

Abstract
Background:
Many diseases are associated with oxidative stress caused by free radicals. The aim of the present study was to evaluate the antidiabetic, antioxidant and antiglycation properties of Eysenhardtia polystachya (EP) bark methanol-water extract.
Materials and Methods
: The antioxidant capacities were evaluated by studying in vitro the scavenging of DPPH and ABTS free radical, reactive oxygen species such as RO2, O2·-, H2O2, OH., H2O2, ONOO-, NO, HOCl,1 O2, chelating ability, ORAC, β-carotene-bleaching and lipid peroxidation. The antiglycation activities of EP were evaluated by haemoglobin, bovine serum albumin (BSA)-glucose, BSA-methylglyoxal and BSA-glucose assays. Oral administration of EP at the doses of 100 mg/kg, 200 mg/kg and 400 mg/g was studied in normal, glucose-loaded and antidiabetic effects on streptozotocin-induced mildly diabetic (MD) and severely diabetic (SD) mice.
Results:
EP showed Hdonor activity, free radical scavenging activity, metal chelating ability and lipid peroxidation Antioxidant activity may be attributed to the presence of phenolic and flavonoid compounds. EP is an inhibitor of fluorescent AGE, methylglyoxal and the glycation of haemoglobin. In STZ-induced diabetic mice, EP reduced the blood glucose, increased serum insulin, body weight, marker enzymes of hepatic function, glycogen, HDL, GK and HK while there was reduction in the levels of triglyceride, cholesterol, TBARS, LDL and G6Pase.
Conclusions:
Eysenhardtia polystachya possesses considerable antioxidant activity with reactive oxygen species (ROS) scavenging activity and demonstrated an anti-AGEs and hepatoprotective role, inhibits hyperglycemic, hyperlipidemic and oxidative stress indicating that these effects may be mediated by interacting with multiple targets operating in diabetes mellitus.
Keywords: Antidiabetic, antiglycation, antioxidant, Eysenhardtia polystachya
INTRODUCTION
It is increasingly being realised that many of today's diseases are due to the “oxidative stress” that results from an imbalance between the formation and neutralisation of pro-oxidants. Oxidative stress is initiated by free radicals, that seeks stability through electron pairing with biological macromolecules such as proteins, lipids and DNA in healthy human cells and cause protein and DNA damage, along with lipid peroxidation.[1] All human cells protect themselves against free radical damage by enzymes such as superoxide dismutase (SOD) and catalase or compounds such as ascorbic acid, tocopherol and glutathione.[2] Sometimes, these protective mechanisms are disrupted by various pathological processes and antioxidant supplements are vital to combat oxidative damage. Recently, much attention has been directed towards the development of ethnomedicines with strong antioxidant properties but low cytotoxicities. Antioxidants are compounds that can delay or inhibit the oxidation of lipids and other molecules and by doing so inhibit the initiation and propagation of oxidative chain reactions. They act by one or more of the following mechanisms: reducing activity, free radical-scavenging, potential complexing of pro-oxidant metals and quenching of singlet oxygen.
Diabetes mellitus is the most prevalent metabolic disorder that is principally characterized by insulin resistance (IR) and elevated blood glucose levels.[3] Prolonged hyperglycemia contributes importantly to the pathogenesis of diabetic complications by increasing protein glycation, leading to the gradual buildup of advanced glycation end products (AGEs) in body tissue.[4] The complex, fluorescent AGE molecules formed during Maillard reaction can lead to protein cross-linking, which contributes to the development and progression of various diabetic complications.[5] Many researchers have discussed the pathological features of diabetes, that are caused to a great extent by the accelerated formation of AGEs promoted by hyperglycaemia in tissues.[6] Inhibition of the formation of AGEs has been shown to be an effective way of retarding the full range of diabetes complications, such as nephropathy, neuropathy, retinopathy and vasculopathy.
The tree Eysenhardtia polystachya, (Ortega) Sarg, belonging to the Leguminosae family, is known as “palo azul” and has wide use for the treatment of nephrolithiasis, as a blood depurative, diuretic and anti-rheumatic and bladder disorders developing during diabetes.[7] Phytochemical studies indicate that E. polystachya contains polyphenols, beside these compounds, 7-hydroxy-2’,4’,5’-trimethoxyisoflavone was isolated as the principal fluorescent phenolic constituent of the heartwood.[8] In another study, (3S)-7-hydroxy-2’, 3’, 4’, 5’, 8-pentamethoxyisoflavan, (3S)-3’, 7-dihydroxy-2’, 4’, 5’, 8-tetramethoxyisoflavan and soduartin displayed moderate cytotoxic activity against KB cell lines.[9] Further studies isolated chalcones coatline A, B and (αR)-α,3, 4, 2’, 4’-penta-hydroxydihydrochalcone, (αR)-3’-C-D-xilopyranosyl-α, 3, 4, 2’, 4’- pentahydroxy-dihydro chalcone and (αR)-3’-C-β-D-xylopyrano- syl-α, 3, 4, 2’, 4’- pentahydroxydihydrochalcone from the bark and trunks.[10,11] The methanolic extract of branches displayed hypoglycaemic activity and their chemical analysis allowed the isolation of 3-O-acetyl-11α, 12α–epoxy-oleanan-28, l3β-olide, (+)-catechin and (+)-catechin 3-O-β-D-galactopyranoside.[12] In another report, methanolic bark extract was further separated by column chromatography, yielding four known substances: (-)-epicatechin, (+)-afzelechin, eriodictyol, (+)-quercetin-3-O-p-D-galacto-pyranoside, all of which showed scavenging properties against DPPH. Subcoriacin, displayed strong antioxidant activity in pancreatic homogenate.[13] Other Eysenhardtia as E. platycarpa, E. punctate and E. subcoriaceae protected the pancreas and displayed antioxidant activity.[12] 3-O-acetyloleanolic acid identified as the major constituent of E. platycarpa, showed a significant decrease (31 mg/kg of body weight, P < 0.05) in the glucose level of STZ-induced diabetic rats.[14] This study was performer to evaluate the hypoglycemic, antioxidant potential and AGEs inhibition capacity of the methanol-water extract from the bark of E. polystachya in in vitro assays and also using diab etes-induced oxidative damage in the liver, kidney and pancreas.
MATERIALS AND METHODS
Plant material and preparation of extracts
Fresh bark of E. polystachya was collected in Mexico State. A voucher specimen (No. 7345) was deposited in the Herbarium of the UAM-Xochimilco, for further reference. Bark was dried at room temperature and powdered (300 g). The powdered material was extracted using 900 ml of methanol-H2O 1:1 v/v consecutively using soxhlet apparatus. These extracts (EP) were filtered and concentrated by rotary vacuum evaporator and kept in a vacuum desiccator for the complete removal of solvent.
Estimation of total phenolic content
Total soluble phenolics of the extracts were determined with Folin-Ciocalteau reagent using gallic acid as the standard.[15]
Antioxidant activity in vitro
1, 1-Diphenyl-2-picrylhydrazyl (DPPH) Assay
The ability of the extract to scavenge DPPH radicals was assessed as described by Gyamfi et al.[16] using one of the most extensively used antioxidant assays for plant samples. This method is based on the scavenging of DPPH radicals by the antioxidants, which produces a decrease in absorbance at 517 nm. When a solution of DPPH is mixed with a substance that can donate a hydrogen atom, the reduced form of the radical is generated, accompanied by a loss of colour. This delocalisation is also responsible for the deep violet colour, characterized by an absorption band in ethanol solution at about 517 nm. A 50 μl of aliquot of extract or control was mixed with 450 μl PBS (10 mM/l, pH 7.4) and 1.0 ml of methanolic DPPH (0.1 mM/l) solution. After a 30 min reaction, the absorbance was recorded at 517 nm.
Trolox equivalent antioxidant capacity assay
The antiradical properties of the extracts were determined using the TEAC assay. The TEAC assay is based on the reduction of the 2, 2’-azinobis (3-ethylbenzothiazoline-6-sulfonic acid (ABTS) radical-cation by antioxidants and was adapted with minor modifications.[17] The ABTS+. radical-cation was prepared by mixing ABTS stock solution (7 mM in water) with 2.45 mM potassium persulphate (K2S2O8). This mixture was left for 12 to24 h in the dark until the reaction was complete and the absorbance was stable [Abs734 nm to 0.700 (±0.030)]. Plant extracts (1 ml) were allowed to react with 1ml of the ABTS solution and the absorbance was taken at 734 nm after 7 min using a spectrophotometer. Appropriate solvent blanks were run in each assay. The scavenging capacity of the extract was compared with that of α-tocopherol and percentage inhibition was calculated as ABTS radical scavenging activity.
Oxygen radical absorbance capacity (ORAC) assay
The assay was performed as per the method described by Cao et al.[18] All solutions were prepared in 75 mM phosphate buffer (pH 7.4). Samples were diluted by a factor of 30 in 5% randomly methylated cyclodextrin solution before being assayed. Twenty microlitres of diluted extracts were manually pipetted into sample wells of the microplate which was then placed in the microplate reader and incubated for 15 min at 37°C. During the first cycle, 120 μl of fluorescein solution (disodium salt, 20 μM) was injected into each well using the first automated reagent injector; each injection was followed by a 1's mixing cycle. During the second cycle, 60 μl of 45 mM of AAPH (2, 2’-azobis-2-methyl-propanimidamide, dihydrochloride) was injected into each well using the second automated reagent injector, followed by a 1 s mixing cycle. Following mixing, the initial fluorescence was read; fluorescence readings were then taken every 30's. with a total assay time of 30 min. A standard curve was prepared with α-tocopherol using a concentration range of 12.5-150 μM. The relative fluorescence versus time graph of each sample was recorded from which the area under curve (AUC) of each sample was calculated. The AUC of each sample was used along with the standard curve to calculate the oxygen radical absorbance capacity (ORAC) of each extract, expressed as μmol α-tocopherol equivalents/g extract equivalent. Fluorescence filters with excitation and emission wavelengths of 485 and 520nm, respectively, were used; these conditions correspond to the fluorescence properties of fluorescein.
