancer invasion and metastasis.
EMT is triggered either by environmental stresses such as inflammation, reactive oxygen species, hypoxia and anoxia/repxygenation or by a number of extracellular mediators, including TGF-β, fibroblast growth factor-2 and epidermal growth factor.(31,32) In studies conducted by Xu et al.,(23,24) it was demonstrated that hyperthermia inhibits both TGF-β-induced and hypoxia-induced EMT in HepG2 hepatocellular carcinoma cells. However, the mechanism by which hyperthermia attenuates the expression of Snail using these two intracellular signaling pathways was not made clear. What is known, is that the Smad pathway, a major transducer of TGF-β signaling,(33) is important in TGF-β-induced EMT, and the hypoxia-induced factor-1 (HIF-1) is the primary factor mediating hypoxia-induced EMT. In our study, we have demonstrated that heat treatment attenuated the phosphorylation of Smad2 induced by TGF-β1 in PANC-1 cells. While TGF-β-induced phosphorylation of Smad2 in PANC-1 cells was not so pronounced in this study, Horiguchi et al.(34) have recently reported that Ras signaling is important for the TGF-β-induced Snail expression in PANC-1 cells. Thus, we hypothesize that this inhibition of the phosphorylation of Smad2 in TGF-β signaling is one of the mechanisms by which heat treatment suppresses TGF-β1-induced EMT.
Heat stress elicits a wide spectrum of stress responses, including an induction of heat shock proteins (HSPs), DNA and RNA damage and reactive oxygen species production in mammalian cells. Most HSPs play important roles as protein chaperones, regulators of protein folding, supporters of the formation of protein complexes and regulators of protein degradation.(35,36) Recently it has been revealed that several HSPs are involved in EMT of renal tubular epithelial cells,(37–39) lung cells,(40) and prostate cancer cells.(41) While there is some agreement as to the characteristics of HSPs, their effect on EMT is still controversial. For example, HSP72 has been shown to inhibit TGF-β1-induced EMT in renal epithelial cells by preventing TGF-β1-induced phosphorylation and nuclear translocation of Smad and p-Smad.(37,38) In contrast, Noh et al.(39) have demonstrated that HSP90 inhibitor blocks TGF-β1-induced Smad phosphorylation and induces the degradation of TGF-β type II receptor (TβRII), thereby preventing TGF-β-stimulation from inducing EMT. HSP27 is considered to be a component of several pathways, including the IL-6/STAT3 pathway, known to induce EMT, in prostate cancer.(41) Additionally, the inhibition of HSP27 blocks EMT features by promoting Snail degradation in lung cells.(40) In each study the observed effects of HSPs on EMT was different and this may have resulted in part, not only from different kind of HSPs addressed in the studies, but also from the different types of cells that were used. Since heat can strongly induce HSP72, it is possible that heat-induced HSP72 is involved in blocking TGF-β1-induced Smad phosphorylation in the present study. However, hyperthermia can induce not only a multitude of HSPs but also various cellular responses. We have shown that hyperthermia inhibits the activation of NF-κB, which can regulate EMT-inducing transcription factors, in pancreatic cancer cells.(42) Several molecular mechanisms might underlie the inhibitory effect of hyperthermia on EMT. However additional study is required to clarify the precise mechanisms underlying the inhibitory effects of hyperthermia on EMT.
In conclusion, this study has demonstrated for the first time that hyperthermia inhibits TGF-β1-induced EMT by blocking Smad2 phosphorylation in pancreatic cancer cells. EMT is more than a feature of metastatic cells; it is a characteristic of cancer stem cells and is associated with treatment resistance.(18–21,43) Our previous studies have shown the potential of hyperthermia for overcoming gemcitabine resistance in pancreatic cancer.(42) These results combined with the results presented in this study strongly suggest that hyperthermia could improve the prognosis of pancreatic cancer by suppressing cell metastasis.
Acknowledgments
This study was partially supported by Grant-in-Aid for Scientific Research (No. 23590891) from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Conflict of Interest
No potential conflicts of interest were disclosed.
