

Abstract
The mammary ducts of humans and mice are comprised of two main mammary epithelial cell (MEC) subtypes: a surrounding layer of basal MECs and an inner layer of luminal MECs. Breast cancer subtypes show divergent clinical behavior that may reflect properties inherent in their MEC compartment-of-origin. How the response to a cancer-initiating genetic event is shaped by MEC subtype remains largely unexplored. Using the mouse mammary gland, we designed organotypic 3D culture models that permit challenge of discrete MEC compartments with the same oncogenic insult. Mammary organoids were prepared from mice engineered for compartment-restricted co-expression of oncogenic H-RASG12V together with a nuclear fluorescent reporter. Monitoring of H-RASG12V-expressing MECs during extended live cell imaging permitted visualization of Ras-driven phenotypes via video microscopy. Challenging either basal or luminal MECs with H-RASG12V drove MEC proliferation and survival, culminating in aberrant organoid overgrowth. In each compartment, Ras activation triggered modes of collective MEC migration and invasion that contrasted with physiological modes used during growth factor-initiated branching morphogenesis. Although basal and luminal Ras activation produced similar overgrowth phenotypes, inhibitor studies revealed divergent use of Ras effector pathways. Blocking either the phosphoinositide 3-kinase (PI3K) or the mammalian target of rapamycin (mTOR) pathway completely suppressed Ras-driven invasion and overgrowth of basal MECs, but only modestly attenuated Ras-driven phenotypes in luminal MECs. We show that MEC subtype defines signaling pathway dependencies downstream of Ras. Thus, cells-of-origin may critically determine the drug sensitivity profiles of mammary neoplasia.
Keywords: Mammary Gland, Ras, Basal, Luminal, 3D culture
Introduction
Mammary epithelium is comprised of two concentric cell compartments that are functionally distinct. The inner luminal MEC layer harbors cells responsible for milk production, whereas the outer basal MEC layer forms a sheath of myoepithelial cells whose contractility enables milk delivery (1). The ontogeny of these compartments remains incompletely defined. Landmark studies showed that a small subset of basal-like MECs can give rise to all MEC subtypes in mammary gland reconstitution assays, suggesting a precursor-product relationship between basal and luminal MECs (2, 3). However, recent lineage tracing studies show that the basal and luminal MEC lineages diverge during embryonic development, after which MECs remain lineage-confined throughout postnatal mammary development (4). Thus, the bulk of mammary gland growth, development, and maintenance likely depends on compartment-restricted MECs.
Distinct breast cancer subtypes have been proposed to arise from malignant transformation of distinct MEC subtypes. It follows that the divergent clinical behavior of breast cancer subtypes may be attributable in part to epigenetic traits inherited from diverse cells of origin (5). Intriguingly, distinct MEC subsets can yield biologically distinct cancers under some experimental conditions (6, 7). How the biology of MEC subtypes shapes the clinical behavior of descendant mammary cancers remains elusive due to the complexity inherent in multi-step tumor progression. Recent reports caution against assuming that breast cancers faithfully maintain biological features of their cell of origin. For example, both human and mouse studies suggest that basal-type breast cancers may arise from a luminal progenitor MEC population (8, 9), indicating that clonal evolution can obscure lineage history.
Since clonal evolution confounds efforts to uncover MEC subtype-specific mechanisms of transformation, we sought a strategy that would allow discrete MEC compartments to be challenged with a single initiating oncogenic lesion, then monitored prospectively during the initiation of neoplastic growth. Mammary organoids cultured under defined media conditions in 3D retain the bilayered architecture of the native mammary gland and remain competent to recapitulate complex developmental programs ex vivo when stimulated with growth factors (10). Adding transgene-encoded fluorescent reporters renders these organoids amenable to time-lapse imaging at single-cell resolution (11). Illustrating the power of this methodology, live MEC monitoring during organoid branching morphogenesis revealed a novel mode of cell migration and invasion termed multilayered epithelial elongation (11). Whether this physiological mode of invasion is co-opted by oncogenes to render MECs invasive remains unknown.
To elucidate early steps in the transformation of discrete MEC subsets, we generated mice harboring transgene combinations designed to permit MEC compartment-restricted expression of inducible oncogenes in organoid culture. As a test case, we chose to examine the impact of challenging discrete MEC compartments with the same oncogenic H-RASG12V allele. Though mutations in H-RAS or the closely-related K-RAS and N-RAS genes are rare in primary human breast cancers, they are common in established breast cancer cell lines (12) and frequently arise as cooperating lesions in mouse breast cancer models (13–15). More importantly, a large fraction of breast cancers harbor mutations that activate signaling pathways linked to Ras. HER2/neu-dependent signaling lies upstream of Ras and provides a validated drug target in more than 20% of breast cancers (16). In addition, roughly 30% of breast cancers harbor PIK3CA mutations that activate a key effector pathway downstream of Ras (17).
Materials and Methods
Transgenic mice
Mice were housed in a barrier facility with access to water and chow (Harlan Tekland, HT2018) ad libitum. All experimental protocols were approved by the Penn State College of Medicine IACUC Committee. Transgenic mouse strains used were as follows: tetO-H2B-eGFP (Jackson Labs), MMTV-rtTA (gift from Lewis Chodosh, University of Pennsylvania), K5-rtTA (Adam Glick, Pennsylvania State University), TRAS, FVB-Tg(tetO-HRAS)65Lc (NCI Mouse Repository). Mice were genotyped by performing PCR on tail clip-derived genomic DNA. PCR primers are available on request. Both Keratin-5-rtTA/TRAS/TGFP and MMTV-rtTA/TRAS/TGFP tri-transgenic mice remained healthy while Dox-naive, but became ill within 24 to 48 hours of Dox treatment. This TRAS transgene-associated morbidity precluded study of Ras-mediated phenotypes in vivo.