Ferrous ion chelating ability
The method of Decker and Welch was used to investigate the ferrous ion chelating ability of extracts.[19] Brief1y, a given volume of the extract (0.1222 mg/ml), ascorbic acid (0.1564 mg/ml), or butylatedhydroxytoluene (BHT) (0.1890 mg/ml) was added to 50 μl of 2.0 mM aqueous FeSO4 in 5.0 ml test tube, then ethanol was added to 4.0 ml. After 5 min incubation, the reaction was initiated by 1.0 ml of 5.0 mM ferrozine. After 10 min of equilibrium, the absorbance at 562 nm was recorded. The controls contained all reaction reagents except the extract or positive control substance.
Nitric oxide radical scavenging assay
This assay was performed according to the method described by Sreejayan et al.[20] Nitric oxide generated from sodium nitroprusside in aqueous solution at a physiological pH interacts with oxygen to nitrite ions, that was measured by Griess reagent. The reaction mixture (3 ml) containing 10mM nitroprusside in phosphate buffered saline and the fractions or the extracts at different concentrations (50-800 μg/ml) were incubated at 25°C for 150 min. Aliquots of 0.5 ml of incubated sample were removed at 30 min intervals and 0.5 ml Griess reagent was added. The absorbance of the chromosphore formed was measured at 546 nm. Inhibition of the nitric oxide generated was measured by comparing the absorbance values of control and extracts.
Peroxynitrite scavenging
Peroxynitrite (ONOO-) was synthesised by the method described by Beckman et al.[21] An acidic solution (0.6 M HCl) of 5ml H2O2 (0.7 M) was mixed with 5 ml 0.6M KNO2 on an ice bath for 1's and 5 ml of ice-cold 1.2 M NaOH was added. Excess H2O2 was removed by treatment with granular MnO2 prewashed with 1.2 M NaOH and the reaction mixture was left overnight at -20° C. Peroxynitrite solution was collected from the top of the frozen mixture and the concentration was measured spectrophotometrically at 302nm (ε = 1670 M-1 cm-1). An Evans Blue bleaching assay was used to measure peroxynitrite scavenging activity. The reaction mixture contained 50 mM phosphate buffer (pH 7.4), 0.1 mM DTPA (diethylene triamine pentaacetic acid), 90 mM NaCl, 5 mM KCl, 12.5 μM Evans Blue, various doses of plant extract (0-200 μg/ml) and 1mM peroxynitrite in a final volume of 1ml. After incubation at 25° C for 30 min, the absorbance was measured at 611nm. The percentage scavenging of ONOO- was calculated by comparing the results of the test and blank samples. All tests were performed six times. Gallic acid was used as the reference compound.
β-Carotene-linoleic acid assay
A solution of β-carotene was prepared by dissolving 2 mg of β-carotene in 10ml chloroform and 1.0 ml of this solution was then pipetted into a flask containing 20 mg of linoleic acid and 200 mg of Tween-40 emulsifier. Chloroform was completely evaporated using a vacuum evaporator. Aliquots of 5.0 ml of this emulsion were transferred into a series of tubes containing various concentrations of the fractions (25-400 µg/ml) or tocopherol. The absorbance of the extracts and the standard was measured immediately (t = 0) and after 90 min at 470 nm. The tubes were incubated at 50°C in a water bath during the test. The antioxidant activities (AA) of the samples were evaluated in terms of bleaching of β-carotene.[22,23]
Superoxide radical scavenging assay
The scavenging activity against chemically generated superoxide radicals (O2·-) of the crude extracts was measured by means of spectrophotometric measurement of the product following the reduction of nitroblue tetrazolium (NBT). Superoxide anions were generated in a non-enzymatic PMS/NADH system.[24] The reaction mixture contained 1ml of test solution, 1.9 ml of 0.1M phosphate buffer (pH 7.4), 1 ml of 20 μM phenazine methosulphate (PMS), 156 μM nicotine adenine dinucleotide (NADH) and 25 μM NBT in phosphate buffer (pH-l 7.4). After 2 min of incubation at 25°C, the colour was read on a spectrophotometer at 560 nm against blank samples that contained no particle mass spectrometry (PMS).
Hydroxyl radical scavenging
This was assayed as described by Elizabeth and Rao.[25] with a slight modification. The assay is based on quantification of the degradation product of 2-deoxyribose by condensation with TBA. Hydroxyl radical was generated by the Fe3+-ascorbate-EDTA-H2O2 system (the Fenton reaction). The reaction mixture contained, in a final volume of 1ml, 2-deoxy-2-ribose (2.8mM), KH2P04-KOH buffer (20 mM, pH 7.4), FeCl3 ( 100 μM), EDTA (100 μM); H2O2 (1.0 mM), ascorbic acid (100 μM) and various concentrations (0-200 μg/ml) of the test sample or reference compound. After incubation for 1 h at 37° C, 0.5 ml of the reaction mixture was added to 1 ml 2.8% TCA, then 1 ml 1% aqueous TBA was added and the mixture was incubated at 90° C for 15 min to develop the colour. After cooling, the absorbance was measured at 532 nm against an appropriate blank solution. All tests were performed six times. Mannitol, a classical OH-scavenger, was used as a positive control. Percentage inhibition was evaluated by comparing the test and blank solutions.
Hydrogen peroxide scavenging
An aliquot of 50 mM H2O2 and various concentrations (0-2 mg/ml) of samples were mixed (1:1 v/v) and incubated for 30 min at room temperature. After incubation, 90 μl of the H2O2-sample solution was mixed with 10 μl methanol and 0.9ml FOX reagent was added (prepared in advance by mixing 9 volumes of 4.4 mM BHT in methanol with 1 volume of 1 mM xylenol orange and 2.56 mM ammonium ferrous sulphate in 0.25 M H2S04). The reaction mixture was then vortexed and incubated at room temperature for 30 min. The absorbance of the ferric-xylenol orange complex was measured at 560 nm. All tests were carried out six times and sodium pyruvate was used as the reference compound.[26]
Singlet oxygen scavenging
The production of singlet oxygen (1 O2) was determined by monitoring N, N-dimethyl-4-nitrosoaniline (RNO) bleaching, using a previously reported spectrophotometric method.[27] Singlet oxygen was generated by a reaction between NaOCl and H2O2 and the bleaching of RNO was monitored at 440 nm. The reaction mixture contained 45 mM phosphate buffer (pH 7.1), 50 mM NaOCl, 50 mM H2O2 50 mM histidine, 10 μM RNO and various concentrations (0-200 μg/ml) of sample in a final volume of 2 ml. It was incubated at 30°C for 40 min and the decrease in RNO absorbance was measured at 440 nm. The scavenging activity of sample was compared with that of lipoic acid, which was used as a reference compound. All tests were performed six times.
Hypochlorous acid scavenging
Hypochlorous acid (HOCl) was prepared immediately before the experiment by adjusting the pH of a 10% (v/v) solution of NaOCl to 6.2 with 0.6 M H2SO4 and the concentration of HOCl was determined by measuring the absorbance at 235 nm using the molar extinction coefficient of 100 M-1 cm-1. The assay was carried out as described by Aruoma and Halliwell.[28] with minor changes. The scavenging activity was evaluated by measuring the decrease in absorbance of catalase at 404 nm. The reaction mixture contained, in a final volume of 1 ml, 50mM phosphate buffer (pH 6.8), catalase (7.2 μM), HOCl (8.4 mM) and increasing concentrations (0-100 μg/ml) of plant extract. The assay mixture was incubated at 25° C for 20 min and the absorbance was measured against an appropriate blank. All tests were performed six times. Ascorbic acid, a potent HOCl scavenger, was used as a reference.
Assay for low-density lipoprotein (LDL) oxidation and measurement of lipid peroxidation
LDL was kept at -8°C and a working suspension (200 μg of protein/ml of phosphate-buffered saline) was prepared just before use. The method of LDL oxidation by copper ions was applied, as has been previously described.[29] To measure the resulting lipid peroxidation, we performed the chromogenic thiobarbituric acid assay. Thiobarbituric acid-reactive substances (TBARS) concentrations were evaluated using a standard curve of standard MDA at different concentrations versus absorbance at 532 nm. All experiments were carried out in triplicate. The formation of conjugated diene (CD), a lipid oxidation product, in low-density lipoprotein (LDL) also was determined according to the method described by Esterbauer et al.[30] The lipid oxidation of an LDL solution containing 200 to 500 μg of EP was initiated at 37°C by 0.1 mM CuC12. Absorbance at 234 nm was continuously recorded for 60 min at 37°C by a Hitachi U-2001 spectrometer with a constant re-circulating temperature. The lag phase, expressed in minutes, was defined as the period where no oxidation occurred. A longer lag phase indicated less CD formation.
Hypoglycemic activity assay in vivo
Animals
The study was conducted in male mice, weighing about 30-35 g. Before and during the experiment, animals were fed a standard laboratory diet (Mouse Chow 5015, Purina) with free access to water. Mice were procured from the bioterium of ENCB and were housed in microloan boxes in a controlled environment (temperature 25 ± 2° C). Animals were acclimatised for a period of three days in their new environment before the initiation of the experiment. Litter in cages was renewed three times a week to ensure hygiene and maximum comfort for animals. The experiments reported in this study were performed following the guidelines stated in Principles of Laboratory Animal Care (NIH publication 85-23, revised 1985) and the Mexican Official Normativity (Norma official Mexicana NOM-062-Z00-1999). Food consumption and weight gain were measured daily.