References
- 1.Dorado J, Lonardo E, Miranda-Lorenzo I, Heeschen C. Pancreatic cancer stem cells: new insights and perspectives. J Gastroenterol. 2011;46:966–973. doi: 10.1007/s00535-011-0422-x. [DOI] [PubMed] [Google Scholar]
- 2.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Kim R. FOLFIRINOX: a new standard treatment for advanced pancreatic cancer? Lancet Oncol. 2011;12:8–9. doi: 10.1016/S1470-2045(10)70237-0. [DOI] [PubMed] [Google Scholar]
- 5.Engelhardt R. Hyperthermia and drugs. Recent Results Cancer Res. 1987;104:136–203. doi: 10.1007/978-3-642-82955-0_5. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa T, Kokura S, Sakamoto N, et al. Phase II trial of combined regional hyperthermia and gemcitabine for locally advanced or metastatic pancreatic cancer. Int J Hyperthermia. 2012;28:597–604. doi: 10.3109/02656736.2012.695428. [DOI] [PubMed] [Google Scholar]
- 7.Falk MH, Issels RD. Hyperthermia in oncology. Int J Hyperthermia. 2001;17:1–18. doi: 10.1080/02656730150201552. [DOI] [PubMed] [Google Scholar]
- 8.Wust P, Hildebrandt B, Sreenivasa G, et al. Hyperthermia in combined treatment of cancer. Lancet Oncol. 2002;3:487–497. doi: 10.1016/s1470-2045(02)00818-5. [DOI] [PubMed] [Google Scholar]
- 9.Issels RD, Lindner LH, Verweij J, et al. Neo-adjuvant chemotherapy alone or with regional hyperthermia for localised high-risk soft-tissue sarcoma: a randomised phase 3 multicentre study. Lancet Oncol. 2010;11:561–570. doi: 10.1016/S1470-2045(10)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Philip PA, Mooney M, Jaffe D, et al. Consensus report of the national cancer institute clinical trials planning meeting on pancreas cancer treatment. J Clin Oncol. 2009;27:5660–5669. doi: 10.1200/JCO.2009.21.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blackstock AW, Tepper JE, Niedwiecki D, Hollis DR, Mayer RJ, Tempero MA. Cancer and leukemia group B (CALGB) 89805: phase II chemoradiation trial using gemcitabine in patients with locoregional adenocarcinoma of the pancreas. Int J Gastrointest Cancer. 2003;34:107–116. doi: 10.1385/ijgc:34:2-3:107. [DOI] [PubMed] [Google Scholar]
- 12.Ishii H, Okada S, Tokuuye K, et al. Protracted 5-fluorouracil infusion with concurrent radiotherapy as a treatment for locally advanced pancreatic carcinoma. Cancer. 1997;79:1516–1520. [PubMed] [Google Scholar]
- 13.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hugo H, Ackland ML, Blick T, et al. Epithelial—mesenchymal and mesenchymal—epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–383. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 15.Min C, Eddy SF, Sherr DH, Sonenshein GE. NF-kappaB and epithelial to mesenchymal transition of cancer. J Cell Biochem. 2008;104:733–744. doi: 10.1002/jcb.21695. [DOI] [PubMed] [Google Scholar]
- 16.Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene. 2005;24:7443–7454. doi: 10.1038/sj.onc.1209091. [DOI] [PubMed] [Google Scholar]
- 17.Wu Y, Zhou BP. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim Biophys Sin. 2008;40:643–650. doi: 10.1111/j.1745-7270.2008.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiscox S, Jiang WG, Obermeier K, et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Caner. 2006;118:290–301. doi: 10.1002/ijc.21355. [DOI] [PubMed] [Google Scholar]
- 19.Wang Z, Li Y, Kong D, et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–2407. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ponti D, Zaffaroni N, Capelli C, Daidone MG. Breast cancer stem cells: an overview. Eur J Cancer. 2006;42:1219–1224. doi: 10.1016/j.ejca.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 21.Arumugam T, Ramachandran V, Fournier KF, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–5828. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uchiyama K, Naito Y, Takagi T, et al. Carbon monoxide enhance colonic epithelial restitution via FGF15 derived from colonic myofibroblasts. Biochem Biophys Res Commun. 2010;391:1122–1126. doi: 10.1016/j.bbrc.2009.12.035. [DOI] [PubMed] [Google Scholar]
