

Abstract
Opioids are commonly used for pain relief, but their strong rewarding effects drive opioid misuse and abuse. How pain affects the liability of opioid abuse is unknown at present. In this study, we identified an epigenetic regulating cascade activated by both pain and the opioid morphine. Both persistent pain and repeated morphine upregulated the transcriptional regulator MeCP2 in mouse central nucleus of the amygdala (CeA). Chromatin immunoprecipitation analysis revealed that MeCP2 bound to and repressed the transcriptional repressor histone dimethyltransferase G9a, reducing G9a-catalyzed repressive mark H3K9me2 in CeA. Repression of G9a activity increased expression of brain-derived neurotrophic factor (BDNF). Behaviorally, persistent inflammatory pain increased the sensitivity to acquiring morphine-induced, reward-related behavior of conditioned place preference in mice. Local viral vector-mediated MeCP2 overexpression, Cre-induced G9a knockdown, and CeA application of BDNF mimicked, whereas MeCP2 knockdown inhibited, the pain effect. These results suggest that MeCP2 directly represses G9a as a shared mechanism in central amygdala for regulation of emotional responses to pain and opioid reward, and for their behavioral interaction.
Keywords: G9a, MeCP2, opioid, pain, reward
Introduction
Opioid analgesics are widely used for relieving pain in clinical pain management. Opioids also have strong rewarding and addictive effects after repeated use. In clinical practice, it has long been a disturbing issue how pain affects abuse liability of prescription opioids in patients taking repeated opioids for pain control. In fact, non-medical abuse of prescription opioids is rapidly arising in recent years, including incidences of illicit opioid use and behaviors of problematic opioid misuse among chronic pain patients under opioid therapy (Woolf and Hashmi, 2004; Ballantyne and LaForge, 2007; Passik and Kirsh, 2011). However, few preclinical or clinical studies have addressed the interaction of pain and rewarding effects of prescription opioids. Previous animal studies on a related topic were mainly intended to use opioid consumption as an alternative measurement for different dimensions of pain behaviors (Martin and Ewan, 2008; King et al., 2009). The neurobiological mechanisms by which behavioral responses to pain and opioid reward are regulated remain unexplored.
A potentially important link between pain and opioid reward is emotion processing, as pain is often associated with a negative affective state whereas drug reward induces positive euphoric emotion (Koob et al., 2004; Hyman et al., 2006; Neugebauer et al., 2009). The amygdalar complex, including the central nucleus of the amygdala (CeA), is a major brain structure for control and integration of emotional responses to positive (e.g., reward) and aversive (e.g., fear and pain) environmental stimuli (Pitkänen et al., 1997). Indeed, CeA mediates both drug reward-related positive emotional behaviors (Baxter and Murray, 2002; Gottfried et al., 2003; See et al., 2003) and negative emotional states of affective pain (Fields, 2004; Neugebauer et al., 2009). Thus, removal of pain-associated aversive state by analgesics can be rewarding (negative reinforcement) and induce reward-related behavior of conditioned place preference (CPP) in animals (Fields, 2004; King et al., 2009), a process regulated likely through emotion interaction involving CeA.
The methyl CpG-binding protein 2 (MeCP2), which binds to methylated CpG sites of DNA, is originally known as a prominent transcriptional repressor and disruptive mutations in MeCP2 cause the neurodevelopmental disorder Rett syndrome. Recent studies show that MeCP2 can act as a transcriptional repressor or activator and has a diverse role in the pathogenesis of several neurological diseases (Chahrour et al., 2008; Guy et al., 2011). For instance, MeCP2 plays an important role in regulating motivational effects of cocaine and psychostimulants (Deng et al., 2010; Im et al., 2010); it also has been implicated in modulation of pain behaviors in animals (Géranton et al., 2007; Tochiki et al., 2012). The molecular mechanisms for these MeCP2 roles are just beginning to be understood and importantly, the direct targets of MeCP2 in its transcription regulation remain largely unclear.
In this study, we investigated the role of MeCP2 in CeA regulation of responses to pain and to opioid reward in animal models of chronic pain, and determined molecular targets of CeA MeCP2 in its regulation of pain and opioid reward.
Materials and Methods
Animals.
MeCP2-TG female mice on FVB background containing one copy of the human MeCP2 transgene were mated to wild-type (WT) males on C57BL6 background. G9a-floxed mice (The Jackson Laboratory) were fully backcrossed to C57BL/6J mice. All procedures involving the use of animals conformed to the guidelines by the University of Texas MD Anderson Cancer Center Animal Care and Use Committee.
Animal model of inflammatory pain.
Complete Freund's adjuvant (CFA; 40 μl; Sigma-Aldrich) or saline was injected into the plantar surface of one hindpaw of a mouse under brief halothane anesthesia. Pain thresholds were measured by the paw-withdrawal test with the Hargreaves' analgesia apparatus (Stoelting) for thermal hyperalgesia, or with von Frey filaments for mechanical allodynia, on a freely moving mouse. The antinociceptive effect of an infused drug was measured 10–20 min post infusion.
Microinjection.
Under Nembutal anesthesia, a mouse was implanted with a 26 gauge guide cannula (Plastics One) aiming CeA (anteroposterior: − 0.94 mm from the bregma; lateral: ± 2.55 mm; ventral: 4.75 mm from dura). Lentivirus vectors expressing GFP (lenti-controls and lenti-sh-MeCP2, with viral supernatant concentrations ranging from 3 × 107 to 5 × 109 infection units per milliliter, 1 μl) were bilaterally infused into CeA through a 33 gauge injector with an infusion pump (0.05 μl/min) 4 weeks before experiments. Adeno-associated virus (AAV)-EF1a-mCherry-IRES-WGA-Cre (1 μl; University of North Carolina vector core facility) was similarly infused 3 weeks before. UNC0224 (0.7 ng/side), BIX01294 (60 ng/side), BDNF (1 ng/side), or TrkB-IgG (50 ng/side) in 0.5 μl was infused into CeA 2–4 h before tests. All infusion sites in CeA were historically verified afterward by injecting a blue dye in 0.5 μl and off-site controls (n = 3 mice) were performed to confirm site specificity, as we described previously (Bie et al., 2009; Cai et al., 2013).
CPP.
In a two-chamber CPP apparatus (MED Associates), a mouse was habituated and then conditioned for 30 min with saline or morphine (0.1–1 mg/kg, i.p.) in a single daily session of saline paired with one chamber in the morning and morphine paired with the other chamber in the afternoon for 3 d to induce CPP behavior. A CPP test (15 min) before the conditioning (pretest) determined the baseline preference, and mice that spent >60% of total time in one chamber (equipment bias) were excluded from the study for an unbiased CPP paradigm. After the conditioning sessions, CPP was measured by a CPP test (post-test, 15 min) and CPP scores were calculated by subtracting the time of pretest from that of post-test in the morphine-paired chamber. CFA was injected 3 d before the conditioning.
Open field test.
Mice were habituated in an open field apparatus consisting of a square area (81 cm × 81 cm) for 1 week before tests. In an open field test, a mouse was placed in the same corner and locomotor activity was observed for 5 min to record the distance it traveled. Seventy-five percent ethanol was used to remove the cues in the apparatus during the interval of tests. The test was conducted 1 d before and 3 d after intraplantar injection of saline or CFA.
Western blotting.
CeA tissues were homogenized in 100 μl RIPA lysis buffer with fresh protease inhibitors. The lysates were centrifuged and the supernatant was used for SDS-PAGE. Membranes were incubated in solutions containing an antibody to MeCP2 (1:2000; Cell Signaling Technology), G9a (1:1000; Millipore), histone 3 dimethyl lys9 (H3K9me2; 1:500; Active Motif), EHMT1/GLP1 (1:1000; Millipore), Enhancer of zeste homolog 2 (EZH2; 1:1000; Cell Signaling Technology), REST (1:500; Millipore), BDNF (1:200; Santa Cruz Biotechnology), NGF (1:200; Santa Cruz Biotechnology), κ opioid receptor (1:1000; Abcam), Dynorphin A (1:200; Abcam), K+/Cl− cotransporter (1:2000; Millipore), corticotropin-releasing factor (1:200; Santa Cruz Biotechnology), β-tubulin (1:2000; Cell Signaling Technology), or GAPDH (1:2000; Cell Signaling Technology). Membranes were incubated in secondary antibody to rabbit HRP (1:10,000) or to mouse Ig HRP (1:20,000; Calbiochem).