Streptozotocin-induced diabetic severe
Severe diabetes mellitus was induced in overnight fasted male mice by a single intra-peritoneal injection of streptozotocin, at a dose of 50 mg/kg body weight dissolved in cold citrate buffer (pH 4.5).[31] The mice with diabetic symptoms such as polydipsia and polyuria, as well as fasting blood glucose concentration higher than 13 mmol/l after 7 days of STZ injection, were selected for use as experimental animals. In some cases, an STZ injection may trigger massive insulin release and result in fatal hypoglycaemia. This hypoglycaemic period was followed by hyperglycaemia which then became permanent. To prevent death induced by STZ, the mice were fed with a 3% glucose solution for 24 h.
Induction of mildly diabetes
Mild diabetes was induced in overnight fasted mice by administering a single dose of freshly prepared solution of streptozotocin (STZ), 45 mg/kg b. w. i. p) in 0.1 mol/l cold citrate buffer (pH 4.5), 15 min after the intra-peritoneal administration of 120mg/kg nicotinamide (Sigma Chemical Company, St. Louis, MO, USA). The STZ treated animals were allowed to drink 5% glucose solution over night to overcome drug induced hypoglycemia. After 10 days of development of diabetes, mice with moderate diabetes having persistent glycosuria and hyperglycaemia (blood glucose >250 mg/dl) were used for further experimentation.[32]
Experimental design in diabetes mice
Effect of single oral administration of extract of E. polystachya in glucose level in normal, severe and mild diabetic mice
After the rat had been denied access to food/water overnight, they were randomly divided into twelve groups (six rats per group) matched for body weight. The test groups were orally administered 100, 200 and 400 mg/kg body weight (b. w.) of EP suspended in Tween 80, 1% via gavage. Glibenclamide (GB) at the dose of 5 mg/kg b. w. as standard drug. Blood samples were collected from the tail vein at 0, 2, 4, 6, 8 and 12 h after the administration. The plasma glucose concentration was determined by an enzymatic colorimetric method using a commercial kit (Sigma Aldrich, USA).
Antidiabetic test in chronic severe and mild streptozotocine-induced diabetic mice
In a parallel study seven groups (n = 10) of diabetic mice were used to determine the chronic effect of methanol: water 1:1 extract. Each group was submitted to a specific treatment, as follows. Normal control and severe and mild diabetic mice, groups, were fed with normal diet and drinking water ad libitum, and were given saline by gastric gavage. Severe and mild diabetic mice that received extracts by gastric gavage (400 mg per kg of body weight) every day were designated as SD + EP and MD + EP. Two groups with severe (SD + GB) and mild diabetes (MD + GB) rat were administered with glibenclamide (GB) 5 mg/kg as positive control.
Determination of body and food intake
Body weights of mice and the intake of food and water and were taken prior to the induction of hyperglycemia, at day 0 of treatment, and on a daily basis thereafter for 4 weeks.
Oral glucose tolerance test in diabetic mice
Animals of each group were orally administered EP extract at doses of 400 mg/kg body weight on a daily basis for 30 days. At the end of the experiment, an oral glucose tolerance test (OGTT) was performed to assess the animals’ sensitivity to a high glucose load. Overnight-fasted rat were fed orally 2 g glucose/kg b.w. Blood samples were collected from the caudal vein from a small incision at the end of the tail at 0 min (immediately after glucose load), 30, 60, 90 and 120 min after glucose administration.
Oral glucose tolerance test in normal mice
Oral glucose tolerance test was performed in overnight (16 h) starved normal mice. The mice were randomly divided into three groups (n = 6). Glucose 2 g/kg was fed 30 min after the administration of 400 mg/kg of extract and glibenclamide. Blood was withdrawn from the tail vein 0, 30, 60, 90 and 120 min, blood glucose level were appraised by commercial kit (Sigma Aldrich, USA).
Collection of organ tissues
At the end of chronic diabetes experiments all mice were anesthetized with 1.0% pentobarbital sodium and blood was obtained from the retro-orbital plexus of each animal following the injection of heparin (100 IU kg-1 body weight) into a tail vein for 10 min. The liver and kidney were removed according to defined anatomical landmarks.
Plasma biochemical analysis
Blood samples were collected from tail vein of the mice into micro centrifuge tubes containing heparin (10 μl, 1000 IU ml-1). The blood samples were then centrifuged at 1600 × g for 15 min at 4ºC for the preparation of plasma. Concentrations of plasma glucose, total cholesterol (TC), triglycerides (TG) and HDL-cholesterol, were measured with enzymatic assay kit (Genzyme Diagnostics), LDL-cholesterol was calculated as the remaining difference of total cholesterol and HDL. Blood glucose levels were measured employing the glucose oxidase-peroxidase (GOD-POD) method. Lipid peroxidation, i.e., thiobarbituric acid reactive substances (TBARS) was estimated by the method of Fraga et al.,[33] and expressed as μM/g of liver and kidney tissue. Serum glutamate oxaloacetate transaminase (SGOT), glutamate pyruvate transaminase (SGPT), serum alkaline phosphatase (SALP) and total protein, using a commercial Diagnostic Kit Biocompare, BioVision, Biocompare and Thermo scientific respectively. Malondialdehyde (MDA) as thiobarbituric acid reactive substances was measured at 532nm spectrophotometrically.
Antioxidant parameters levels in serum, liver, pancreas and kidney
Activity of serum superoxide dismutase (SOD) was measured by the xanthine oxidase method using commercial kits with the absorbance measured using spectrophotometer at 550 nm. Serum catalase (CAT) and glutathione peroxidase (CSH-Px) activities were measured by the colorimetric method measuring absorbances at 405 nm and 412 nm respectively. Glutathione reductase (GSH) by measuring the rate of NADPH oxidation at 340 nm. All the assay kits were purchased from Cayman Chemical (Michigan, USA) and the procedures were according to the kits instructions. In the pancreas the protein concentration was determined by the Bradford method as described in the Bio-Rad protein assay kit.
Estimation al glucose metabolic enzymes activities in liver tissues
When the mice were sacrificed, the liver tissues were removed and immediately frozen by liquid nitrogen and stored at -80°C for further study. The activity of glucokinase and glucose-6-phosphatase was assayed by the color change of therapeutic massage and bodywork (TMB) substrate using commercial EUSA kits purchased from R and O system (USA) and the color change was measured spectrophotometrically at the wavelength of 450 nm. Protein concentration and liver tissue glycogen were estimated using commercial kits purchased from Cayman Chemical (Michigan, USA). All the tests were carried out according to the kit instructions, respectively.
Anti-AGES activity assay in vitro
Bovine serum albumin-glucose assay
The methodology was based on that of Brownlee et al.[34] BSA (l0 mg/ml) was incubated with glucose (500 mM) in phosphate buffered-saline (PBS) (5 ml total volume, pH 7.4) and extract containing 0.02% sodium azide at 37°C. All the reagent and samples were sterilised by filtration through 0.2 μm membrane filters. The protein, the sugar and the prospective inhibitor were included in the mixture simultaneously. Aminoguanidine was used as an inhibitor positive control. Reactions without any inhibitor were also set up. Each solution was kept in the dark in a capped tube. After 15 days of incubation, fluorescence intensity (excitation wavelength of 370 nm and emission wavelength of 440 nm) was measured for the test solutions.
BSA-methylglyoxal assay
This assay was modified based on a published method.[35] The assay evaluates the middle stage of protein glycation. BSA and methylglyoxal were dissolved in phosphate buffer (100 mM, pH 7.4) to a concentration of 20 mg/ml and 60 mM, respectively. Extract or fractions were dissolved in the same phosphate buffer. One millilitre of the BSA solution was mixed with 1ml of methylglyoxal solution and 1 ml of OM extract. The mixture was incubated at 37ºC. Sodium azide (0.2 g/l) was used as an aseptic agent. Phosphate buffer was used as a blank. Aminoguanidine and phloroglucinol were used as positive controls. After seven days of incubation, fluorescence of the samples was measured using an excitation of 340nm and an emission of 420nm, respectively.
Glycation of haemoglobin
Glycosylated haemoglobin (HbA1c) was estimated using a commercial diagnostic kit from Sigma-Aldrich (Human haemolysate [glycated haemoglobin (HbA1c)] Kit).
Anti-AGES activity assay In vivo
Thiobarbituric acid-reactive substance level and AGE level in kidneys
TBARS levels in serum were determined using the method of Naito and Yamanaka.[36] Mitochondria were prepared from kidney homogenate by differential centrifugation (800 × g and 12000 × g, respectively) at 4°C according to the methods of Jung and Pergande[37], with minor modifications. Each pellet was resuspended in preparation medium and the concentration of TBA-reactive substance was determined by the method of Mihara and Uchiyama.[38]
The renal AGE level was determined by the method of Nakagawa.[39] Briefly, minced kidney tissue was degreasing with chloroform and methanol (2:1, v/v) overnight. After washing, the tissue was homogenised in 0.1 N NaOH, followed by centrifugation at 8000 × g for 15 min at 4°C. The amounts of AGEs in these alkali-soluble samples were determined by measuring the fluorescence at an emission wavelength of 440 nm and an excitation wave length of 370 nm. A native BSA preparation (l mg/ml of 0.1 N NaOH) was used as a standard and its fluorescence intensity was defined as one unit of fluorescence. The fluorescence values of samples were measured at a protein concentration of 1 mg/ml and expressed in arbitrary units (AU).