Mammary organoid preparation and culture
Organoids were generated from mammary tissue using mechanical dissociation and enzymatic digestion, followed by filtration and differential centrifugation steps as described (10, 11). Pelleted organoids were resuspended in Growth Factor Reduced Matrigel (GFR Matrigel, BD Biosciences) for plating. Wells of an optical-grade 96-well plate were pre-coated with 10 uL of GFR Matrigel underlay and permitted to set for 20 min at 37°C. A 45 μl suspension of organoids plus GFR-Matrigel was placed atop the underlay and permitted to set for 30 min at 37°C prior to overlaying 200 μL of organoid culture media. Dox-free organoid culture media consisted of the following: DMEM/F12 plus 2X antibiotic/antimycotic plus ITS (10 μg/mL insulin, 5.5 μg/mL transferrin and 2 ng/mL sodium selenite, all from Sigma). Organoid culture plates were typically allowed to equilibrate for 24 hours in a 37°C incubator before initiating experiments and acquiring images. Culture media was changed every other day. Media additives and growth factors at their typical final concentrations were as follows: Dox (Sigma, 1000 ng/mL); Fibroblastic growth factor, basic (Sigma, 50 ng/mL in TRIS, pH=7.0); PD0325901 (Cayman Chemicals, 100 nM in DMSO); LY294002 Calbiochem, 10 uM, in DMSO); Rapamycin (gift from Leonard Jefferson, Penn State College of Medicine, 5 nM). In experiments using inhibitors, inhibitor-free control wells were administered equivalent doses of DMSO or ethanol carrier. Organoid recovery for confocal microscopy was performed at room temperature as described (10), and slides were stored at −20°C until analysis.
Microscopy
Time-lapse imaging was performed using a Zeiss Axiovision Observer microscope with a 20X objective lens and an AxioCam MRM camera. Temperature was held at 37°C and CO2 concentration at 5% using a Zeiss Temp Module S, CO2 Module S and Heating Unit XL. Images of each organoid were acquired every 15 minutes using a robotic stage and Axiovision Multidimensional Imaging Modules, with Time-Lapse and Mark and Find functions. Time-lapse images were collected for up to 7 days, with images acquired and saved in 12–24 hour blocks. After each daily acquisition period, the z-axis was reset to maximize image focus.
For confocal microscopy, z-stacks were acquired using a Leica TCS SP2 AOBS inverted stage confocal microscope with laser lines 405, 476, and 543 (to detect nuclear GFP, Hoechst, and rhodamine phalloidin, respectively). Images were captured using either a 40X or a 63X objective lens.
Image and Statistical Analysis
To determine changes in organoid size and morphology, wide-field microscopy images were measured for 2D area and circularity. For area measurements, 2D organoid borders were traced. Area and perimeter measurements were determined using Zeiss Axiovision software. Circularity is a unitless measurement of the compactness of a shape, and ranges from zero to one. A perfect circle results in a circularity of one. Using perimeter and area measurements, circularity was measured using the formula: 4π(area)/(perimeter)2. Statistical significance on these measurements was determined by using ANOVA or repeated measures ANOVA analysis, where applicable. Individual group differences were then determined using the Tukey-Kramer Procedure. Repeated measures ANOVAs were used when organoid area or circularity was measured over time.
Mitotic and cell death events were quantified by tallying events observed during frame-by-frame analysis of time-lapse image sequences acquired between 24 and 72 hrs of Dox treatement. For statistical analysis, ANOVA analysis was performed, followed by the Tukey-Kramer Procedure for individual group differences. When only the difference between two groups was important, two-tailed Student t-tests were performed.
Results
First, we tested transgene combinations designed to restrict expression of identical tet-regulated responder genes to distinct MEC compartments. For luminal-restricted expression, a previously characterized MMTV-rtTA transactivator line was used (18). For basal-restricted expression, we tested a Keratin-5-rtTA (K5-rtTA) transactivator line (19). Each transactivator line was crossed to a tet-O-H2BGFP (hereafter TGFP) reporter line (20) and bitransgenic K5-rtTA/TGFP (basalGFP) and MMTV-rtTA/TGFP (luminalGFP) offspring were identified. Then, mammary organoids were prepared from Dox-naïve females, placed in 3D Matrigel culture, and subjected to live cell imaging before and after Dox addition.
Both basalGFP and luminalGFP organoids consistently lacked detectable GFP fluorescence in the absence of Dox treatment when visualized by wide-field fluorescent microscopy. Within 24 hrs of Dox treatment, organoids of both genotypes showed robust labeling with numerous H2BGFP-positive cells (Fig. 1A; Videos 1 and 2). Most H2BGFP-labeled MECs resided near the periphery of basalGFP organoids, but near the center of luminalGFP organoids (Fig. 1A), consistent with transactivator-specific labeling of spatially distinct MEC subsets. Similarly, these transgene combinations enable Dox-dependent labeling of spatially distinct MEC compartments in vivo (Mathers, J.M. and Gunther, E.J., unpublished data). To better define the cell compartments labeled by each transactivator in vitro, organoids collected at the conclusion of live cell imaging were counterstained and visualized by confocal microscopy (Fig. 1B). Optical sections through basalGFP organoids revealed H2BGFP-labeledcells exclusively in the outermost basal MEC layer. By contrast, luminalGFP organoids never contained labeledcells within this outer layer; instead, labeled cells exclusively resided internally. Labeling, was highly compartment-restricted but incomplete in that each transactivator labeled only a subset of the cells in its respective compartment. When basalGFP and luminalGFP organoids were subjected to extended live cell imaging with image capture every 15 minutes, nuclear H2BGFP fluorescence typically permitted tracking of individual MECs over many hours of imaging (Videos 1 and 2). Moreover, frame-by-frame analysis of time-lapse videos routinely permitted visualization of MEC mitoses and cell death events (Fig. 1C).