Chromatin immunoprecipitation assays.
CeA tissues were harvested and immediately cross-linked in 1% formaldehyde for 15–20 min. After washes, the CeA tissue was homogenized 10–30 strokes in a cell lysis buffer. The homogenate was centrifuged and the supernatant was removed. The extracted chromatin was sheared by sonication into 200–500 bp fragments and was diluted tenfold in chromatin immunoprecipitation (ChIP) dilution buffer. Normal mouse IgG immunoprecipitates with a mouse polyclonal anti-IgG antibody were used as control to normalize appropriate enrichment of signal amplification, and the data were presented after being normalized to saline/WT control groups. Samples were incubated with an antibody to MeCP2 (Cell Signaling Technology), G9a (Millipore), or H3K9me2 (Novus Biologicals). DNA and histones were dissociated with reverse buffer. Binding buffer was used for DNA precipitation and purification, and elution buffer was used to elute purified DNA from the columns. All buffers were provided in the ChIP kit.
DNA quantification.
Quantitative real-time (RT)-PCR with SYBR Green Master kit (Applied Biosystems) was used to measure the amount of MeCP2-, G9a-, and H3K9me2-associated DNA with adenine phosphoribosyltransferase (house-keeping mRNA) as negative control. Signal difference was calculated by: ΔCt = (Nexp − Nave) × Ctave (Nexp, normalized Ct value of the target or Cttarget/Ctinput; Nave, mean N value for control; and Ctave, mean Ct value for control).
Quantitative RT-PCR.
RNA was extracted with the RNAqueous-4PCR Kit and reverse transcribed with the RETROscript Kit (Applied Biosystems). cDNA was quantified by RT-PCR and specific cDNA regions of the transcripts were amplified with custom-designed primers (Invitrogen). Fold differences of mRNA levels over controls were calculated by ΔCt. The following primers were used: G9a, forward 5′ TGCCTATGTGGTCAGCTCAG-3′, reverse 5′-GGTTCTTGCAGCTTCTCCAG-3′; Bdnf, forward 5′-GAGGGCTCCTGCTTCTCAA-3′, reverse 5′-GCCTTCATGCAACCGAAGT-3′; Mecp2, forward 5′-CGCTCCGCCCTATCTCTGA-3′, reverse 5′-ACAGATCGGATAGAAGACTC-3′; Gapdh, forward 5′- AGGTCGGTGTGAACGGATTTG-3′, reverse 5′-TGTAGACCATGTAGTTGAGGTCA-3′.
Data analysis and materials.
ANOVA (one-way and two-way) and post hoc analysis were used to statistically analyze experimental data between treatment groups with multiple comparisons. Simple comparisons of data between two groups were made with the unpaired Students' t test. Behavioral data with multiple measurements were statistically analyzed by two-way ANOVA for repeated measures with the Bonferroni method for post hoc tests. Data are presented as mean ± SEM and p < 0.05 was considered statistically significant. All statistical analyses were performed with the Prism software version 5.04 (GraphPad Software). Drugs were purchased from Sigma-Aldrich or Tocris Bioscience.
Results
Acquisition of reward behavior is facilitated in pain conditions
We used a mouse model of persistent inflammatory pain induced by an injection of CFA (40 μl) into a hindpaw and measured by thermal and mechanical threshold of sensory pain (Fig. 1A; two-way ANOVA, time: F(16,96) = 7.219, p < 0.001; CFA: F(1,6) = 111.9, p < 0.001; interaction: F(16,96) = 8.442, p < 0.001). To determine pain-induced changes in opioid reward at different stages of opioid exposure, we assessed opioid reward with the CPP paradigm (Tzschentke, 2007), which allows studies on initial acquisition of reward-related behavior and its reacquisition (reinstatement) after re-exposure to opioids. In naive mice, three daily sessions of morphine conditioning induced CPP behavior in a dose-dependent manner (Fig. 1B; one-way ANOVA, F(3,15) = 23.08, p < 0.001). In mice with the pain condition (3 d post-CFA injection), a subthreshold dose of morphine (0.1 mg/kg, i.p.), ineffective in control mice, induced significant CPP after three conditioning sessions (Fig. 1C; t(1,14) = 3.641, p = 0.0027), suggesting increased response of CPP behavior to opioid reward under the pain condition. This low dose of morphine (0.1 mg/kg, i.p.) had no effect on CFA-induced sensory pain (Fig. 1D).
Figure 1.
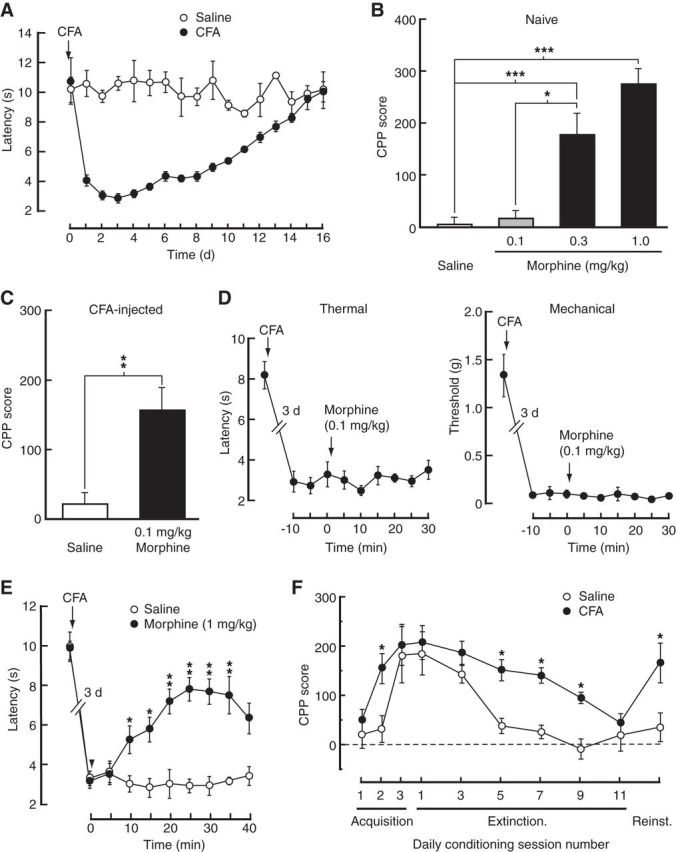
Acquisition of reward behavior is facilitated in pain conditions. A, Time course for the development of persistent sensory pain induced by CFA and measured by the paw-withdrawal test (n = 6 mice each group). B, Behavior of CPP in naive mice conditioned with saline or three doses of morphine (n = 4–6 mice each group). C, CPP after conditioning with saline or a subthreshold dose of morphine in CFA-injected mice (n = 8 each group). D, Effects of 0.1 mg/kg morphine on CFA-induced sensitization of thermal and mechanical pain. E, Effects of 1 mg/kg morphine on CFA-induced sensitization of thermal pain. F, Time course of CPP acquisition, extinction, and reinstatement (reinst.) in mice (n = 6 each group) injected with saline or CFA 3 d before the first conditioning session; *p < 0.05, **p < 0.01, ***p < 0.001.