The renal glucose level was determined by the method of Momose.[40] with some modifications. In brief, frozen kidney tissue was homogenised with ice-cold physiological saline and, after being deproteinised, the content of glucose was determined using the Wako kit described above. Minced kidney tissue was delipidated with chloroform and methanol (2: 1, v/v) overnight. After washing with methanol and distilled water, the tissue was homogenised in 0.1 NaOH, followed by centrifugation at 8000 × g for 15 min at 4°C. The amounts of AGEs in these alkali-soluble samples were determined by measuring fluorescence at an emission wavelength of 440 nm and an excitation wavelength of 370 nm. A native BSA preparation (1 mg/ml of 0.1 N NaOH) was used as a standard and its fluorescence intensity was defined as one unit of fluorescence. The fluorescence values of samples were measured at a protein concentration of 1 mg/ml and expressed in arbitrary units (AU) compared with the native BSA preparation.
Statistical analysis
All experiments were performed in triplicate (n = 3) and results were expressed as mean ± SEM. Statistical analysis was carried out by using OriginPro 7.5 software. One way ANOVA was applied to data and results were compared by using Tukey test. A difference was considered to be statistically significant when the (P < 0.05).
RESULTS AND DISCUSSION
Due to the complex nature of the extract derived from EP, it is not possible to evaluate the antioxidant activity of the extract by employing only a single method. Therefore, several radical-scavenging assays were performed, to determine the abilities of the extract to inhibit oxidation.
The total phenolic (TP) and total flavanoid (TF) content in methanol-water extract were expressed as chemical equivalents of gallic acid and catechin, respectively, since different phenolic compounds contribute differently to the readings using the Folin-Ciocalteau reagent. TP and TF values were 856.50 ± 13.53 and 368.29 ± 16.3, respectively, indicating that the mean TF value corresponds to 43% of the mean TP value.
The DPPH· radical is considered to be a model of a stable lipophilic radical. This reaction has been widely used to test the ability of compounds to act as free-radical scavengers or hydrogen donors and to evaluate the antioxidative activity of plant extracts, In this test, the free radical scavenging activity of EP was found to be 90.34% at 0.1 mg/ml while showed 98.32% and ascorbic acid 86.53% at the same concentrations [Figure 1].
Figure 1.
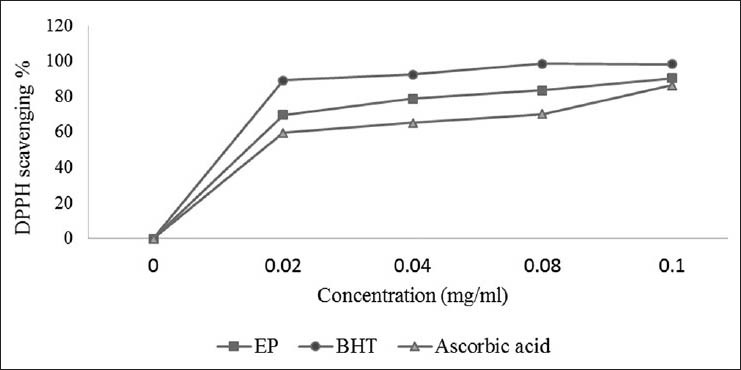
Free radical (DPPH) scavenging activity of the methanol/ water extract of the Bark of E. polystachya compared to ascorbic acid and BHT. Values are mean ± SEM and significantly P < 0.05
The free-radical scavenging activity of the extract was confirmed in the TEAC assay, which is based on the ability of antioxidants to quench the ABTS+ radical-cation. The methanol/water extract of EP was fast and effective scavengers of the ABTS radical [Figure 2] and this activity was comparable to that of BHT. At 0.02 mg/ml, the extract exhibited higher activity than BHT, but at 0.1 mg/ml the activity of the extracts was similar to that of BHT. The percentage inhibition was 98.34% for the extract and 99.42% for BHT at 0.1 mg/ml concentration. Proton radical scavenging is an important attribute of antioxidants.
Figure 2.
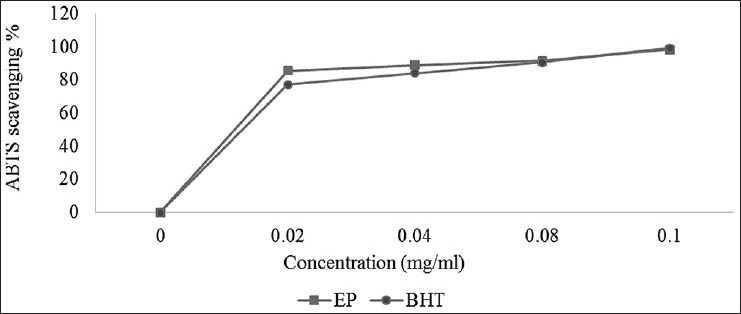
ABTS•+ scavenging activity of the methanol-water extract of the leaves of E. polystachya. Values are mean ± SEM and significantly P < 0.05
Peroxyl radicals are the most common radicals found in the human body, but ORAC measurements should be more biologically relevant. EP showed significant antioxidant potential and the values represent ORACROO + activities of the tested extract equivalent to Trolox. The results showed EP with the ORACROO + value of 5.12 ± 1.28 μmol α-tocopherol [Table 1].
Table 1.
Antioxidant activities of methanol/water (EP) extract of E. polystachya
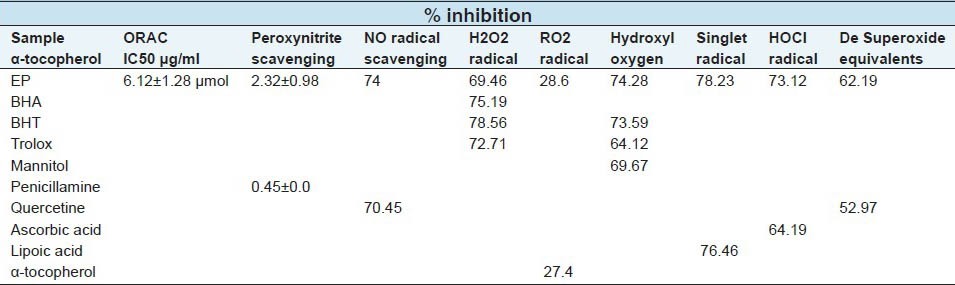
Both assays (TEAC and ORAC) are inhibition methods: TEAC reflects the relative ability of hydrogen or electron donating antioxidants to scavenge the ABTS radical-cation compared with Trolox, while ORAC is a method used to measure the scavenging activity of peroxyl radicals. The ORAC, ABTS and DPPH values indicated that EP possesses significant radical quenching properties.
Iron is the most important lipid pro-oxidant. It is known that Fe2+-accelerates lipid peroxidation by breaking down hydrogen and lipid peroxides formed by Fenton free radical reaction. As shown in Figure 3, EP has the best antioxidants of ascorbic acid and BHT, suggesting that EP is able to chelate metals. The median inhibitory concentration values for the EP, ascorbic acid and BHT were 23.67, 50.1 and 47.2 μg/ml, respectively. EP and the standard antioxidant ascorbic acid and BHT competes for the metal with ferrozine, suggesting that they have chelating activity, capturing the ferrous ion before it can form a complex with ferrozine.[41]
Figure 3.
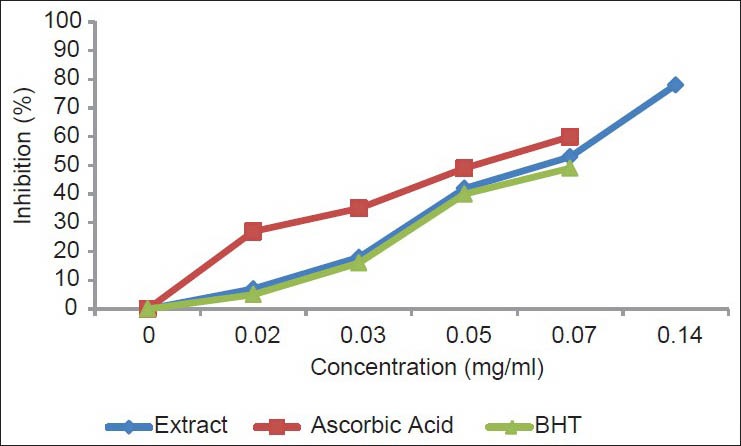
Formation of the Fe2+-ferrozine complex of methanol-water extract of the bark of E. polystachya and EDTA. Values are mean ± SEM and significantly P <0.05
Table 1, illustrates the decrease in the concentration of nitric oxide free radicals due to the scavenging ability of EP and quercetin. A 50 μg/ml solution of EP and quercetin exhibited 74% and 70.45% inhibition, respectively. NO scavenging capacity of extract was higher than that of quercetin. In this study, the ONOO- scavenging ability of EP was also investigated. The result showed that EP possesses a moderate scavenging activity with IC50 = 2.02 ± 0.98 μg/ml compared to penicillamine, with an IC50 = 0.45 ± 0.0 μg/ml. Nitric oxide (NO) is a particular oxygen reactive species; although its excess can produce a harmful effect in the organism, it is a very important cell mediator that regulates a number of functions and cellular processes in the organism.[42] As a member of reactive species, ONOO- has been implicated in several major chronic diseases such as diabetes, Alzheimer's disease, rheumatoid arthritis, cancer and atherosclerosis. In the current research, EP was shown to have a moderate scavenging activity against ONOO-.