Figure 1. MMTV-rtTA and K5-rtTA restrict transgene expression to opposing MEC compartments.
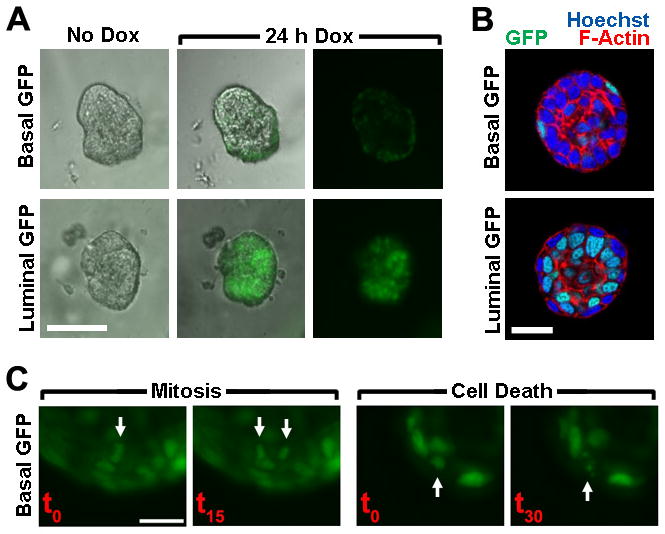
Organoids prepared from Dox-naïve bi-transgenic mice engineered for basal MEC-restricted labeling (K5-rtTA/TGFP; BasalGFP) or luminal MEC-restricted labeling (MMTV-rtTA/TGFP; LuminalGFP) were cultured in 3D. (A) Compartment-restricted MEC labeling. Live cell images (20X) of representative organoids captured before and after Dox-induced reporter gene expression are shown. Bar, 100 μm. (B). Confirmation of compartment-restricted labeling by confocal microscopy. Organoids were recovered after 7 d Dox treatment in 3D culture. Equatorial confocal images (63X) of representative BasalGFP and LuminalGFP organoids are shown. Bar, 20 μm. (C) Live cell imaging of MEC proliferation and death. Time-lapse images (20X) depict typical MEC mitosis and cell death events captured during extended imaging of a Dox-treated BasalGFP organoid. Bar, 20 μm.
To challenge discrete MEC compartments with the same oncogenic insult, crosses were performed to introduce a tet-regulated H-RASG12V transgene (21) (hereafter TRAS) into each model, yielding K5-rtTA/TRAS/TGFP (basalRAS/GFP) and MMTV-rtTA/TRAS/TGFP (luminalRAS/GFP) tri-transgenic mice. Mammary glands derived from Dox-naïve transgenic mice showed normal histomorphology, and sections from transgenic mammary glands showed normal MEC compartmentalization when subjected to immunohistochemistry using basal and luminal MEC markers (Fig. 2A). Organoids prepared from these glands were cultured in 3D and subjected to live cell imaging as before. In the absence of Dox treatment, basalRAS/GFP and luminalRAS/GFP organoids lacked detectable H2BGFP label and were morphologically indistinguishable from wild-type organoids, indicating tight regulation of the responder transgenes. In contrast, Dox treatment of both basalRAS/GFP and luminalRAS/GFP organoids triggered markedly aberrant organoid overgrowth (Fig. 2B; Videos 3 and 4). Genetic control organoids lacking the TRAS transgene (derived from basalGFP and luminalGFP mice) cultured in neighboring wells underwent Dox-dependent H2BGFP labeling but lacked Ras-mediated perturbations in organoid morphology and size, demonstrating that Dox treatment on its own did not perturb growth.
Figure 2. H-RASG12V expressed in either MEC compartment drives organoid overgrowth.

(A) Normal histomorphology of Dox-naïve transgenic mammary glands. Representative images of camine-stained mammary gland whole mounts and immunohistochemically stained tissue sections are shown. White bar, 500 μm; black bar, 100 μm. (B) Time-lapse imaging of organoid overgrowth. Organoids prepared from Dox-naïve mice engineered for Ras pathway activation in either basal MECs (K5-rtTA/TGFP/TRAS; BasalRAS/GFP) or luminal MECs (MMTV-rtTA/TGFP/TRAS; LuminalRAS/GFP) were cultured concurrently in 3D along with genetic control organoids lacking the TRAS transgene (BasalGFP and LuminalGFP). Sequential images of representative organoids of each genotype captured during a one week live-cell imaging experiment are shown. Times denote hrs after Dox addition. Bar, 50 μm. (C) Ras-mediated changes in organoid size and shape. 2D organoid images captured after 6 d Dox treatment were subjected to morphometric analysis to determine mean organoid area (left) and circularity (right). Mean values reflect analysis of 23 to 111 organoids for each genotype and treatment condition. (D) Ras-mediated increases in MEC proliferation and survival. Organoid videos were viewed frame-by-frame to score mitotic (left) and cell death (right) events occurring during the first 2 days of Dox treatment. Mean values reflect indices (events per organoid, normalized to a 24 hour acquisition period) derived from analyzing videos from 8 to 38 organoids of each genotype. *, p < 0.01; **, p < 0.05; Kruskal-Wallis analysis of variance, Tukey-Kramer method. All error bars indicate SEM.