We then assessed this pain effect on CPP behavior at different stages of CPP acquisition, extinction, and reinstatement with a higher, analgesic dose of morphine at 1 mg/kg intraperitoneally (Fig. 1E; two-way ANOVA, time: F(9,45) = 16.41, p < 0.001; morphine: F(1,5) = 11.80, p = 0.0185; interaction: F(9,45) = 3.847, p = 0.0011). During the initial acquisition period, mice with pain acquired CPP for morphine more rapidly than control mice. Specifically, control mice required a minimum of three daily morphine-conditioning sessions to acquire CPP, but only two such sessions were sufficient in mice with pain (Fig. 1F; two-way ANOVA, time: F(9,72) = 9.69, p < 0.001; CFA: F(1,8) = 12.63, p = 0.0075; interaction: F(9,72) = 1.18, p = 0.3201). However, after the acquisition, both groups displayed CPP of similar amplitudes, suggesting that pain may mainly affect the sensitivity to acquiring CPP behavior. During CPP extinction induced by daily sessions of conditioning with saline only, CPP extinguished far less slowly in CFA-injected mice when compared with controls (Fig. 1F) while CFA-induced sensory pain was diminishing. After CPP extinction, the subthreshold dose of morphine (0.1 mg/kg), ineffective in control mice, reinstated the CPP in CFA-injected mice (Fig. 1F) when CFA-induced sensory pain had fully recovered. These behavioral results suggest that the sensitivity to acquiring the opioid-induced preference behavior is increased under persistent pain conditions.
MeCP2 is important for increased sensitivity to reward behavior
Recent studies have shown that MeCP2 plays an important role in regulating drug reward-related motivational effects of cocaine and psychostimulants (Deng et al., 2010; Im et al., 2010). MeCP2 also has been implicated in modulation of pain behaviors in animals (Tochiki et al., 2012). Therefore, we examined the underlying molecular mechanisms for the pain effect, focusing on the role of MeCP2 in CeA regulation of pain and opioid reward. We found that MeCP2 expression in CeA was significantly increased in mice with persistent pain (Fig. 2A,B; t(1,12) = 2.266, p = 0.0428), but not with acute pain (4 h post-CFA injection; Fig. 2C,D). MeCP2 also was increased in mice with morphine (0.3 mg/kg)-induced CPP (Fig. 2A,B; t(1,6) = 3.917, p = 0.0078), but not in mice conditioned with a subthreshold morphine dose (0.1 mg/kg) without CPP (Fig. 2C,D). Repeated home-cage injections of morphine (0.3 mg/kg) without conditioning treatment did not alter the MeCP2 expression (Fig. 2C,D). In addition, the MeCP2 expression was closely linked to the acquisition of CPP behavior, which was increased after initial CPP acquisition (t(1,14) = 2.200, p = 0.0451), decreased after CPP extinction (t(1,14) = 2.417, p = 0.0299), and increased again after CPP reinstatement (t(1,14) = 2.300, p = 0.0373) in CFA-injected mice (Fig. 2E,F). Using the transgenic line of MeCP2-overexpressing mice (MeCP2-TG; Chahrour et al., 2008), we found that the MeCP2 level was ∼2.5-fold higher in the CeA of MeCP2-TG mice compared with WT controls (Fig. 2A,B; t(1,14) = 3.719, p = 0.0023). Consistently, MeCP2-TG mice displayed sensitized pain behavior with lower baseline pain threshold (Fig. 2G; t(1,22) = 6.496, p < 0.001) and their sensitivity to morphine-induced CPP was also increased (Fig. 2H; two-way ANOVA; MeCP2-TG: F(1,20) = 8.374, p = 0.0090; morphine dose: F(1,20) = 26.24, p < 0.0001; interaction: F(1,20) = 1.578, p = 0.2235), similar to the CFA-injected mice. Noticeably, the small increase in the magnitude of CPP induced by the higher morphine dose (0.3 mg/kg) in MeCP2-TG mice was not statistically significant (Fig. 2H), indicating a possibility that the sensitivity increase for acquired CPP is masked by increasing doses of morphine.
Figure 2.
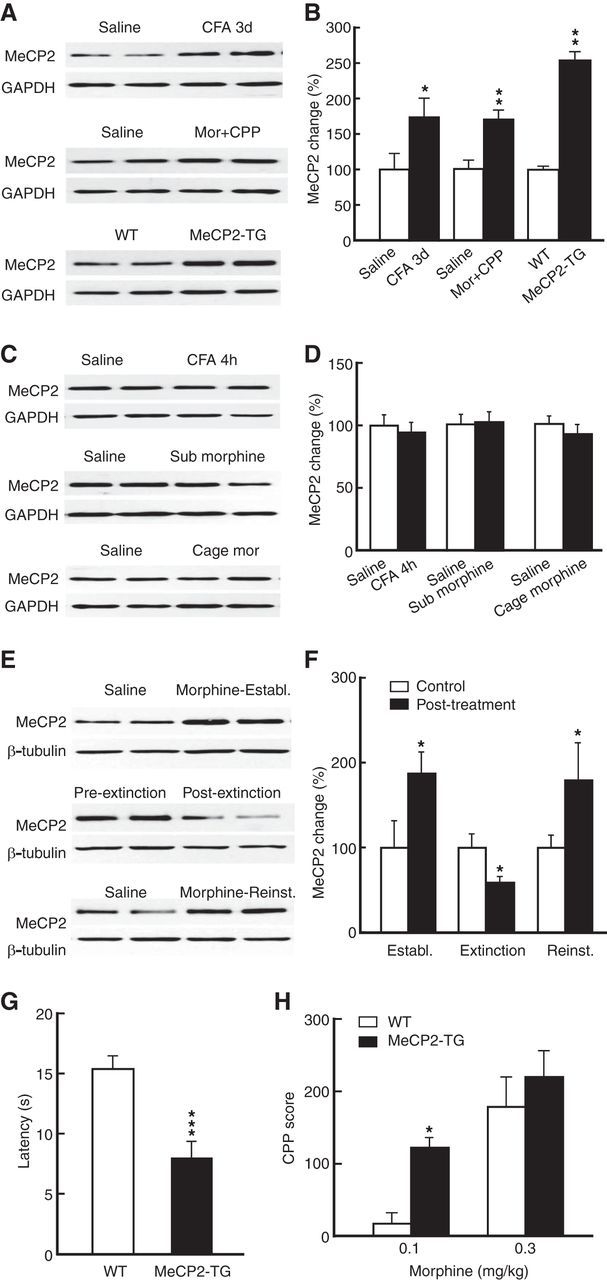
Persistent pain and morphine reward upregulate MeCP2 in CeA. A, B, Representative Western blots (A) and summarized results (B) of MeCP2 protein in the CeA from mice with persistent pain (CFA 3 d, n = 7 mice per group), with 0.3 mg/kg morphine-induced CPP (n = 4 mice per group), and from MeCP2-overexpressing mice (MeCP2-TG, n = 8 mice per group). C, D, Similar data of MeCP2 protein in the CeA from mice with acute pain (CFA 4 h), mice conditioned by a subthreshold dose of morphine (0.1 mg/kg, i.p.) with no CPP, and mice receiving injections of morphine (0.3 mg/kg, i.p.) in their home cages (n = 4–7 mice per group). E, F, Expression of CeA MeCP2 protein at the three CPP stages of establishment (establ.; after acquisition), extinction, and reinstatement (reinst.) in control mice and in CFA-injected mice (n = 8 mice per group). CPP was reinstated by 0.1 mg/kg morphine. G, Baseline pain threshold in WT (n = 5) and MeCP2-TG mice (n = 12). H, CPP behavior induced by two doses of morphine in WT (n = 5) and MeCP2-TG mice (n = 9). Mor, morphine.
To further determine the MeCP2 role, we used a recently described lentivirus vector expressing a short-hairpin interfering RNA against MeCP2 (lenti-sh-MeCP2; Im et al., 2010) to knockdown CeA MeCP2 by CeA infusion of the vector. Lenti-sh-MeCP2 reduced CeA MeCP2 level by ∼60% (Fig. 3A; t(1,6) = 3.810, p = 0.0089). CFA-induced pain sensitization was significantly inhibited in mice with CeA infusions of lenti-sh-MeCP2 when compared with control mice receiving CeA infusions of a control empty lentivirus vector (lenti-control; Fig. 3B,C; two-way ANOVA; Thermal, time: F(14,112) = 31.61, p < 0.001; sh-MeCP2: F(1,8) = 13.20, p = 0.0142; interaction: F(14,112) = 1.73, p = 0.0592. Mechanical, time: F(14,112) = 21.58, p < 0.001; sh-MeCP2: F(1,8) = 5.96, p = 0.0405; interaction: F(14,112) = 2.01, p = 0.023). CeA infusions of lenti-sh-MeCP2 had no effect on baseline pain threshold in WT mice (Fig. 3D). Lenti-sh-MeCP2-mediated knockdown of CeA MeCP2 greatly inhibited the reward response to morphine-induced CPP in naive mice (t(1,8) = 3.110, p = 0.0144); and it also reversed the sensitized response to subthreshold morphine-induced CPP in CFA-injected mice (t(1,8) = 3.706, p = 0.006; Fig. 3E,F). Representative positions of the cannula tips within the CeA for effective vector infusions are shown in Figure 3G and photomicrograph of a brain slice with CeA infusion of a dye is illustrated in Figure 3H. Together, these results provide several lines of evidence suggesting that amygdalar MeCP2 regulates behavioral responses to pain and opioid reward, and is important for the increased CPP sensitivity in pain conditions.