The antioxidant compounds in the plant extract prevent the oxidation of β-carotene [Figure 4]. The extract showed high antioxidant activity (80.9 ± 9.7%) by inhibiting the formation of conjugated dienes. It can be concluded that the methanolic/water extract had a similar activity to those of the standards BHA (81.3 ± 1.7%) and BHT (84.2 ± 1.1%) (P < 0.05). EP exhibited the highest antioxidant activity of β-carotene remaining after 20 min. Hydrophobic antioxidants are reported to perform more efficiently than hydrophilic antioxidants in the β-carotene bleaching test, because they have an affinity for the lipid phase, where they can react with peroxyl radicals, avoiding β-carotene oxidation. The strong activity of EP components may be due to their higher level of hydrophobic antioxidants.
Figure 4.
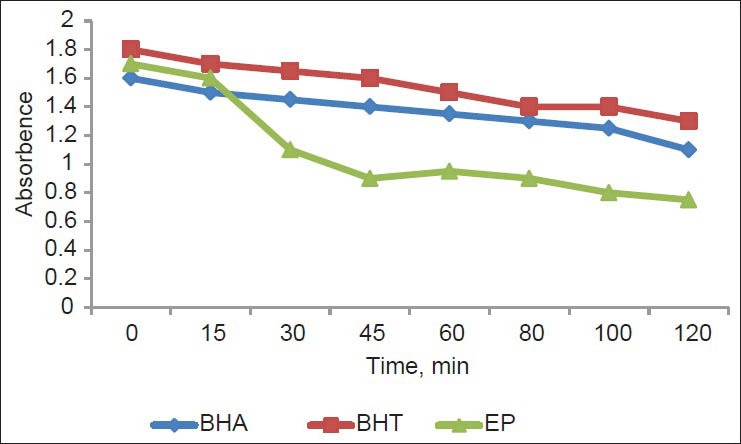
Absorbance change of β- carotene at 490 nm in the presence of EP and standards, BHT and BHA. Values are mean ± SEM and significantly P <0.05
The production of highly reactive oxygen species such as superoxide anion radicals is catalysed by free iron through Haber-Weiss reactions;[43] superoxide anions are also implicated as harmful ROS, as they have a detrimental effect on the cellular components in a biological system. Superoxide anions indirectly initiate lipid oxidation by generating singlet oxygen.[41]
Table 1 presents the superoxide radical scavenging activity of 50 μg/ml EP in comparison with the same dose of quercetin. EP had a superoxide radical scavenging activity (62.19% inhibition), which was greater than that of reference antioxidant (52.97% inhibition) at the same concentrations (P < 0.05).
Another ROS, singlet oxygen, which is a high energy form of oxygen, is generated in the skin upon UV-irradiation. Singlet oxygen induces hyperoxidation, oxygen cytoxicity and decreases the antioxidant activity.[44] ROS scavenging activities of the extract are shown in Table 1. Methanol/water extract showed the radical scavenging activities against HOCl, ONOO-, NO, O2-, OH, H2O2 and ORAC. Hydroxyl radical is one of the ROS formed in biological systems, causing DNA strand breakage, which brings about carcinogenesis, mutagenesis and cytotoxicity.[45] The addition of EP to the reaction mixture removes hydroxyl radicals and prevents further damage. The EP observed value indicates that the bark extract is a better hydroxyl radical scavenger than the standards BHT, Trolox and mannitol with values of 74.28, 73.59, 64.12 and 69.67%, respectively. EP is a source of antioxidant compounds, especially for scavenging the highly reactive hydroxyl radicals. It has been observed that H2O2 through the Fenton reaction is an active oxygen species and has the potential to produce the highly reactive hydroxyls radical which are often involved in free radical chain reactions known to cause damage to biological macromolecules. This result suggests that antioxidants in the methanol/water extract were more reactive toward O2-. Butylated hydroxyanisole (BHA), butylated hydroxyl-tolueno (BHT), quercetin, penicillamine, lipoic acid and ascorbic acid were used as standards. The extract was an effective scavenger of ROS and this activity was comparable to that of the standards used.
At the sites of inflammation, the oxidation of Cl- ions by the neutrophil enzyme myeloperoxidase results in the production of another harmful ROS, hypochlorous acid.[44] HOCl has the ability to inactivate the antioxidant enzyme catalase through the breakdown of the heme-prosthetic group. From the results obtained, it is anticipated that EP is a more efficient scavenger than standard ascorbic acid.
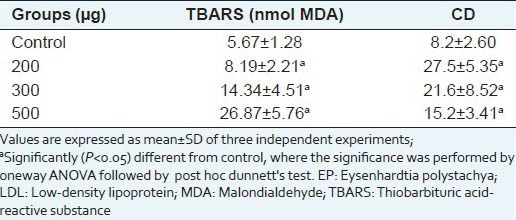
Table 2 shows the oxidative modification of LDL by CuSO4, as well as the antioxidant activity of EP. The extract inhibited the chemically-induced LDL oxidation. The effect of EP on LDL may be due to the polyphenol content, since these compounds have been found to act against LDL oxidation and their capacity is related to their chemical structures.[46]
Table 2.
Effects of EP on LDL oxidation induced-CuSO4 in vitro
In order to determine the inhibitory effect of the methanol/water extract from Eysenhardtia polystachya on the formation of AGEs, several assay methods have been proposed, including assays based on the inhibition of specific fluorescence generated during the course of glycation and AGE formation and assays based on the inhibition of AGE-protein cross-linking. Table 3 displays the inhibitory effects of EP on AGE formation in BSA-glucose and BSA-methylglyoxal models. EP, phloglucinol and aminoguanidine exhibited higher inhibitory activity against AGE formation after the incubation at 37°C for 15 days, with IC50 values of 0.204, 0.070 and 0.323 mg/ml, respectively. Methylglyoxal-mediated protein glycation inhibition was evaluated for EP, which exhibited substantial activity compared with methylglyoxal; this has received considerable attention as a mediator of advanced glycation end-product formation and are known to react with lysine, arginine and cysteine residues in proteins to form glycosylamine protein crosslinks.[47]
Table 3.
Effects of EP on glyco-oxidative damage to BSA by glucose and methylglioxal
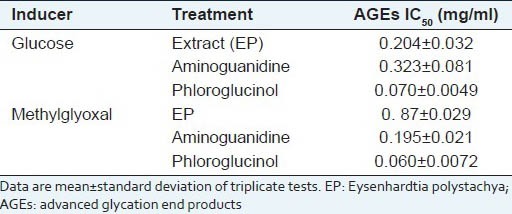
In this study, we found that EP inhibited the formation of methylglyoxal-derived advanced glycation end-products in a bovine serum-albumin-methylglyoxal system and may also act by blocking the conversion of dicarbonyl intermediates to advanced glycation end-products.
Haemoglobin A1C, a biomarker for chronic exposure to high concentrations of glucose, was also significantly decreased in STZ-induced diabetic mice. Table 4 shows the amount of glycated haemoglobin (%GHb). When haemoglobin was used alone (NC), the amount of glycated haemoglobin was 9.5%. This noticeably increased with the addition of glucose to a 27.6% (PC). Nonetheless, it decreased significantly with the treatment of EP (23.8%) and dropped further with the treatment of glutathione (8.1%). The amount of haemoglobin A1c (%HbA1c) corresponds to a specific sub-fraction of glycated haemoglobin; it is lower than the amount of glycated haemoglobin. However, it showed a similar tendency in the percentage of glycation. This result indicates that EP demonstrates the most potent glycation inhibition in the early stages of protein glycation at a concentration of 10 mg/ml. The plant, therefore, can effectively prevent HbA1C formation. The formation and accumulation of AGEs in various tissues increases rapidly in chronic diabetes. We found that EP decreased the in vitro formation of fluorescent AGE and HbA1C, a kind of AGE. Thus, EP may possess specific antiglycation properties that contribute to the reduction in HbA1C levels. EP could directly decrease the formation of glycated haemoglobin, possibly as a result of the antioxidant activity.
Table 4.
The inhibitory activities of EP on glycation protein and hemoglobin. Effect of EP on glucose, weight and ages levels in kidney
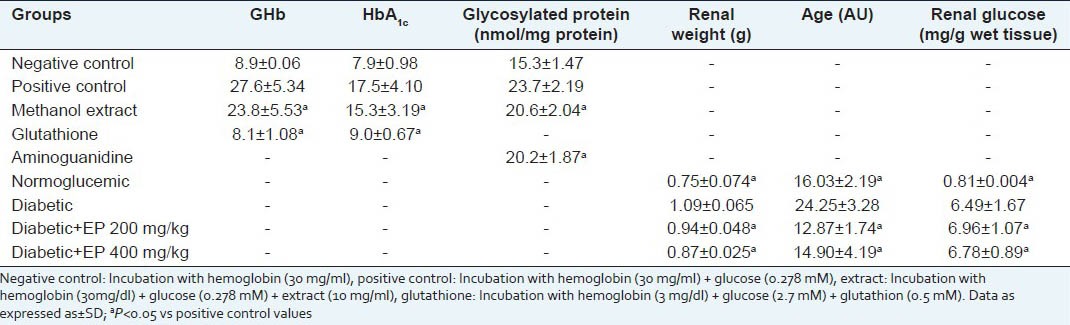
The renal glucose level in diabetic control mice at the end of the experiment was significantly increased compared with normal mice (0.81 ± 0.004 mg/g wet tissue). However, treatment with 200 and 400 mg of EP led to a slight increase to 6.96 and 6.49 mg/g wet tissue, respectively [Table 4]. Additionally, diabetic control mice showed an increase in the fluorescent AGE level compared with that of normal mice (P < 0.01). However, in mice treated with EP for 30 days, this was significantly decreased in the 400 mg-treated group [Table 4]. These effects may be caused by the antioxidant activity possessed by EP, because we found that EP scavenged free radicals. Another possible cause may be the direct inhibitory activity of EP on glycation, resulting in a decrease in free radical release.