To quantify Ras-mediated changes in organoid size and morphology, wide-field microscopy images of individual organoids were acquired after one week of culture and subjected to software-based image analysis. Organoid size was quantified by measuring organoid area in 2D. Organoid morphology was quantified by using 2D measurements to calculate circularity values indicative of organoid “roundness” (derived circularity values approach one as objects more closely approximate a perfect circle but decline as objects become less round, see Materials and Methods). Size and circularity measurements for representative sets of organoids were then compared across genotypes and Dox treatment conditions.
For both basalRAS/GFP and luminalRAS/GFP organoids, Dox treatment reproducibly increased size and decreased circularity in comparison to genetic and Dox-naïve controls (Fig. 2C). Changes occurring in luminalRAS/GFP organoids (9-fold increase in size vs. controls; 0.46 decrease in circularity) were modestly more pronounced than those occurring in basalRAS/GFP organoids (6-fold increase in size; 0.31 decrease in circularity). These differences may be attributable in part to the luminal transactivator targeting a larger MEC population at the outset (Fig. 1B).
To examine the cellular mechanisms of Ras-mediated overgrowth, live cell imaging videos from the first 72 hrs of Dox treatment were viewed frame-by-frame to identify MEC mitoses and cell death events. Ras expression in either compartment markedly increased the number of cell divisions and decreased the number of cell death events. Compared against GFP-only genetic controls, mitoses in basalRAS/GFP and luminalRAS/GFP organoids were elevated 20-fold and 11-fold respectively, whereas cell death events decreased approximately 4-fold and 3-fold respectively (Fig. 2D). These findings are consistent with Ras imparting both pro-proliferative and pro-survival signals in a cell autonomous manner, whether expressed basally or luminally.
Next, Ras-driven perturbations in morphogenesis were compared with a well-defined morphogenesis program triggered by growth factor supplementation. Organoids stimulated with Fgf2 use a mode of collective cell migration termed multilayered epithelial elongation when completing a morphogenesis program closely akin to physiological mammary branching (11). We confirmed that Fgf2-induced multilayered epithelial elongation proceeds without basally directed MEC extensions and protrusions and absent solitary “leader” cells at the forefront of invasion (Fig. 3A). Moreover, MECs at sites of Fgf2-induced branching aligned to form a smooth multicellular border at the invasion front. By contrast, for both basalRAS/GFP and luminalRAS/GFP organoids, Ras activation triggered focal invasion initiated by solitary leader cells, which typically extended one or more basal cellular protrusions into the surrounding Matrigel (Fig. 3B). Neighboring MECs migrated behind these leader cells, forming solid strands of invading cells. Typically, leader cells emerged at multiple sites along the organoid periphery, initiating numerous invasion foci and generating an irregular organoid border. This mode of invasion was clearly Ras-initiated, since nearly all basalRAS/GFP and luminalRAS/GFP organoids developed invasive foci during Dox treatment, whereas both Dox-naïve and genetic control organoids almost never did (Fig. 3C). To contrast the growth factor-driven and Ras-driven modes of collective cell migration within the same organoid, luminalRAS/GFP organoids were first stimulated with Fgf2 under Dox-free conditions for one week, permitting branching morphogenesis. Upon subsequent Dox addition, Ras activation triggered the formation of multiple invasive foci on each organoid, with basal cellular protrusions and migrating leader cells evident within 24 hours (Fig. 3D).
Figure 3. H-RASG12V, but not Fgf2, triggers focal invasion into Matrigel.

(A) Fgf2-induced multilayered epithelial elongation. Still image from a live cell imaging experiment depict a representative luminalGFP organoid 7 d after initiating Fgf2-induced branching morphogenesis. (B) Ras-mediated focal invasion. Time-lapse images depict leader cells initiating invasion. Arrowheads mark protrusions emanating from leader cells indicated by white arrows. Red Arrows indicate follower “stalk” cells. Bar, 25μm. (C) Quantification of Ras-mediated focal invasion. Organoids of the indicated genotypes were cultured in the presence and absence of Dox for 6 d, then scored for the presence of focal invasion as described in Materials and Methods. Mean values reflect analysis of 20 to 77 organoids for each genotype and treatment condition. (D) Contrasting modes of collective MEC invasion triggered by Fgf2 versus H-RASG12V. Organoids of the indicated genotypes underwent Fgf2-induced branching prior to Dox treatment. Panels depict morphology of a representative organoid before and after Dox-induced transgene expression.
To determine the downstream signaling pathways engaged during Ras-mediated MEC overgrowth, pharmacologic inhibitors of well-known Ras effector pathways were employed at standard doses in time-lapse imaging experiments. Blockade of the mitogen-activated protein kinase (MAPK) cascade using the MEK inhibitor PD0325901 (PD) only modestly suppressed Ras-driven phenotypes in both MEC compartments (Fig. 4; Videos 5 and 6). Notably, for both basalRAS/GFP and luminalRAS/GFP organoids, Dox-dependent initiation of focal invasion was unimpeded by PD, and gross organoid morphology as quantified by circularity measurements was only partially preserved. Likewise, PD only partially suppressed overgrowth of both basalRAS/GFP and luminalRAS/GFP organoids, in each case reducing Ras-mediated increases in organoid size approximately two-fold. While these findings indicate that MAPK pathway activation contributes to Ras-mediated phenotypes in both MEC compartments, they underscore likely roles for additional Ras effectors in the invasion and overgrowth of both basal and luminal MECs.
Figure 4. MEK inhibition partially blocks Ras phenotypes in both MEC compartments.