Figure 3.

Knockdown of CeA MeCP2 inhibits persistent pain and morphine reward. A, Expression of CeA MeCP2 protein in mice (n = 4 per group) after CeA infusion of a lentivirus vector expressing a short-hairpin interfering RNA against MeCP2 (lenti-sh-MeCP2) or a control empty lentivirus vector (lenti-control). B, C, Time course of CFA-induced changes in thermal (B) and mechanical (C) pain thresholds in mice with CeA infusion of lenti-sh-MeCP2 or control vector (n = 5 mice per group). D, Baseline pain thresholds in mice after CeA infusion of lenti-sh-MeCP2 or control vector (n = 4 mice per group). E, CPP behaviors in naive mice conditioned with 0.3 mg/kg morphine and in CFA-injected mice conditioned with 0.1 mg/kg morphine after CeA infusion of lenti-sh-MeCP2 or control vector (n = 5 mice per group). F, Preference behaviors before (pretest) and after (post-test) morphine conditioning in mice with CeA infusion of lenti-sh-MeCP2, showing no significant CPP after MeCP2 knockdown in both groups (n = 5). G, Schematic drawing showing representative positions of cannula tips within the CeA for the vector infusions. H, Photomicrograph of a brain slice with similar infusion of a dye in CeA. Scale bars: G, 0.5 mm; H, 1 mm.
MeCP2 represses G9a
Next, we determined the targets of MeCP2 in the pain effect by examining changes in the expression of possible candidate proteins, focusing on those that are related to MeCP2-mediated gene repression and are involved in neurological diseases from the list of genes that have been shown to be regulated by MeCP2 (Chahrour et al., 2008). Consistent with the previous report (Chahrour et al., 2008), we found that a range of proteins known to be involved in pain and drug-induced neuroplasticity (Pan et al., 1997; Basbaum et al., 2009; Robison and Nestler, 2011) were in fact upregulated (not repressed) in the CeA of MeCP2-overexpressing mice (Fig. 4A; KCC2: t(1,12) = 3.899, p = 0.0021; NGF: t(1,6) = 2.895, p = 0.0275; BDNF: t(1,6) = 4.426, p = 0.0044). Of note is the unchanged expression of repressor element 1-silencing transcription factor (REST), another general neuronal transcriptional repressor (Ooi and Wood, 2007). Among those proteins that were downregulated by MeCP2 overexpression, we found that the expression of the histone dimethyltransferase G9a showed a profound decrease (>50%) in the CeA of MeCP2-TG mice (Fig. 4B; t(1,14) = 2.442, p = 0.0285). G9a specifically catalyzes the dimethylation of Histone 3 at lysine 9 (H3K9me2), an epigenetic mark for transcriptional repression, and plays an important role in MeCP2-related mechanism of gene repression in neurological diseases (Li et al., 2007; Maze et al., 2010; Robison and Nestler, 2011). This indicates that MeCP2 may regulate pain responses and morphine reward through transcriptional repression of G9a in CeA. Consistently, protein level of G9a in CeA was significantly reduced in mice with persistent pain (Fig. 4C; t(1,8) = 2.380, p = 0.0445), but not with acute pain (Fig. 4D). The G9a protein level was also reduced in mice displaying morphine-induced CPP (Fig. 4E; t(1,12) = 3.334, p = 0.0059), but not in mice receiving morphine injections in their home-cages (Fig. 4F). Levels of H3K9me2 in CeA were changed accordingly along with G9a protein (Fig. 4B–F; MeCP2-TG: t(1,14) = 2.785, p = 0.0146; CFA: t(1,10) = 2.533, p = 0.0297; morphine: t(1,7) = 2.880, p = 0.0237). Corresponding changes also were found at G9a mRNA levels (Fig. 4G; MeCP2-TG: t(1,14) = 3.379, p = 0.0045; CFA: t(1,14) = 3.258, p = 0.0057; morphine: t(1,14) = 3.276, p = 0.0055).
Figure 4.
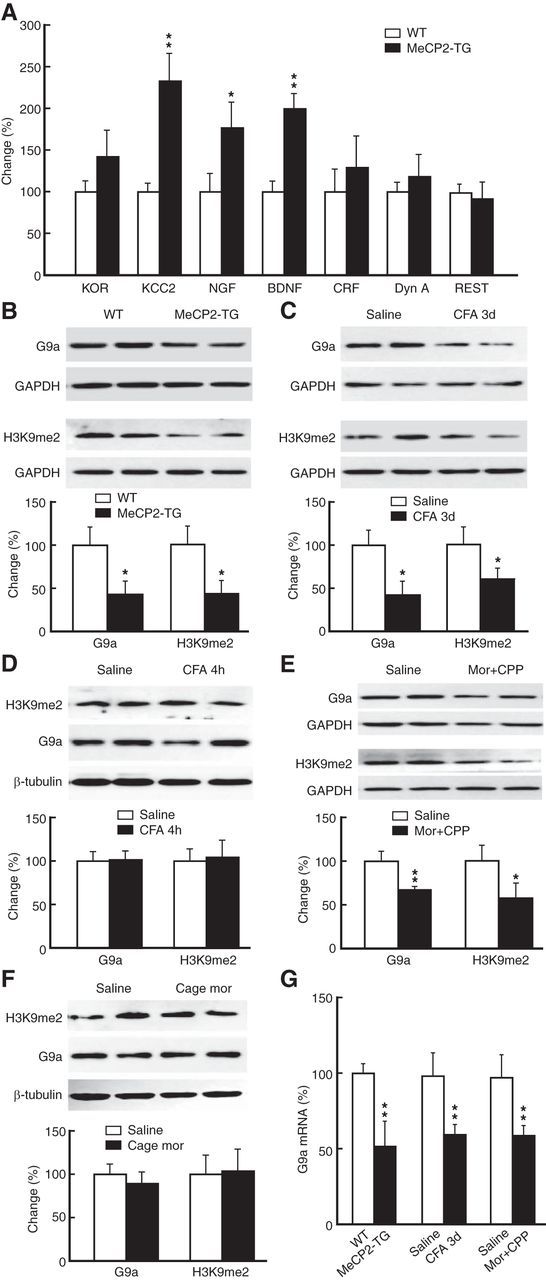
MeCP2 represses G9a expression. A, Expression of selected proteins analyzed by Western blotting in the CeA from WT and MeCP2-TG mice (n = 4–7 mice per group). KOR, κ opioid receptor; KCC2, K+/C− cotransporter; NGF, nerve growth factor; CRF, corticotropin-releasing factor; Dyn A, Dynorphin A. B–F, Levels of G9a protein and H3K9me2 in the CeA from MeCP2-TG mice (n = 8 per group; B), from mice with persistent pain (n = 4–6 per group; C), from mice with acute pain (n = 4 per group; D), from mice with morphine-induced CPP (n = 4–7 per group; E), and from mice receiving injections of morphine (0.3 mg/kg, i.p.) in their home cages (n = 4–7 per group; F). G, Levels of G9a mRNA determined by quantitative RT-PCR in the CeA from mice with MeCP2 overexpression, with persistent pain, or with morphine (Mor)-induced CPP (n = 6–8 per group).