Effect of single and repeated oral administration of EP on blood glucose levels in STZ-diabetic and normoglycemic mice are presented in Table 5 and Table 6 respectively. EP, administered at three different doses of 100, 200 and 400 mg/kg, to normoglycemic and STZ-treated diabetic mice (SD and MD) caused significant reduction of blood glucose levels which was related to dose and duration of treatment. STZ produced significant loss in body weight as compared to normal animals during the study. Diabetic control continued to lose weight till the end of the study while EP at dose of 400 mg/kg, showed significant improvement (P < 0.05) in body weight compared to diabetic control [Table 10]. All the diabetic mice consumed more feed than normal mice throughout the experiment compared to their respective control groups, indicating the probable appetite-enhancing property or a decreased efficiency in feed utilization.
Table 5.
Effect of a single oral administration of EP on fasting blood glucose level of in non-diabetic and diabetic mice
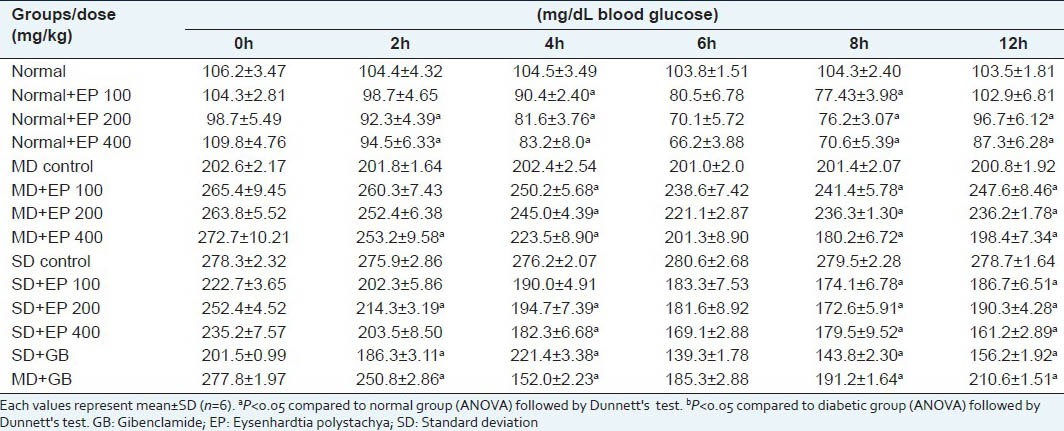
Table 6.
Effect of EP on blood glucose level and oral glucose tolerance test in diabetic mice after 30 day treatment
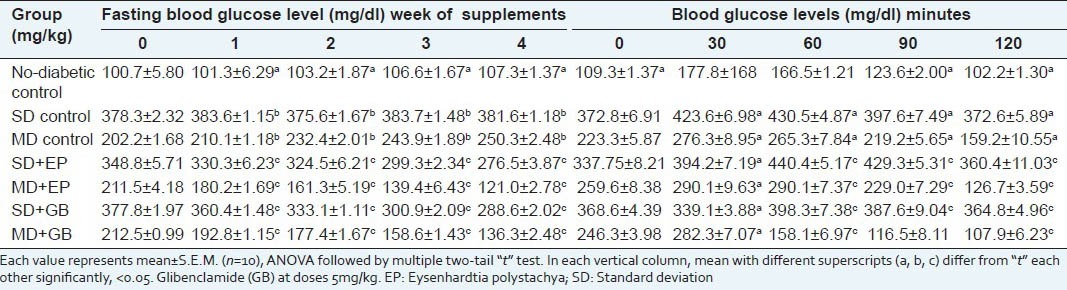
Table 10.
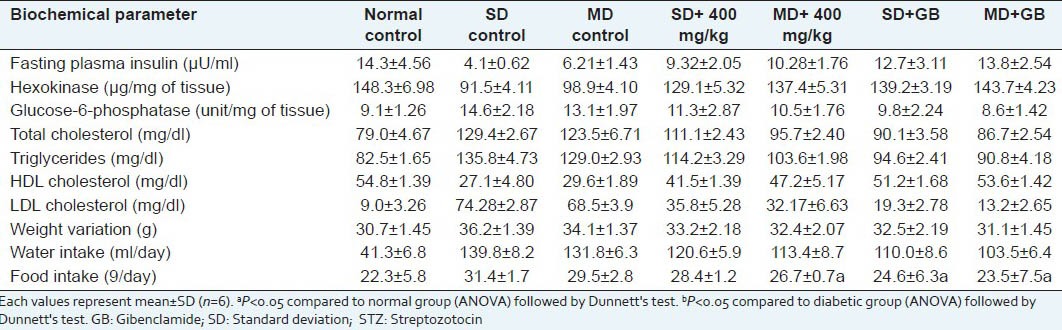
Effect of EP on lipid profile, renal and hepatic functions. Body weight, water and food intake in streptozotocin-induced diabetic mice
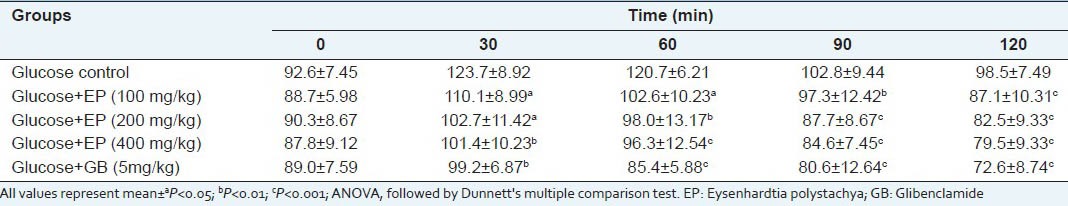
In the oral glucose tolerance test, the blood glucose levels of glucose treated diabetic mice were increased markedly at 30 min. EP at dose of 400 mg/kg inhibited the increasing blood sugar level significantly (P < 0.05) at the 60 min and 120 min when compared with disease control [Table 6]. Effect of EP on oral glucose tolerance test in normal mice is shown in Table 7. The different doses (100, 200 and 400 mg/kg) produced a significant reduction in blood glucose level at 120 min when compared to the vehicle control.
Table 7.
Effect of EP on oral glucose tolerance test in normal mice
Free radicals are formed disproportionately in diabetes mellitus by glucose degradation, non-enzymatic glycation of proteins and the subsequent oxidative degradation.[48] Increased oxidative stress is involved in diabetes. There is evidence that glycation itself induces the generation of oxygen-derived free radicals in diabetic condition.[49] The generatian of free radicals may lead to lipid peroxidation in diabetes mellitus.[48]
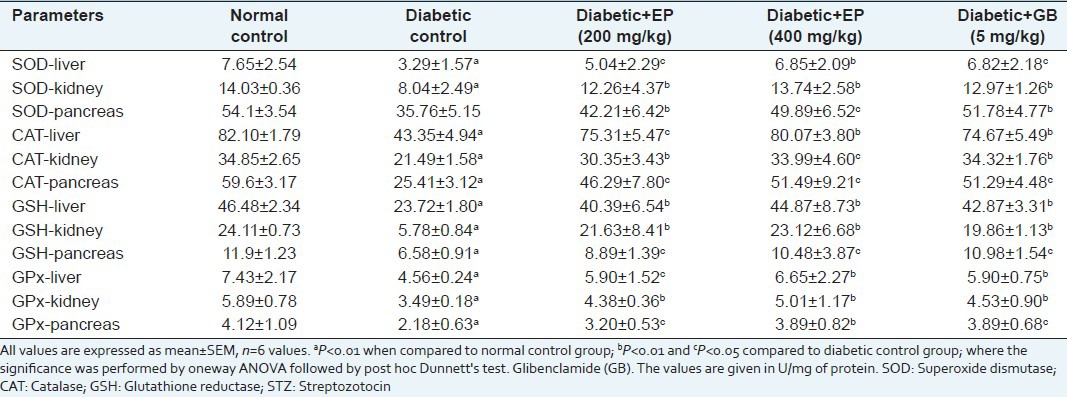
Diabetic mice showed a significant reduction in SOD, CAT, GSH and GPx in hepatic, pancreatic and renal tissues. Levels of these enzymes reverted close to normal values after treatment with EP extract [Table 8]. These results suggest that EP prevents oxidative stress, acts as a suppressor of liver, kidney and pancreas cell damage and inhibits the progression of dysfunction induced by chronic hyperglycaemia. In total, these results suggest that the protection shown by EP extract may be due to its antioxidant properties.
Table 8.
Effect EP on antioxidant enzyme activities in liver, pancreas and kidney in diabetic mice
STZ induction of diabetes in mice leads to lipid peroxidation. TBARS are an indication of endogenous lipid peroxidation and oxidative stress as a result of intensified free radical production. Increased TBARS suggests an increase in oxygen radicals that could be caused either by increased production or by decreased destruction. These elevated TBARS levels in diabetic mice might be due to the stimulation of hepatic triglyceride synthesis as a result of free fatty acid influx. Daily administration of extract at a dose of 400 mg/kg to diabetic mice for 30 days significantly reduced TBARS levels by 50.4% in the liver and 57.3% in the pancreas [Table 9].
Table 9.