BasalRAS/GFP and luminalRAS/GFP organoids were cultured under the indicated treatment conditions for 6 d. PD denotes MEK inhibitor PD0325901 at 100 nM. Left panels depict day 6 images (10X) of representative organoids for each genotype and treatment condition (see also Videos 5 and 6). Morphometry of 2D images captured at days 0, 2, 4 and 6 were used to determine the prevalence of focal invasion, as well as the mean organoid circularity and area at each time point. Each data point reflects the analysis of 37 to 55 organoids. Error bars denote SEM.
Whereas both basalRAS/GFP and luminalRAS/GFP organoids responded similarly to MAPK pathway inhibition, outcomes for each compartment following PI3K inhibition with LY294002 (LY) were strikingly divergent (Fig. 5A; Videos 7 and 8). For luminalRAS/GFP organoids, LY failed to suppress Ras-initiated focal invasion or preserve circularity, and partially suppressed organoid overgrowth by reducing Ras-mediated increases in organoid size approximately two-fold. In contrast, for basalRAS/GFP organoids, LY completely suppressed all Ras-mediated phenotypes initiated by Dox induction (Fig. 5A). Indeed, basalRAS/GFP organoids treated with both Dox and LY were, by morphometry, indistinguishable from Dox-naïve controls. Notably, despite abrogating Ras-mediated overgrowth, LY treatment of Dox-induced basalRAS/GFP organoids did not prevent H2BGFP labeling of basal MECs (Fig. 5A, lower left panels), demonstrating that LY does not act by inhibiting Dox-induced transgene expression.
Figure 5. PI3K/mTOR inhibition fully blocks Ras phenotypes in basal but not luminal MECs.

BasalRAS/GFP and luminalRAS/GFP organoids were cultured under the indicated treatment conditions for 6 d. Left panels depict day 6 images (10X) of representative organoids for each genotype and treatment condition (see also Videos 7–10). Morphometry of 2D images captured at days 0, 2, 4 and 6 were used to determine the prevalence of focal invasion, as well as the mean organoid circularity and area at each time point. (A) PI3K pathway inhibition. LY denotes PI3K inhibitor LY294002 at 10 μM. Each data point reflects the analysis of 24 to 47 organoids. (B) mTOR pathway inhibition. Rap denotes rapamycin at 5nM. Each data point reflects the analysis of 18 to 114 organoids. Error bars denote SEM.
To confirm and extend these findings, rapamycin (Rap) was used to inhibit mTOR, a known downstream effector of PI3K pathway activation (Fig. 5B). Strikingly, Rap treatment provided a close phenocopy of LY treatment. Like LY, Rap completely suppressed all Ras-mediated phenotypes in basalRAS/GFP organoids, including overgrowth, without precluding the H2BGFP labeling indicative of trangene induction (Fig. 5B; videos 9 and 10). Taken together, the LY and Rap treatment outcomes show that basal MECs, unlike their luminal counterparts, strictly depend on PI3K/mTOR signaling during Ras-mediated invasive overgrowth.
To examine the cellular mechanisms whereby inhibitors suppress Ras-mediated overgrowth, we again scored live cell imaging videos from the first 72 hrs of Dox treatment to quantify MEC fates. For both basalRAS/GFP and luminalRAS/GFP organoids, MEK inhibition with PD yielded no discernible changes in the frequency of mitoses and cell death events (Fig. 6A), in keeping with the relatively modest impact of PD on organoid morphology and size. In contrast, inhibition of PI3K/mTOR signaling by either LY or Rap treatment altered MEC fates in a compartment-specific manner (Fig. 6B). Whereas the frequency of mitotic events for luminalRAS/GFP organoids was unchanged by LY and reduced only modestly by Rap (38% reduction), mitoses in basalRAS/GFP organoids were reduced 3-fold by each inhibitor (Fig. 6B, left panel). Neither LY nor Rap treatment changed the frequency of cell death events in luminalRAS/GFP organoids. Interestingly, although both LY and Rap completely suppressed Ras-mediated phenotypes in basalRAS/GFP organoids, LY increased the number of MEC death events 8-fold whereas Rap treatment yielded no detectable change (Fig. 6B, right panel). Thus, treatments that suppressed invasive overgrowth of basal MECs consistently decreased cell proliferation but variably impacted cell death. These findings suggest that proliferation arrest may be the primary means by which PI3K/mTOR pathway inhibition counters the Ras-mediated overgrowth of basal MECs.
Figure 6. Suppressing Ras-driven phenotypes involves decreased proliferation and preservation of MEC compartments.

BasalRAS/GFP and luminalRAS/GFP organoids were cultured under the indicated treatment conditions in 3D. Mitotic and cell death events were scored during the first 2 days of Dox treatment. (A) MEK inhibitor effects on MEC proliferation and survival. PD denotes MEK inhibitor PD0325901 at 100 nM. (B) PI3K/mTOR inhibitor effects on MEC proliferation and survival. Mean values reflect indices (events per organoid, normalized to a 24 hour acquisition period) derived from analyzing videos from 11 to 34 organoids of each genotype. Error bars indicate SEM. (C) PI3K/mTOR inhibitor effects on MEC compartmentalization. Organoids were recovered after 3D culture for 7 d, then fixed and stained with Hoechst and phalloidin. Panels depict equatorial confocal microscopy images (63X) of representative organoids of each indicated genotype and treatment condition. Bars, 50 μm. LY denotes PI3K inhibitor LY294002 at 10 μM. Rap denotes rapamycin at 5nM.