Further supporting a repressive role of MeCP2 on G9a expression, lenti-sh-MeCP2 knockdown of MeCP2 increased levels of G9a protein (t(1,14) = 2.312, p = 0.0393), H3K9me2 marks (t(1,14) = 3.097, p = 0.0079), and G9a mRNA (t(1,14) = 3.123, p = 0.0088) in CeA (Fig. 5A,B). To determine whether MeCP2 directly regulated G9a transcription, we examined the physical association of MeCP2 with the G9a promoter in CeA tissues by ChIP assays. Using primers spanning the region from −100 bp to −3 kb upstream of the transcription start site (TSS) of G9a, we found selective enrichment of MeCP2 occupancy in the proximal promoter regions upstream of TSS in mouse CeA tissues, and this binding was markedly reduced in mice with virus-mediated knockdown of MeCP2 in CeA (Fig. 5C; t(1,14) = 3.670, p = 0.0025). In the G9a promoter region of −316 to −251 bp where MeCP2 binding was decreased by local MeCP2 knockdown, ChIP analysis showed that MeCP2 occupancy was significantly increased in the CeA of mice under behavioral conditions where MeCP2 expression was increased (Fig. 2B), including mice with MeCP2 overexpression (t(1,6) = 2.721, p = 0.0346), mice with persistent pain (t(1,12) = 3.164, p = 0.0082), and mice with morphine-induced CPP (t(1,6) = 3.318, p = 0.016; Fig. 5D). Conversely, MeCP2 knockdown decreased the MeCP2 occupancy on the G9a promoter (Fig. 5D; t(1,12) = 3.482, p = 0.0045). These findings suggest a direct MeCP2 repression of G9a transcription in CeA.
Figure 5.

MeCP2 binds to and represses G9a. A, B, Western blots (A) and summarized data (B) of levels of G9a protein, H3K9me2, and G9a mRNA in mice with CeA infusion of lenti-control (n = 7) or lenti-sh-MeCP2 vector (n = 9). C, Levels of relative occupancy of MeCP2 on a selected region (−3 kb to −100 bp) upstream of the transcription start site of G9a in mice after CeA infusion of lenti-control or lenti-sh-MeCP2 vector (n = 8 mice per group). D, Levels of MeCP2 on a proximal promoter region of G9a (−316 to −251 bp) in the CeA from MeCP2-TG mice (n = 4 per group), from mice with persistent pain (n = 6–8 per group), from mice with morphine-induced CPP (n = 4 mice per group), and from mice with CeA infusion of lenti-sh-MeCP2 (n = 6–8 per group).
G9a knockdown increases pain and morphine reward
Based on the above findings, it appears that direct repression of G9a by MeCP2 in CeA contributes to the pain-increased sensitivity to morphine-induced CPP. To test this hypothesis, we examined pain effects on behavior of opioid reward after knockdown of G9a in CeA, using an AAV-EF1a-mCherry-IRES-WGA-Cre (AAV-Cre) vector (Fig. 6A). Infusion of AAV-Cre into the CeA of G9afl/fl mice significantly reduced the levels of G9a protein (t(1,10) = 6.889, p < 0.0001) and H3K9me2 (t(1,10) = 3.370, p = 0.0071), but not G9a-like protein (GLP) and EZH2, two other repressive histone methyltransferases (Fig. 6B,C). Supporting a localized effect, G9a expression in the basolateral amygdala (BLA) was not affected by the transgene (Fig. 6B,C). We then determined the behavioral effect of this G9a knockdown and found that AAV-Cre-injected G9afl/fl mice displayed sensitized pain response (t(1,12) = 2.243, p = 0.0446) and increased sensitivity to morphine-induced CPP (t(1,10) = 2.503, p = 0.0313) when compared with AAV-GFP-injected control G9afl/fl mice (Fig. 6D)—findings similar to those observed in MeCP2-overexpressing mice (Fig. 2G,H).
Figure 6.
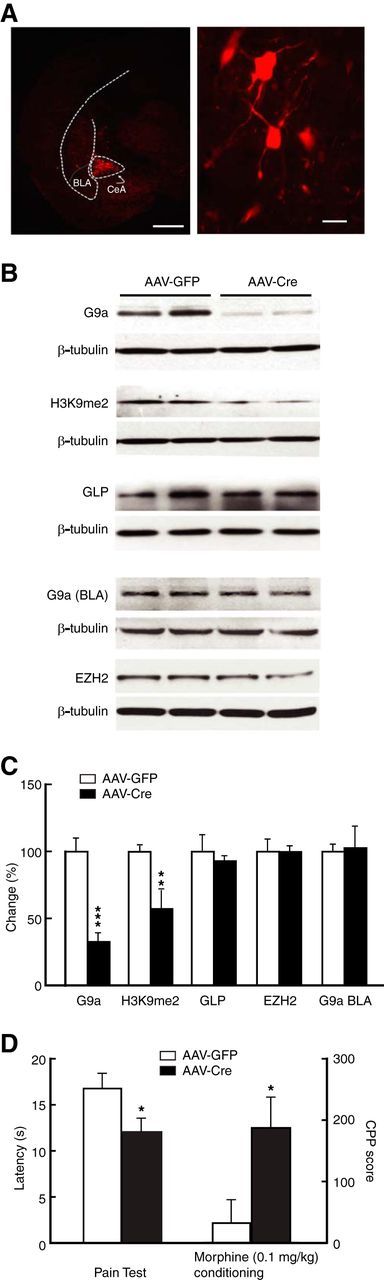
Knockdown of CeA G9a facilitates behavior of morphine reward. A, Representative images of expression of an AAV-Cre vector infused in the CeA (left) and in the CeA neurons (right) from a G9afl/fl mouse. Scale bars: Left, 500 μm; right, 15 μm. B, C, Western blots (B) and summarized data (C) of CeA levels of G9a, H3K9me2, GLP, and EZH2 (n = 5–6 mice per group), and G9a in the BLA (n = 4 mice per group) in G9afl/fl mice after CeA infusion of AAV-GFP (control) or AAV-Cre. D, Pain thresholds and CPP behaviors induced by a subthreshold dose of morphine (0.1 mg/kg) in the AAV-GFP-treated and AAV-Cre-treated G9afl/fl mice (n = 6–8 mice per group).
G9a represses BDNF
It has been shown that MeCP2 controls BDNF expression with positive correlation in expression and through homeostatic interaction with microRNA-212 (Sun and Wu, 2006; Im et al., 2010), but the epigenetic mechanism by which MeCP2 upregulates BDNF expression is unclear. Considering our current findings and previous reports that BDNF has promoting roles in both chronic pain and drug addiction (Pezet and McMahon, 2006; Pickens et al., 2011), we hypothesized that MeCP2 may regulate BDNF indirectly through G9a in CeA and BDNF may be an important target gene in the regulating cascade of MeCP2–G9a. Consistent with this hypothesis, we found that BDNF protein level was increased in the CeA of mice with persistent pain or morphine-induced CPP (Fig. 7A,B; CFA: t(1,19) = 2.208, p = 0.0397; morphine: t(1,13) = 2.744, p = 0.0167). In contrast to previous reports on direct MeCP2 repression of Bdnf transcription in cultured cortical cells (Chen et al., 2003; Martinowich et al., 2003), we found that BDNF was upregulated in the CeA of MeCP2-overexpressing mice and was downregulated by MeCP2 knockdown in vivo (Fig. 7A,B; MeCP2-TG: t(1,12) = 5.753, p = 0.0001; Lenti-sh-MeCP2: t(1,6) = 3.372, p = 0.015). However, MeCP2 occupancy at various Bdnf promoters in CeA was unchanged in mice with persistent pain (Fig. 7C), indicating an indirect interaction of MeCP2 and Bdnf. In contrast, G9a occupancy on the Bdnf axon II and IV promoters was reduced in mice with persistent pain (Fig. 7D; promoter II: t(1,6) = 3.145, p = 0.0199; promoter IV: t(1,6) = 3.130, p = 0.0203). Additionally, both the G9a binding of Bdnf promoter II and H3K9me2 levels were reduced in mice under conditions of increased MeCP2 expression by persistent pain (G9a: t(1,6) = 3.130, p = 0.0203; H3K9me2: t(1,6) = 3.847, p = 0.0085), by morphine-induced CPP (G9a: t(1,6) = 2.639, p = 0.0386; H3K9me2: t(1,6) = 3.268, p = 0.0171), and by MeCP2 overexpression (G9a: t(1,6) = 4.320, p = 0.005; H3K9me2: t(1,6) = 4.363, p = 0.0048), but were increased by MeCP2 knockdown (G9a: t(1,6) = 2.465, p = 0.0488; H3K9me2: t(1,6) = 3.128, p = 0.0204) in CeA (Fig. 7E). As shown in Figure 7F, the G9a inhibitor UNC0224 or the G9a/GLP inhibitor BIX01294 infused into CeA also reduced the H3K9me2 level at the Bdnf promoter (UNC0224: t(1,8) = 2.850, p = 0.0215; BIX01294: t(1,10) = 3.069, p = 0.0119) and increased BDNF protein level (UNC0224: t(1,6) = 4.052, p = 0.0067; BIX01294: t(1,6) = 4.052, p = 0.0067). These data support a direct G9a repression of Bdnf expression and suggest that MeCP2 may upregulate BDNF by repressing G9a expression in CeA.