Effect of EP on TBA-reactive substance levels in liver, pancreas, serum and renal Mitochondrial in experimental mice
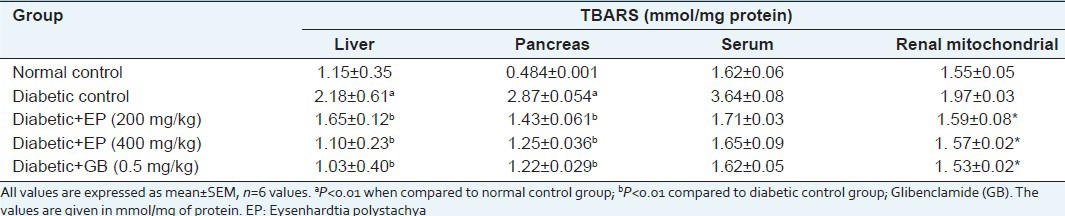
The effects of EP on TBARS levels were also evaluated in serum and renal mitochondria. As shown in Table 9, the serum TBARS level in diabetic control mice was significantly elevated compared to that of normal (2.3-times) and the oral administration of EP at concentrations of 200 and 400 mg significantly inhibited this increase compared to the diabetic control group (P < 0.05). The renal mitochondrial TBARS level was also significantly increased in diabetic control mice (1.97 ± 0.03 nmol/mg protein) compared with normal mice (1.55 ± 0.05 nmol/ml), but levels in groups treated with EP were significantly reduced to the normal level (1.59 ± 0.08 and 1. 53 ± 0.02 nmol/mg protein, respectively (P < 0.05). Therefore, we hypothesised that EP might mitigate oxidative stress in the liver, pancreas and kidney in STZ-induced diabetic mice. The present investigation showed that the methanol/water extract of bark of EP contains a high amount of flavonoids and phenolics, possesses considerable antioxidant activity with ROS scavenging activity and has the ability to reduce lipid peroxidation. It also has iron chelating, TEAC and DPPH activities and we also have proven that the oral administration of EP could decrease oxidative stress associated with diabetes mellitus in the liver, pancreas and kidney, enhancing the generation of typical antioxidant enzymes.
To evaluate the effect of EP on lipid profile level, the level of cholesterol, triglyceride and LDL (low density cholesterol) were increased and the level of HDL (high density cholesterol) was decreased in the STZ-induced diabetic mice. Oral administration of EP was showed reduction in cholesterol, triglyceride and LDL compared to the diabetic control mice group and the level of HDL was increased compared to the diabetic control mice in a significant manner (P < 0.001) [Table 10].
Insulin deficiency is associated with hypercholesterolaemia and hypertriglyceridaemia. STZ-induced diabetes showed increased plasma levels of cholesterol, triglyceride, free fatty acid and phospholipids. Insulin deficiency or insulin resistance could be responsible for dyslipidaemia because insulin increases fatty acid as well as triglyceride synthesis in adipose tissue and liver.[50] Insulin deficiency leads to fall in lipoprotein lipase activity. In our study, STZ-mice showed hypercholesterolaemia and hypertriglyceridaemia and the treatment with EP significantly decreased both cholesterol and triglyceride levels.
There was a significant (P < 0.05) decrease in the level plasma insulin in untreated diabetic mice compared to normal control mice. Oral administration of EP (400 mg/kg) daily for a period of 30 days to diabetic mice significantly (P < 0.05) increased the level of serum insulin compared to diabetic mice [Table 10].
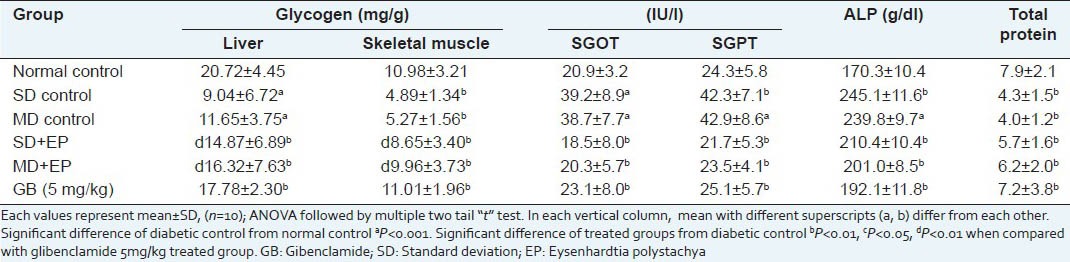
Table 10 shows the effect of the methanol extract on G6Pase, GK, HK activity and glycogen content of liver and skeletal muscle. Administration of EP at 400 mg/kg body weight, increased the content of hepatic glycogen, GK and HK in diabetic rat while G6Pase decreased. Biochemical parameters like SGOT, SGPT, SALP and proteins in the STZ control group were significantly (P < 0.001) elevated as compared with the normal control group. Treatment with EP at the dose of 400 mg/kg b.w. significantly (P < 0.001) brought the SGOT, SGPT, SALP and serum protein toward the normal values [Table 11]. In increase in activities of these enzymes might be mainly due to the leakage from the liver cytosol into blood stream which gives an indication of the hepatotoxic effect of STZ.[51] Reductions in the activity of these enzymes in EP treated diabetic mice indicated the hepato protective role in preventing diabetic complications.
Table 11.
Effect of EP on glycogen level and hepatic enzyme activities in non-diabetic and diabetic mice
CONCLUSION
Our results in this experiment showed that E. polystachya has an antidiabetic, antihyperlipidemic, a significant ability to reduce the formation of AGEs and antioxidant activities, which are considered to play important roles in the development of diabetes complications. Therefore, this plant may have relevance in the prevention and treatment of diseases in which oxidants or free radicals or AGEs are implicated. As a result, chemical studies are now being undertaken to characterise these bioactivities.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Ames BN. Dietary carcinogens and anticarcinogens: Oxygen radicals and degenerative diseases. Science. 1983;221:1256–64. doi: 10.1126/science.6351251. [DOI] [PubMed] [Google Scholar]
- 2.Sumino M, Sekine T, Ruangrungsi N, Igarashi K, Ikegami F. Ardisiphenols and other antioxidant principles from the fruits of Ardisia colorata. Chem Pharm Bull. 2002;50:1484–7. doi: 10.1248/cpb.50.1484. [DOI] [PubMed] [Google Scholar]
- 3.Annadurai T, Muralidharan AR, Joseph T, Hsu MJ, Thomas PA, Geraldine P. Antihyperglycemic and antioxidant effects of a flavanone, naringenin, in streptozotocin-nicotinamide-induced experimental diabetic rats. J Physiol Biochem. 2012;68:307–18. doi: 10.1007/s13105-011-0142-y. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Liu D. Flavonol kaempferol improves chronic hyperglycemia-impaired pancreatic beta-cell viability and insulin secretory function. Eur J Pharmacol. 2011;670:325–32. doi: 10.1016/j.ejphar.2011.08.011. [DOI] [PubMed] [Google Scholar]
- 5.Lin CY, Ni CC, Yin MC, Lii CK. Flavonoids protect pancreatic beta-cells from cytokines mediated apoptosis through the activation of PI3-kinase pathway. Cytokine. 2012;59:65–71. doi: 10.1016/j.cyto.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 6.Rahbar S, Figarola JL. Novel inhibitors of advanced glycation endproducts. Arch Biochem Biophys. 2003;419:63–79. doi: 10.1016/j.abb.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 7.Perez RM, Vargas R, Perez GS, Zavala S. Antiurolithiatic activity of Eysenhardtia polystachya aqueous extract on rats. Phytother Res. 1998;12:144–5. [Google Scholar]
- 8.Burns DT, Dalgarno BG, Ggargan P, Grimshaw J. An isoflavone and a coumestan from Eysenhardtia polystachya-robert boyle's fluorescent acid-base indicator. Phytochemistry. 1984;3:167–9. [Google Scholar]
- 9.Alvarez L, Rios MY, Esquivel C, Chávez MI, Delgado G, Aguilar MI, et al. Cytotoxic isoflavans from Eysenhardtia polystachya. J Nat Prod. 1998;61:767–70. doi: 10.1021/np970586b. [DOI] [PubMed] [Google Scholar]
- 10.Beltrami E, Bernardi M, Fronzat G, Mellerio G, Vidari G, Vita-finzi P. Coatline A and B, two C-glucosyl-α-hydroxydihydrochalcones from Eysenhardtia polystachya. Phytochemistry. 1982;21:2931–3. [Google Scholar]
- 11.Alvarez L, Delgado G. C- and O-Glycosyl-cxhydroxydihydrochalcones from Eysenhardtia polystachya. Phytochemistry. 1999;50:681–7. [Google Scholar]
- 12.Narvaez-Mastache JM, Soto C, Delgado G. Antioxidant evaluation of Eysenhardtia species (Fabaceae):Relay synthesis of 3-O-Acetyl- 11α,12α-epoxy-oleanan-28,13β-olide isolated from E. platycarpa and Its Protective Effect in Experimental Diabetes. Biol Pharm Bull. 2007;30:1503–10. doi: 10.1248/bpb.30.1503. [DOI] [PubMed] [Google Scholar]
- 13.Narvaez-Mastache JM, Novillo F, Delgado G. Antioxidant aryl-prenylcoumarin, flavan- 3- ols and flavonoids from Eysenhardtia subcoriacea. Phytochemistry. 2008;69:451–6. doi: 10.1016/j.phytochem.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 14.Narvaez-Mastache JM, Garduño-Ramirez ML, Alvarez L, Delgado G. Antihyperglycemic activity and chemical constituents of Eysenhardtia platycarpa. J Nat Prod. 2006;69:1687–91. doi: 10.1021/np060166z. [DOI] [PubMed] [Google Scholar]
- 15.Kahkonen MM, Kylli P, Ollilainen V, Salminen J, Heinonen M. Antioxidant activity of isolated ellagitannins from red raspberries and cloudberries. J Agric Food Chem. 2012;60:1167–74. doi: 10.1021/jf203431g. [DOI] [PubMed] [Google Scholar]