In our inhibitor studies, treatment conditions that robustly suppressed Ras-driven increases in organoid size also preserved gross organoid morphology by preventing focal invasion. Since disruptions in cell polarity and tissue architecture promote mammary tumorigenesis (22), we examined whether treatments that abrogate Ras-driven invasion and overgrowth also preserve the organization of MEC compartments. Organoids collected at the conclusion of inhibitor experiments were counterstained and visualized by confocal microscopy (Fig. 6C). Examination of optical sections from basalRAS/GFP and luminalRAS/GFP organoids after 6 days of Dox treatment in the absence of inhibitors revealed H2BGFP-labeled MECs dispersed throughout both the periphery and center of organoids, indicating Ras-driven loss of MEC compartmentalization. For basalRAS/GFP organoids, when either LY or Rap was co-administered with Dox, PI3K/mTOR inhibition not only blocked overgrowth but also preserved MEC compartments, since H2BGFP-labeled cells continued to reside exclusively in the outermost MEC layer. These findings link Ras-driven organoid overgrowth to a breakdown in MEC compartmentalization.
Next, we considered whether the failure to block Ras-driven overgrowth of luminalMECs with PI3K/mTOR pathway inhibitors might be a trivial consequence of achieving higher levels of Ras pathway activation using our luminal transactivator. To test this possibility, both luminalRAS/GFP and basalRAS/GFP organoids were treated for 7 days with a range of Dox doses designed to drive varying levels of TRAS transgene expression. In pilot experiments, no changes in organoid size or morphology were apparent below a threshold Dox dose of 25 to 50 ng/ml (data not shown). Escalating Dox doses beyond this threshold yielded monotonic increases in organoid size up to 1000 ng/ml, indicating graded responses to a dynamic range of Ras activation levels (Fig. 7A). For basalRAS/GFP organoids, LY treatment completely abrogated Ras-mediated size increases over the full range of Ras activation levels tested. By contrast, for luminalRAS/GFP organoids, LY treatment reduced Ras-mediated size increases by a fixed proportion (approximately two-fold) at each of the three Dox treatment doses tested (Fig. 7A). Notably, LY treatment only partially blocked Ras-mediated overgrowth of luminalRAS/GFP organoids when co-administered with lower Dox doses that yielded sub-maximal size increases in the absence of inhibitor. Thus, even in the context of sub-maximal Ras pathway activation, LY only partially suppressed overgrowth of luminal MECs.
Figure 7. Pi3k/mTOR blockade abrogates basal but not luminal MEC overgrowth across a range of Ras activation levels and inhibitor doses.
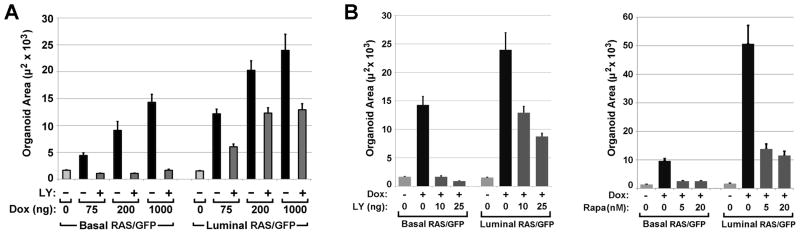
BasalRAS/GFP and luminalRAS/GFP organoids were cultured under the indicated treatment conditions for 6 d. 2D image morphometry was used to determine mean organoid area. (A) LY effects on organoid size across varied Dox doses. When administered, the LY dose was held constant at 10 μM. (B) Inhibitor effects on organoid size across varied drug doses. When administered, the Dox dose was held constant at 1000 ng/ml. Mean values reflect analysis of 18 to 34 organoids for each genotype and treatment condition. Error bars denote SEM.
Finally, we tested whether higher doses of Pi3k/mTOR pathway inhibitors would suffice to abrogate Ras-driven overgrowth of luminal MECs. Neither a 2.5-fold increase in LY dose (from 10 to 25 ng/ml) nor a 4-fold increase in Rapamycin dose (from 5 to 20 nM) abrogated Ras-driven overgrowth of luminalRAS/GFP organoids (Fig. 7B). Furthermore, MEC toxicity became apparent in control organoids when LY was dosed at 25 ng/ml (data not shown), indicating that Ras-driven overgrowth of luminal MECs persists even at the maximum tolerated LY dose.
Discussion
Numerous studies have explored how different cell subsets within a tissue respond to long-term challenge with activated Ras alleles. For example, directing expression of H-RASG12V transgenes to distinct skin compartments yielded models with markedly different rates of tumor onset and progression (23). Furthermore, some, but not all, skin compartments gave rise to tumors after undergoing cre-mediated activation of a latent K-RasG12D allele (24, 25). Similarly, activation of latent K-Ras alleles in distinct cell compartments within the pancreas drove tumorigenesis with different rates of onset and different requirements for cooperating tissue injury (26–28). By comparison, the short-term impact of cell type on Ras-initiated phenotypes remains largely unexplored.
Challenging basal or luminal MECs with the same oncogenic H-RASG12V allele triggered an MEC invasion program that differed markedly from the growth factor-initiated invasion program previously shown to recapitulate branching morphogenesis in 3D culture (11). The specific signaling events that lead to focal invasion upon H-RASG12V expression rather than multilayered epithelial elongation remain unclear. Mammary organoids engage known Ras effectors, including the MAPK pathway, when stimulated with growth factors (10), raising the possibility that focal invasion involves activating these same pathways to supraphysiologic levels. Alternatively, focal invasion may result from engaging Ras effector pathways in the wrong MECs or at the wrong time. The precise MEC subset that normally responds to FGF stimulation during branching morphogenesis remains undefined and may not be targeted by the transactivators employed in our study. Furthermore, proper initiation of the branching morphogenesis program may depend upon a specific duration of Ras pathway activation, as suggested by others (10).