Figure 7.

G9a represses BDNF. A, B, Western blot (A) and summarized data (B) of CeA BDNF protein in mice with CFA-induced pain, with morphine-induced CPP, with overexpression of CeA MeCP2, and with CeA infusion of lenti-sh-MeCP2 vector (n = 4–12 mice per group). C, D, Levels of MeCP2 (C) and G9a (D) on different Bdnf promoters in saline- and CFA-injected mice (n = 4–8 mice per group). E, Levels of G9a (n = 4–8 mice per group) and H3K9me2 (n = 4–6 mice per group) on the Bdnf promoter in mice groups as in B. F, CeA levels of H3K9me2 and BDNF protein in mice (n = 4–6 per group) treated by CeA infusion of the G9a inhibitors. Mor, morphine.
We then examined the roles of G9a and BDNF in behaviors of pain and morphine reward in mice in vivo. In contrast to the pain-inhibiting effect of MeCP2 knockdown (Fig. 3B,C), CeA-infusion of the G9a inhibitors enhanced CFA-induced pain response (Fig. 8A, two-way ANOVA; time: F(6,78) = 62.87, p < 0.001; inhibitor: F(2,13) = 3.753; p = 0.0517; interaction: F(12,78) = 2.317, p = 0.0137). Mimicking the pain effect of increased response to morphine-induced CPP (Fig. 1C), this G9a inhibition also rendered the subthreshold dose of morphine (0.1 mg/kg) effective in inducing CPP (Fig. 8B; UNC0224: t(1,9) = 2.994, p = 0.0151; BIX01294: t(1,9) = 3.177, p = 0.0112). These findings suggest that G9a inhibition augments pain response and increases response in morphine-induced CPP behavior, similar to conditions of persistent pain where MeCP2 expression is upregulated. Consistent with the effect of G9a inhibition, CeA infusion of BDNF, whose transcription was repressed by G9a, promoted responses to pain and morphine reward (Fig. 8D; two-way ANOVA; time: F(6,108) = 8.794, p = 0.0001; BDNF: F(1,18) = 1.543, p = 0.2301; interaction: F(6,108) = 9.834, p = 0.0001; Figure 8E; morphine 0.1 mg/kg, t(1,9) = 6.258, p = 0.0001). Importantly, CeA-applied BDNF rescued CPP by overcoming the inhibition of morphine CPP induced by MeCP2 knockdown (Fig. 8E; t(1,9) = 3.610, p = 0.0057). Supporting the pain-promoting and reward-facilitating effects of CeA BDNF, blocking BDNF signaling by CeA infusion of TrkB-IgG inhibited CFA-induced pain sensitization (Fig. 8F; two-way ANOVA; time: F(8,112) = 70.62, p < 0.0001; TrkB-IgG: F(1,14) = 13.45, p = 0.0025; interaction: F(8,112) = 7.396, p = 0.0001); it also reduced morphine-induced CPP in WT mice (Fig. 8G; t(1,8) = 4.220, p = 0.0029). CeA-infused TrkB-IgG also blocked the increased response in CPP induced by persistent pain (t(1,9) = 2.748, p = 0.0226) and by MeCP2 overexpression (t(1,13) = 5.192, p = 0.0002; Fig. 8G). Thus, it appears that G9a binds to Bdnf promoters and represses Bdnf expression, and G9a inhibition and activation of BDNF signaling in CeA have the same effect of pain augmentation and reward facilitation. Finally, CPP behavior might be confounded by a pain-induced effect on locomotor activity. In an open field test, we found that mice in saline- and CFA-injected groups displayed similar total distance traveled during the test, and there was also no significant difference in total distance traveled before and after CFA injection (two-way ANOVA; time: F(1,10) = 1.791, p = 0.2104; CFA: F(1,10) = 0.7360, p = 0.4110; interaction: F(1,10) = 1.423, p = 0.2604; Figure 8I). These data indicate little impact of the pain condition on general locomotor activity of the animals, which is consistent with a previous report under similar pain conditions in mice (Urban et al., 2011).
Figure 8.

G9a and BDNF are involved in pain facilitation of CPP. A, Time course of CFA-induced changes in pain threshold in mice with CeA infusion of lenti-sh-MeCP2 followed by vehicle or the G9a inhibitors (n = 5–6 mice per group); *p < 0.05 and #p < 0.05. B, CPP behaviors in mice conditioned with the subthreshold dose of morphine after CeA infusion of vehicle or the G9a inhibitors (n = 5–6 mice per group). C, Schematic drawing showing representative positions of cannula tips within the CeA for infusions of the G9a inhibitors. D, Changes in baseline pain threshold in mice (n = 10 per group) after CeA infusion of saline or BDNF. E, CPP behaviors in mice conditioned with a subthreshold dose (0.1 mg/kg) or an effective dose (0.3 mg/kg) of morphine without or with knockdown of CeA MeCP2 after CeA infusion of saline or BDNF (n = 5–6 mice per group). F, Changes in pain threshold 3 d after CFA injection in mice with CeA infusion of vehicle or the BDNF inhibitor TrkB-IgG (n = 8 per group). G, CPP behaviors in WT mice conditioned with an effective morphine dose, in WT mice with pain and conditioned with a subthreshold morphine dose, and in MeCP2-overexpressing mice conditioned with the subthreshold morphine dose after CeA infusion of vehicle or TrkB-IgG (n = 5–10 per group). H, Schematic drawing showing representative positions of cannula tips within the CeA for infusions of BDNF and TrkB-IgG. I, Total distance traveled in the open field test in mice before (preinjection) and after injection (post injection) with saline or CFA (n = 6 each group). Scale bars: C, 0.5 mm; H, 0.5 mm.
Discussion
The present study has presented several lines of molecular evidence suggesting that G9a is a direct transcriptional target of MeCP2 in the regulating cascade of MeCP2–G9a–BDNF in CeA regulation of pain and morphine reward. This convergence of regulating mechanisms on common epigenetic regulators may provide a molecular base for understanding functional interactions between pain responses and rewarding effects of opioids through emotion regulations by CeA. Our findings indicate that priming of this shared mechanism in CeA may contribute to increased response in behavioral preference for opioids under pain conditions.