- 23.Yuan GJ, Li QW, Shan SL, Wang WM, Jiang S, Xu XM. Hyperthermia inhibits hypoxia-induced epithelial-mesenchymal transition in HepG2 hepatocellular carcinoma cells. World J Gastroenterol. 2012;18:4781–4786. doi: 10.3748/wjg.v18.i34.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu XM, Yuan GJ, Li QW, Shan SL, Jiang S. Hyperthermia inhibits transforming growth factor beta-induced epithelial-mesenchymal transition (EMT) in HepG2 hepatocellular carcinoma cells. Hepatogastroenterology. 2012;59:2059–2063. doi: 10.5754/hge12404. [DOI] [PubMed] [Google Scholar]
- 25.Nagashima K, Takagi R, Hoshina H. Effect of local hyperthermia on metastases in oral squamous cell carcinoma. Int J Oral Maxillofac Surg. 2002;31:84–89. doi: 10.1054/ijom.2001.0176. [DOI] [PubMed] [Google Scholar]
- 26.Tohnai I, Hayashi Y, Mitsudo K, Shigetomi T, Ueda M, Ishigaki T. Prognostic evaluation of preoperative thermochemoradiotherapy for N(3) cervical lymph node metastases of oral cancer. Oncology. 2002;62:234–240. doi: 10.1159/000059571. [DOI] [PubMed] [Google Scholar]
- 27.Sawaji Y, Sato T, Seiki M, Ito A. Heat shock-mediated transient increase in intracellular 3',5'-cyclic AMP results in tumor specific suppression of membrane type 1-matrix metalloproteinase production and progelatinase A activation. Clin Exp Metastasis. 2000;18:131–138. doi: 10.1023/a:1006760021997. [DOI] [PubMed] [Google Scholar]
- 28.Sawaji Y, Sato T, Takeuchi A, Hirata M, Ito A. Anti-angiogenic action of hyperthermia by suppressing gene expression and production of tumour-derived vascular endothelial growth factor in vivo and in vitro. Br J Cancer. 2002;86:1597–1603. doi: 10.1038/sj.bjc.6600268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liang X, Zhou H, Liu X, et al. Effect of local hyperthermia on lymphangiogenic factors VEGF-C and -D in a nude mouse xenograft model of tongue squamous cell carcinoma. Oral Oncol. 2010;46:111–115. doi: 10.1016/j.oraloncology.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 30.Fukao H, Ikeda M, Ichikawa T, et al. Effect of hyperthermia on the viability and the fibrinolytic potential of human cancer cell lines. Clin Chim Acta. 2000;296:17–33. doi: 10.1016/s0009-8981(00)00198-4. [DOI] [PubMed] [Google Scholar]
- 31.Okajima M, Kokura S, Ishikawa T, et al. Anoxia/reoxygenation induces epithelial-mesenchymal transition in human colon cancer cell lines. Oncol Rep. 2013;29:2311–2317. doi: 10.3892/or.2013.2401. [DOI] [PubMed] [Google Scholar]
- 32.Sánchez-Tilló E, Liu Y, de Barrios O, et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69:3429–3456. doi: 10.1007/s00018-012-1122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 34.Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M. Role of Ras signaling in the induction of snail by transforming growth factor-beta. J Biol Chem. 2009;284:245–253. doi: 10.1074/jbc.M804777200. [DOI] [PubMed] [Google Scholar]
- 35.Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 36.Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–579. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- 37.Mao H, Li Z, Zhou Y, et al. HSP72 attenuates renal tubular cell apoptosis and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2008;295:F202–F214. doi: 10.1152/ajprenal.00468.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou Y, Mao H, Li S, et al. HSP72 inhibits Smad3 activation and nuclear translocation in renal epithelial-to-mesenchymal transition. J Am Soci Nephrol. 2010;21:598–609. doi: 10.1681/ASN.2009050552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noh H, Kim HJ, Yu MR, et al. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-β type II receptor. Lab Invest. 2012;92:1583–1596. doi: 10.1038/labinvest.2012.127. [DOI] [PubMed] [Google Scholar]
- 40.Wettstein G, Bellaye PS, Kolb M, et al. Inhibition of HSP27 blocks fibrosis development and EMT features by promoting Snail degradation. FASEB J. 2013;27:1549–1560. doi: 10.1096/fj.12-220053. [DOI] [PubMed] [Google Scholar]
- 41.Shiota M, Bishop JL, Nip KM, et al. Hsp27 regulates epithelial mesenchymal transition, metastasis, and circulating tumor cells in prostate cancer. Cancer Res. 2013;73:3109–3119. doi: 10.1158/0008-5472.CAN-12-3979. [DOI] [PubMed] [Google Scholar]
- 42.Adachi S, Kokura S, Okayama T, et al. Effect of hyperthermia combined with gemcitabine on apoptotic cell death in cultured human pancreatic cancer cell lines. Int J Hyperthermia. 2009;25:210–219. doi: 10.1080/02656730802657036. [DOI] [PubMed] [Google Scholar]
- 43.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]