Previous studies have examined links between Ras effector pathways and MEC invasion. When an inducible Raf construct was used to activate the MAP kinase cascade selectively, the MCF-10A line of immortalized human MECs acquired only a non-invasive mode of motility (29), suggesting that invasion requires input from another Ras effector pathway. However, MCF-10A cells engineered to express breast cancer-associated PIK3CA mutations acquired the capacity for growth factor-independent proliferation, but not invasion (30, 31). Thus, PI3K pathway activation by itself may be necessary but insufficient to confer invasiveness, as others have suggested based on studies performed on epithelial cell monolayers (32). Our study extends this paradigm in two ways. First, we show that the Ras effectors required for invasion and overgrowth differ for distinct MEC compartments. Second, we show that MEC invasion and overgrowth are closely correlated phenotypes. In our inhibitor studies, treatments that suppressed focal MEC invasion and preserved MEC compartment boundaries also robustly suppressed Ras-mediated overgrowth. These findings add to the accumulating evidence that maintaining proper mammary tissue architecture potently suppresses neoplasia.
Why do basal MECs uniquely require PI3K/mTOR signaling for Ras-mediated invasion and overgrowth? The answer may lie in the distinct microenvironments encountered by basal and luminal MECs. Unlike their luminal counterparts, basal MECs reside in direct contact with the extracellular matrix. Recent studies suggest that matrix attachment provides MCF-10A cells with critical cues that promote nutrient uptake and survival (33). For basal MECs, Ras-driven activation of PI3K/mTOR signaling may substitute for growth signals lost upon matrix detachment, thereby enabling aberrant compartment egress and expansion. Ras-mediated invasion of basal MECs may involve cell non-autonomous mechanisms as well. H-RASG12V has been shown to render epithelial cells invasive only when neighboring cells lack mutant Ras, suggesting that invasion can depend on pro-migratory interactions between mutant and wild-type cells (32). Our findings do not preclude roles for additional Ras effectors, such as the Ral/GDS pathway, in MEC overgrowth.
Some studies suggest that exogenous mutant Ras alleles yield more extreme phenotypes than corresponding mutations affecting endogenous Ras alleles. Retroviral transduction of K-RasG12V into either MCF-10A cells or hTERT-immortalized human MECs triggered fulminant invasion and overgrowth in 3D culture, whereas clones carrying K-RasG12V alleles generated by gene targeting showed morphology and growth that were nearly indistinguishable from wild-type (34). It is unknown whether these discrepancies primarily reflect quantitative or qualitative changes in the signaling events downstream of Ras. Supporting the importance of signaling intensity, strong selective pressure favors increased expression of mutant Ras alleles during the progression of both skin and mammary tumors in mice (35, 36). Likewise, human tumor cell lines often selectively overexpress endogenous mutant Ras alleles (34). Therefore, the transgene-mediated Ras pathway activation employed in our study may more closely model a scenario in which mutant Ras activity has been augmented by a secondary genetic event, such as copy number gain.
Chronic expression of activated H-RAS alleles in luminal MECs can drive mammary tumorigenesis in transgenic mouse models(35), whereas the fate of basal MECs similarly challenged remains unknown. Recent studies suggest that basal MECs may infrequently serve as cells-of-origin for highly prevalent breast cancer subtypes. Initiating transformation of luminal MECs from mice(9) (via conditional Brca1 deletion) and from humans(7) (via retroviral transduction) yielded tumors that resembled basal-like ductal carcinomas, a breast cancer subtype commonly encountered in the clinic. By contrast, transformation of basal MECs using identical protocols yielded tumors that resembled extremely rare breast cancer subtypes, namely adenomyoepitheliomas and metaplastic carcinomas. It will be valuable to confirm these findings using models that permit tracing of MEC lineages in situ during oncogene challenge and tumor progression(5). Toward this end, we are developing in vivo methods that build upon those described in this report.
Our studies explore signaling dependencies in the immediate aftermath of an initiating oncogenic event. By contrast, mammary tumorigenesis in mice and humans likely requires the step-wise acquisition of cooperating genetic lesions over months to years. This protracted course leaves ample opportunity for signaling dependencies to change during clonal evolution. However, a recurring theme in molecular oncology is the remarkable degree to which signaling pathway dependencies can remain in force over time, manifesting as oncogene addiction or lineage addiction (37, 38). We find that, at the initiation stage of MEC transformation, signaling pathway dependencies can be profoundly shaped by MEC subtype. It will be important to determine whether compartment-defined signaling dependencies persist during tumor progression.
Acknowledgments
We thank Travis Leonard for expert mouse genotyping and Daniel Beachler for initial characterization of basalGFP and basalRas/GFP mammary glands. We thank Lisa Shantz for advice on inhibitor dosing, Wade Edris for help with confocal microscopy, and Ethan Kuperman for critical review of the manuscript. Animal housing was provided through a facility constructed with support from a Research Facilities Improvement Grant (C06 RR-15428-01) from the National Center for Research Resources. This work was supported by a Department of Defense Predoctoral Traineeship Award (W81XWH-09-1-0123 to KPH), and grants from the National Cancer Institute (R01 CA152222) and the Mary Kay Foundation (Translational Breast Cancer Research Award). We thank Jake Gittlen, Warren Gittlen and the many benefactors of the Gittlen Cancer Research Foundation.
Footnotes
Conflicts of Interest: None of the authors have a conflict of interest to disclose.