The molecular function of MeCP2 as a transcriptional regulator has attracted much research attention in recent years due to its important roles in many neurological diseases (Samaco and Neul, 2011). MeCP2 has been prevailingly regarded as a general repressor by associating with transcriptional corepressors to inhibit gene transcription (Lewis et al., 1992; Bienvenu and Chelly, 2006). However, this original view has been extended by recent evidence suggesting that MeCP2 also functions as a transcriptional activator. In addition to earlier evidence that is inconsistent with an exclusive gene-silencing role of MeCP2 (Chang et al., 2006; Yasui et al., 2007), the study by Chahrour et al. (2008), using mice lacking or overexpressing MeCP2, has provided compelling evidence that the majority of hypothalamus genes are in fact activated by MeCP2, and MeCP2 binds to the promoters of the transcriptional activator cAMP response element binding protein (CREB) on selected genes. This presents a model of direct transcriptional activation by MeCP2. In contrast, the current study in CeA suggests a model of indirect transcriptional activation of BDNF by MeCP2 through de-repression (i.e., repression of the repressor G9a), and provides molecular evidence for the direct interactions of MeCP2–G9a and G9a–BDNF. It is interesting to note that such an indirect MeCP2 activation of target genes appears lacking in hypothalamus, as no repressor was found repressed by MeCP2 as previously described (Chahrour et al., 2008). The present study is in line with previous reports (Chang et al., 2006; Chahrour et al., 2008) in that MeCP2 activates Bdnf transcription in the brain, but direct MeCP2 binding to Bdnf promoters has been reported to date only in cultured cortical neurons in vitro where MeCP2 represses Bdnf expression (Chen et al., 2003; Martinowich et al., 2003). Another reported mechanism of indirect MeCP2 regulation of BDNF is mediated by homeostatic interactions of MeCP2 with microRNA-212 in striatum (Im et al., 2010). Thus, it appears that MeCP2 may repress or activate transcriptional activity of target genes either directly or indirectly through corepressors in different brain regions.
Recent studies have demonstrated critical roles of MeCP2 and G9a in a number of neuropsychiatric disorders and particularly in drug addiction. MeCP2 in striatum and nucleus accumbens (NAc) was found to mediate cocaine intake and behavioral response to psychostimulants (Deng et al., 2010; Im et al., 2010). G9a in NAc was recently reported to inhibit preference behavior for both cocaine and morphine (Maze et al., 2010; Sun et al., 2012). Repressive MeCP2 control of BDNF was characterized in BDNF-promoted behavior of cocaine intake (Im et al., 2010; Sadri-Vakili et al., 2010). Our findings in this study of CeA are in general agreement with these reports on reward-promoting roles of MeCP2 and BDNF, and further suggest repressive interactions between MeCP2 and G9a for control of BDNF expression in the preference behavior of opioid reward.
Evidence for roles of epigenetic regulators in the mechanisms of pain behaviors is just emerging (Denk and McMahon, 2012). Expression of MeCP2 in spinal cord was recently found altered under pain conditions (Tochiki et al., 2012; Kynast et al., 2013). A recent study showed that chronic morphine downregulated G9a in NAc and G9a overexpression promoted analgesic tolerance and withdrawal (Sun et al., 2012). We reported previously that epigenetic mechanism of histone acetylation was involved in pain development (Zhang et al., 2011). The present study provides original evidence that MeCP2 and the histone methyltransferase G9a, by transcriptional de-repression of Bdnf, play an important role in promoting pain behavior through BDNF upregulation, representing a molecular mechanism of MeCP2 and G9a-mediated methylation for the development of pain sensitization. It should be noted that G9a also methylates several nonhistone proteins (Rathert et al., 2008; Shankar et al., 2013). Thus, it is also possible that mechanisms by nonhistone proteins are involved in the behavioral effects after the G9a manipulations in this study.
An important question that remains to be answered is how chronic pain and repeated morphine activate this MeCP2–G9a pathway. As described in cultured cortical neurons, a cellular activity-dependent regulation of MeCP2 activity is MeCP2 phosphorylation, which releases MeCP2 from the promoter of its target gene, resulting in transcription activation via de-repression of the gene (Ebert et al., 2013). MeCp2 phosphorylation has also been attributed to upregulation of several genes in spinal dorsal horn neurons from a rat model of inflammatory pain (Géranton et al., 2007). While it is likely that cellular activity under the pain and opioid conditions also induces MeCP2 phosphorylation in CeA, it is still unclear how MeCP2 phosphorylation for gene de-repression is related to the MeCP2 upregulation in conditions of pain and repeated morphine found in this study. An alternative mechanism may operate for enhanced MeCP2 repression of its target genes, as observed in this study. It would be very intriguing in future studies to identify mechanisms that link disease conditions, such as chronic pain and opioid dependence, to altered MeCP2 activity in the CNS.
The effects of pain on opioid reward and consequently opioid use are still unclear, not to mention the underlying mechanisms. Indeed, sustained pain or noxious stimuli can activate the brain's reward circuitry in humans (Becerra et al., 2001; Zubieta et al., 2001). Previous animal studies provide conflicting results regarding pain effects on behaviors of opioid reward. While some studies reported that animals with pain consumed less opioids in a self-administration model or displayed reduced CPP behavior (Ozaki et al., 2003; Narita et al., 2005; Martin and Ewan, 2008), other studies showed that pain increased opioid self-administration and enhanced CPP behavior (Sufka, 1994; Kupers and Gybels, 1995; Colpaert et al., 2001; Cahill et al., 2013). In this regard, an important factor to consider is the sensory and affective dimensions of pain. Behaviors of drug seeking and drug use generally involve both a positive reinforcing effect of drugs and removal of negative reinforcing effect by drug withdrawal (Koob et al., 2004). The increased sensitivity in opioid-induced CPP behavior under pain condition we observed in this study may result from pain-induced priming of the reward-regulating cascade in CeA. As pain is often associated with an aversive state and pain relief is rewarding (Fields, 2004; King et al., 2009; Neugebauer et al., 2009), it is also possible that morphine inhibition of the affective component of pain (i.e., pain relief-induced rewarding effect) contributed to the increased sensitivity, providing that CFA-induced affective pain outlasts the sensory pain (Fig. 1F) and that the low dose of morphine (0.1 mg/kg), ineffective on sensory pain (Fig. 1D), is effective in inhibiting CFA-induced affective pain. Further studies are warranted to assess this pain relief-induced rewarding effect. Interestingly, our results indicate that pain may mainly affect the initial stage of reward for learning and acquiring the behavior and may be less effective on already acquired behavior (e.g., drug self-administration).
From a clinical perspective, how pain affects use and abuse of opioid analgesics has become a pressing issue in recent years (Ballantyne and LaForge, 2007). In addition to multiple systems involved in processing of pain and drug reward, patients with chronic pain commonly develop other comorbid emotional disorders, such as depression, anxiety, and stress, which may differentially alter behaviors of opioid reward (Edlund et al., 2007). Nevertheless, by identifying molecular mechanisms for CeA functions that regulate emotional processes associated with pain and opioid reward, the current study is an initial step toward understanding the multifaceted interactions between pain experience and behaviors of opioid use.
Footnotes
This work was supported by National Institutes of Health–National Institute on Drug Abuse grants DA023069 and DA027541 to Z.Z.P. and DA025983 to P.J.K., and by National Natural Science Foundation of China (31100802 and 91332109) and Development Program of Basic Research of China (2014CB548100) to Z.Z. We thank Dr. Huda Zoghbi for providing the MeCP2-TG mouse strain.
The authors declare no financial conflict of interest.