- 16.Gyamfi MA, Yonamine M, Aniya Y. Free-radical scavenging action of medicinal herbs from Ghana: Thonningia sanguinea on experimentally induced liver injuries. Gen Pharmacol. 1999;32:661–7. doi: 10.1016/s0306-3623(98)00238-9. [DOI] [PubMed] [Google Scholar]
- 17.Mahakunakorn P, Tohda M, Murakami Y, Matsumoto K, Watanabe H. Antioxidant and free radical-scavenging activity of Choto-san and its related constituents. Biol Pharm Bull. 2004;27:38–46. doi: 10.1248/bpb.27.38. [DOI] [PubMed] [Google Scholar]
- 18.Cao G, Alessio HM, Cutler RG. Oxygen-radical absorbance capacity assay for antioxidants. Free Radic Biol Med. 1993;14:303–11. doi: 10.1016/0891-5849(93)90027-r. [DOI] [PubMed] [Google Scholar]
- 19.Yildrim A, Oktay M, Bilaloglu V. The antioxidant activity of the leaves of Cydonia vulgaris. Turk J Med Sci. 2001;31:23–7. [Google Scholar]
- 20.Sreejayan N, Roa MN. Nitric oxide scavenging by cucurminoids. J Pharm Pharmacol. 1997;49:105–7. doi: 10.1111/j.2042-7158.1997.tb06761.x. [DOI] [PubMed] [Google Scholar]
- 21.Beckman S, Chen H, Ischiropulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods Enzymol. 1994;233:229–40. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 22.Singh N, Rajini PS. Antioxidant-mediated protective effect of potato peel extract in erythrocytes against oxidative damage. Chem Biol Interact. 2008;173:97–104. doi: 10.1016/j.cbi.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Ozen T. Investigation of antioxidant properties of Nasturtium officinale (Watercress) leaf extracts. Acta Pol Pharm Drug Res. 2009;66:187–93. [PubMed] [Google Scholar]
- 24.Yu L, Haley S, Perret M, Harris J, Wilson J, Qian M. Free radical scavenging properties of wheat extracts. J Agric Food Chem. 2002;50:1619–24. doi: 10.1021/jf010964p. [DOI] [PubMed] [Google Scholar]
- 25.Elizabeth K, Rao MN. Oxygen radical scavenging activity of curcumin. Int J Pharm. 1990;58:237–40. [Google Scholar]
- 26.Floriáno-Sanchez E, Villanueva C, Medina-Campos ON, Rocha D, Sánchez-González D, Cárdenas-Rodríguez N, et al. Nordihydroguaiaretic acid is a potent in vitro scavenger of peroxynitrite, singlet oxygen, hydroxyl radical, superoxide anion and hypochlorous acid and prevents in vivo tyrosine nitration in lung. Free Radic Res. 2006;40:523–33. doi: 10.1080/10715760500419365. [DOI] [PubMed] [Google Scholar]
- 27.Pedraza-Chaverri J, Barrera D, Maldonado PD, Chirino YI, Macias-Ruvalcaba NA, Medina-Campos ON, et al. S-allylmercaptocysteine scavenges hydroxyl radical and singlet oxygen in vitro and attenuates gentamicininduced oxidative and nitrosative stress and renal damage in vivo. BMC Clin Pharmacol. 2004;4:5–9. doi: 10.1186/1472-6904-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aruoma OI, Halliwell B. Action of hypochlorous acid on the antioxidant protective enzymes superoxide dismutase, catalase and glutathione peroxidase. Biochem J. 1987;248:973–6. doi: 10.1042/bj2480973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menzel EJ, Sobal G, Staudinger A. The role of oxidative stress in the long- term glycation of LDL. Biofactors. 1997;6:111–24. doi: 10.1002/biof.5520060204. [DOI] [PubMed] [Google Scholar]
- 30.Esterbauer H, Gebicki J, Puhl H, Jurgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. 1992;13:341–90. doi: 10.1016/0891-5849(92)90181-f. [DOI] [PubMed] [Google Scholar]
- 31.Maghrani M, Zeggwagh NA, Lemhadri A. Study of the hyperglycemic activity of Fraxinus excelsior and Silybum marianum in an animal model of type 2 diabetes mellitus. J Ethnopharmacol. 2004;91:309–16. doi: 10.1016/j.jep.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 32.Tahara A, Matsuyama-Yokono A, Nakano R. Hypoglycaemic effects antidiabetic drugs in streptozotocin-nicotinamide-induced mildly diabetic and streptozotocin-induced severely diabetic rats. Basic Clin Pharmacol Toxicol. 2008;103:560–8. doi: 10.1111/j.1742-7843.2008.00321.x. [DOI] [PubMed] [Google Scholar]
- 33.Fraga CG, Leibovitz BE, Tappel AL. Lipid peroxidation measured as thiobarbituric acid-reactive substances in tissue slices: Characterization and comparison with homogenates and microsomes. Free Radic Biol Med. 1988;4:155–61. doi: 10.1016/0891-5849(88)90023-8. [DOI] [PubMed] [Google Scholar]
- 34.Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science. 1986;232:1629–32. doi: 10.1126/science.3487117. [DOI] [PubMed] [Google Scholar]
- 35.Peng X, Zheng Z, Cheng KW, Shan F, Ren GX, Chen F, et al. Inhibitory effect of mung bean extract and its constituents vitexin and isovitexin on the formation of advanced glycation end products. Food Chem. 2008;106:475–81. [Google Scholar]
- 36.Naito C, Yamanaka T. Lipid peroxides in atherosclerotic diseases. Nippon Ronen Igakkai Zasshi. 1978;15:187–91. [PubMed] [Google Scholar]
- 37.Jung K, Pergande M. Influence of cyclosporin A on the respiration of isolated rat kidney mitochondria. Fed Eur Biochem Soc Lett. 1985;183:167–9. doi: 10.1016/0014-5793(85)80977-7. [DOI] [PubMed] [Google Scholar]
- 38.Mihara M, Uchiyama M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem. 1978;86:271–8. doi: 10.1016/0003-2697(78)90342-1. [DOI] [PubMed] [Google Scholar]
- 39.Nakagawa S. Immunochemical detection of advanced glycation end products in lens crystallins from streptozotozin-induced diabetic rats. Diabetes. 1993;42:345–50. doi: 10.2337/diab.42.2.345. [DOI] [PubMed] [Google Scholar]
- 40.Momose T, Yano Y, Ohashi K. Organic analysis. XLIV. A new deproteinizing agent for determination of blood sugar. Chem Pharm Bull. 1963;11:968–72. doi: 10.1248/cpb.11.968. [DOI] [PubMed] [Google Scholar]
- 41.Podrez EA, Schmitt D, Hoff HF, Hazen SL. Mye1operoxidase-generated reactive nitrogen species convert LDL into unatherogenic form in vitro. J Clin lnvest. 1999;103:1547–60. doi: 10.1172/JCI5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akaike T, Suga Maeda H. Free radicals in viral pathogenesis: Molecular mechanism, involving superoxide and NO. Proc Soc Esp Biol Med. 1998;21:64–73. doi: 10.3181/00379727-217-44206. [DOI] [PubMed] [Google Scholar]
- 43.Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide and hypochlorous acid. Free Radic Biol Med. 1989;6:593–7. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 44.Kochevar IE, Redrnond RW. Photosensitized production of singlet oxygen. Methods Enzymol. 2000;319:20–8. doi: 10.1016/s0076-6879(00)19004-4. [DOI] [PubMed] [Google Scholar]
- 45.Esterbauer H. Cytotoxicity and genotoxicity of lipid-oxidation products. Am J Clin Nutr. 1993;57(Suppl 5):779S–85. doi: 10.1093/ajcn/57.5.779S. [DOI] [PubMed] [Google Scholar]
- 46.Janisch K, Hippeli S, Dornisch K, Kern S, Elslner EF. Determination of the antioxidative potential of human plasma after supplementation with pycnogenol and whey. Food Res lnt. 2002;35:257–66. [Google Scholar]
- 47.Matsuura N, Aradate T, Sasaki E, Kojima H, Ohara M, Hasegawa J, et al. Screening system for the Maillard reaction inhibitor from natural product extracts. J Health Sci. 2002;48:520–6. [Google Scholar]
- 48.Mahboob M, Rahman MF, Crover P. Serum lipid peroxidation and antioxidant enzyme levels in male and female diabetic patients. Singapore Med J. 2005;46:322–4. [PubMed] [Google Scholar]
- 49.Senthilkumar GP, Arulselvan P, Sathishkurnar D, Subramanian SP. Antidiabetic activity of fruits of Termina/ia chebula an streptozotacin induced diabetie rats. J Health Sci. 2006;52:283–91. [Google Scholar]
- 50.Gohil T, Pathak N, Jivani N. Treatment with extract of Eugenia jambolana seed and Aegle marmelos leaf extracts prevents hyperglycemia and hyperlipidemia in alloxan-induced diabetic. Afr J Pharm Pharmacol. 2010;4:270–5. [Google Scholar]
- 51.Kasetti RB, Rajasekhar MD, Kondeti VK, Fatima SS, Kumar EG, Swapna S, et al. Antihyperglycemic and antihyperlipidemic activities of methanol: Water (4:1) fraction isolated from aqueous extraer of Syzygium altemifolium seeds in streptozotocin induced diabetic rats. Food Chem Toxicol. 2010;48:1078–84. doi: 10.1016/j.fct.2010.01.029. [DOI] [PubMed] [Google Scholar]