References
- 1.Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev. 2009;23:2563–2577. doi: 10.1101/gad.1849509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, et al. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 3.Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, et al. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 4.Van Keymeulen A, Rocha AS, Ousset M, Beck B, Bouvencourt G, Rock J, et al. Distinct stem cells contribute to mammary gland development and maintenance. Nature. 2011;479:189–193. doi: 10.1038/nature10573. [DOI] [PubMed] [Google Scholar]
- 5.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 6.Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell. 2007;12:160–170. doi: 10.1016/j.ccr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Keller PJ, Arendt LM, Skibinski A, Logvinenko T, Klebba I, Dong S, et al. Defining the cellular precursors to human breast cancer. Proc Natl Acad Sci U S A. 2012;109:2772–2777. doi: 10.1073/pnas.1017626108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 9.Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–417. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Fata JE, Mori H, Ewald AJ, Zhang H, Yao E, Werb Z, et al. The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFalpha and FGF7 in morphogenesis of mouse mammary epithelium. Dev Biol. 2007;306:193–207. doi: 10.1016/j.ydbio.2007.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell. 2008;14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollestelle A, Elstrodt F, Nagel JH, Kallemeijn WW, Schutte M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol Cancer Res. 2007;5:195–201. doi: 10.1158/1541-7786.MCR-06-0263. [DOI] [PubMed] [Google Scholar]
- 13.D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235–239. doi: 10.1038/84691. [DOI] [PubMed] [Google Scholar]
- 14.Podsypanina K, Li Y, Varmus HE. Evolution of somatic mutations in mammary tumors in transgenic mice is influenced by the inherited genotype. BMC Med. 2004;2:24. doi: 10.1186/1741-7015-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang JW, Boxer RB, Chodosh LA. Isoform-specific ras activation and oncogene dependence during MYC- and Wnt-induced mammary tumorigenesis. Mol Cell Biol. 2006;26:8109–8121. doi: 10.1128/MCB.00404-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higgins MJ, Baselga J. Targeted therapies for breast cancer. J Clin Invest. 2011;121:3797–3803. doi: 10.1172/JCI57152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gustin JP, Cosgrove DP, Park BH. The PIK3CA gene as a mutated target for cancer therapy. Curr Cancer Drug Targets. 2008;8:733–740. doi: 10.2174/156800908786733504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunther EJ, Belka GK, Wertheim GB, Wang J, Hartman JL, Boxer RB, et al. A novel doxycycline-inducible system for the transgenic analysis of mammary gland biology. FASEB J. 2002;16:283–292. doi: 10.1096/fj.01-0551com. [DOI] [PubMed] [Google Scholar]
- 19.Diamond I, Owolabi T, Marco M, Lam C, Glick A. Conditional gene expression in the epidermis of transgenic mice using the tetracycline-regulated transactivators tTA and rTA linked to the keratin 5 promoter. J Invest Dermatol. 2000;115:788–794. doi: 10.1046/j.1523-1747.2000.00144.x. [DOI] [PubMed] [Google Scholar]
- 20.Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, et al. Defining the epithelial stem cell niche in skin. Science. 2004;303:359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 22.Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, et al. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008;135:865–878. doi: 10.1016/j.cell.2008.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown K, Strathdee D, Bryson S, Lambie W, Balmain A. The malignant capacity of skin tumours induced by expression of a mutant H-ras transgene depends on the cell type targeted. Curr Biol. 1998;8:516–524. doi: 10.1016/s0960-9822(98)70203-9. [DOI] [PubMed] [Google Scholar]
- 24.Lapouge G, Youssef KK, Vokaer B, Achouri Y, Michaux C, Sotiropoulou PA, et al. Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci U S A. 2011;108:7431–7436. doi: 10.1073/pnas.1012720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White AC, Tran K, Khuu J, Dang C, Cui Y, Binder SW, et al. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108:7425–7430. doi: 10.1073/pnas.1012670108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16:379–389. doi: 10.1016/j.ccr.2009.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–18912. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Habbe N, Shi G, Meguid RA, Fendrich V, Esni F, Chen H, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pearson GW, Hunter T. Real-time imaging reveals that noninvasive mammary epithelial acini can contain motile cells. J Cell Biol. 2007;179:1555–1567. doi: 10.1083/jcb.200706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 31.Lauring J, Cosgrove DP, Fontana S, Gustin JP, Konishi H, Abukhdeir AM, et al. Knock in of the AKT1 E17K mutation in human breast epithelial cells does not recapitulate oncogenic PIK3CA mutations. Oncogene. 2010;29:2337–2345. doi: 10.1038/onc.2009.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hogan C, Dupre-Crochet S, Norman M, Kajita M, Zimmermann C, Pelling AE, et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol. 2009;11:460–467. doi: 10.1038/ncb1853. [DOI] [PubMed] [Google Scholar]
- 33.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konishi H, Karakas B, Abukhdeir AM, Lauring J, Gustin JP, Garay JP, et al. Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras mediated transformation. Cancer Res. 2007;67:8460–8467. doi: 10.1158/0008-5472.CAN-07-0108. [DOI] [PubMed] [Google Scholar]
- 35.Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol. 2007;9:493–505. doi: 10.1038/ncb1567. [DOI] [PubMed] [Google Scholar]
- 36.Burns PA, Bremner R, Balmain A. Genetic changes during mouse skin tumorigenesis. Environ Health Perspect. 1991;93:41–44. doi: 10.1289/ehp.919341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garraway LA, Sellers WR. Lineage dependency and lineage- survival oncogenes in human cancer. Nat Rev Cancer. 2006;6:593–602. doi: 10.1038/nrc1947. [DOI] [PubMed] [Google Scholar]
- 38.Haber DA, Gray NS, Baselga J. The evolving war on cancer. Cell. 2011;145:19–24. doi: 10.1016/j.cell.2011.03.026. [DOI] [PubMed] [Google Scholar]