References
- Ballantyne JC, LaForge KS. Opioid dependence and addiction during opioid treatment of chronic pain. Pain. 2007;129:235–255. doi: 10.1016/j.pain.2007.03.028. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter MG, Murray EA. The amygdala and reward. Nat Rev Neurosci. 2002;3:563–573. doi: 10.1038/nrn875. [DOI] [PubMed] [Google Scholar]
- Becerra L, Breiter HC, Wise R, Gonzalez RG, Borsook D. Reward circuitry activation by noxious thermal stimuli. Neuron. 2001;32:927–946. doi: 10.1016/S0896-6273(01)00533-5. [DOI] [PubMed] [Google Scholar]
- Bie B, Zhu W, Pan ZZ. Ethanol-induced delta-opioid receptor modulation of glutamate synaptic transmission and conditioned place preference in central amygdala. Neuroscience. 2009;160:348–358. doi: 10.1016/j.neuroscience.2009.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Xue L, Grenier P, Magnussen C, Lecour S, Olmstead MC. Changes in morphine reward in a model of neuropathic pain. Behav Pharmacol. 2013;24:207–213. doi: 10.1097/FBP.0b013e3283618ac8. [DOI] [PubMed] [Google Scholar]
- Cai YQ, Wang W, Hou YY, Zhang Z, Xie J, Pan ZZ. Central amygdala GluA1 facilitates associative learning of opioid reward. J Neurosci. 2013;33:1577–1588. doi: 10.1523/JNEUROSCI.1749-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Colpaert FC, Tarayre JP, Alliaga M, Bruins Slot LA, Attal N, Koek W. Opiate self-administration as a measure of chronic nociceptive pain in arthritic rats. Pain. 2001;91:33–45. doi: 10.1016/S0304-3959(00)00413-9. [DOI] [PubMed] [Google Scholar]
- Deng JV, Rodriguiz RM, Hutchinson AN, Kim IH, Wetsel WC, West AE. MeCP2 in the nucleus accumbens contributes to neural and behavioral responses to psychostimulants. Nat Neurosci. 2010;13:1128–1136. doi: 10.1038/nn.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron. 2012;73:435–444. doi: 10.1016/j.neuron.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Gabel HW, Robinson ND, Kastan NR, Hu LS, Cohen S, Navarro AJ, Lyst MJ, Ekiert R, Bird AP, Greenberg ME. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499:341–345. doi: 10.1038/nature12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund MJ, Steffick D, Hudson T, Harris KM, Sullivan M. Risk factors for clinically recognized opioid abuse and dependence among veterans using opioids for chronic non-cancer pain. Pain. 2007;129:355–362. doi: 10.1016/j.pain.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5:565–575. doi: 10.1038/nrn1431. [DOI] [PubMed] [Google Scholar]
- Géranton SM, Morenilla-Palao C, Hunt SP. A role for transcriptional repressor methyl-CpG-binding protein 2 and plasticity-related gene serum- and glucocorticoid-inducible kinase 1 in the induction of inflammatory pain states. J Neurosci. 2007;27:6163–6173. doi: 10.1523/JNEUROSCI.1306-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried JA, O'Doherty J, Dolan RJ. Encoding predictive reward value in human amygdala and orbitofrontal cortex. Science. 2003;301:1104–1107. doi: 10.1126/science.1087919. [DOI] [PubMed] [Google Scholar]
- Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Im HI, Hollander JA, Bali P, Kenny PJ. MeCP2 controls BDNF expression and cocaine intake through homeostatic interactions with microRNA-212. Nat Neurosci. 2010;13:1120–1127. doi: 10.1038/nn.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Ahmed SH, Boutrel B, Chen SA, Kenny PJ, Markou A, O'Dell LE, Parsons LH, Sanna PP. Neurobiological mechanisms in the transition from drug use to drug dependence. Neurosci Biobehav Rev. 2004;27:739–749. doi: 10.1016/j.neubiorev.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Kupers R, Gybels J. The consumption of fentanyl is increased in rats with nociceptive but not with neuropathic pain. Pain. 1995;60:137–141. doi: 10.1016/0304-3959(94)00106-O. [DOI] [PubMed] [Google Scholar]
- Kynast KL, Russe OQ, Möser CV, Geisslinger G, Niederberger E. Modulation of central nervous system-specific microRNA-124a alters the inflammatory response in the formalin test in mice. Pain. 2013;154:368–376. doi: 10.1016/j.pain.2012.11.010. [DOI] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-O. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Martin TJ, Ewan E. Chronic pain alters drug self-administration: implications for addiction and pain mechanisms. Exp Clin Psychopharmacol. 2008;16:357–366. doi: 10.1037/a0013597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Maze I, Covington HE, 3rd, Dietz DM, LaPlant Q, Renthal W, Russo SJ, Mechanic M, Mouzon E, Neve RL, Haggarty SJ, Ren Y, Sampath SC, Hurd YL, Greengard P, Tarakhovsky A, Schaefer A, Nestler EJ. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Kishimoto Y, Ise Y, Yajima Y, Misawa K, Suzuki T. Direct evidence for the involvement of the mesolimbic kappa-opioid system in the morphine-induced rewarding effect under an inflammatory pain-like state. Neuropsychopharmacology. 2005;30:111–118. doi: 10.1038/sj.npp.1300527. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Galhardo V, Maione S, Mackey SC. Forebrain pain mechanisms. Brain Res Rev. 2009;60:226–242. doi: 10.1016/j.brainresrev.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi L, Wood IC. Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet. 2007;8:544–554. doi: 10.1038/nrg2100. [DOI] [PubMed] [Google Scholar]
- Ozaki S, Narita M, Narita M, Iino M, Miyoshi K, Suzuki T. Suppression of the morphine-induced rewarding effect and G-protein activation in the lower midbrain following nerve injury in the mouse: involvement of G-protein-coupled receptor kinase 2. Neuroscience. 2003;116:89–97. doi: 10.1016/S0306-4522(02)00699-1. [DOI] [PubMed] [Google Scholar]
- Pan ZZ, Tershner SA, Fields HL. Cellular mechanism for anti-analgesic action of agonists of the kappa-opioid receptor. Nature. 1997;389:382–385. doi: 10.1038/38730. [DOI] [PubMed] [Google Scholar]
- Passik SD, Kirsh KL. Addictions in pain clinics and pain treatment. Ann N Y Acad Sci. 2011;1216:138–143. doi: 10.1111/j.1749-6632.2010.05897.x. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Pickens CL, Airavaara M, Theberge F, Fanous S, Hope BT, Shaham Y. Neurobiology of the incubation of drug craving. Trends Neurosci. 2011;34:411–420. doi: 10.1016/j.tins.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/S0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng X, Jeltsch A. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol. 2008;4:344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Kumaresan V, Schmidt HD, Famous KR, Chawla P, Vassoler FM, Overland RP, Xia E, Bass CE, Terwilliger EF, Pierce RC, Cha JH. Cocaine-induced chromatin remodeling increases brain-derived neurotrophic factor transcription in the rat medial prefrontal cortex, which alters the reinforcing efficacy of cocaine. J Neurosci. 2010;30:11735–11744. doi: 10.1523/JNEUROSCI.2328-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaco RC, Neul JL. Complexities of Rett syndrome and MeCP2. J Neurosci. 2011;31:7951–7959. doi: 10.1523/JNEUROSCI.0169-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See RE, Fuchs RA, Ledford CC, McLaughlin J. Drug addiction, relapse, and the amygdala. Ann N Y Acad Sci. 2003;985:294–307. doi: 10.1111/j.1749-6632.2003.tb07089.x. [DOI] [PubMed] [Google Scholar]
- Shankar SR, Bahirvani AG, Rao VK, Bharathy N, Ow JR, Taneja R. G9a, a multipotent regulator of gene expression. Epigenetics. 2013;8:16–22. doi: 10.4161/epi.23331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sufka KJ. Conditioned place preference paradigm: a novel approach for analgesic drug assessment against chronic pain. Pain. 1994;58:355–366. doi: 10.1016/0304-3959(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Sun H, Maze I, Dietz DM, Scobie KN, Kennedy PJ, Damez-Werno D, Neve RL, Zachariou V, Shen L, Nestler EJ. Morphine epigenomically regulates behavior through alterations in histone H3 lysine 9 dimethylation in the nucleus accumbens. J Neurosci. 2012;32:17454–17464. doi: 10.1523/JNEUROSCI.1357-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YE, Wu H. The ups and downs of BDNF in Rett syndrome. Neuron. 2006;49:321–323. doi: 10.1016/j.neuron.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Tochiki KK, Cunningham J, Hunt SP, Géranton SM. The expression of spinal methyl-CpG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol Pain. 2012;8:14. doi: 10.1186/1744-8069-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol. 2007;12:227–462. doi: 10.1111/j.1369-1600.2007.00070.x. [DOI] [PubMed] [Google Scholar]
- Urban R, Scherrer G, Goulding EH, Tecott LH, Basbaum AI. Behavioral indices of ongoing pain are largely unchanged in male mice with tissue or nerve injury-induced mechanical hypersensitivity. Pain. 2011;152:990–1000. doi: 10.1016/j.pain.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Hashmi M. Use and abuse of opioid analgesics: potential methods to prevent and deter non-medical consumption of prescription opioids. Curr Opin Investig Drugs. 2004;5:61–66. [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Cai YQ, Zou F, Bie B, Pan ZZ. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med. 2011;17:1448–1455. doi: 10.1038/nm.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA, Stohler CS. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315. doi: 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]