

Abstract
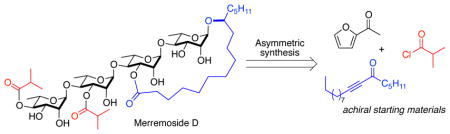
The first synthesis of the purported structure of Merremoside D has been achieved in 22 longest linear steps. The de novo asymmetric synthesis relied on the use of asymmetric catalysis to selectively install all 21 stereocenters in the final compounds from commercially available achiral starting materials. Adiabatic gradient 2D NMR techniques (gHSQCAD, gHMBCAD, gH2BCAD, gHSQCTOXYAD, ROESYAD) were used to completely assign the structure of synthetic Merremoside D. Comparison of our assignments with the limited NMR data reported for natural Merremoside D allows for the tentative confirmation of its structure.
The merremoside family of resin glycoside type natural products were isolated from the fresh tuber of Merremia mammosa (Lour.) Hall. f. (convolvulaceae) by Kitagawa.1 These structurally complex oligosaccharides possess a macrolactone (20 or 21-membered) which consisted of a bis-rhamnose disaccharide bridged at the C-1 and C-2′ or C-3′ by a jalapinolic acid (Fig. 1). The tuber of the Merremia mammosa plant has been traditionally used for the treatment of illnesses associated with the throat and respiratory systems.1a This tradition continues to the present day, where the powder of the tuber is sold as a herbal medicine for treating patients with various maladies (e.g., cancers, appendicitis, swollen veins, typhus). 2 The amphiphilic nature of the resin glycosides has been suggested to be the source of its ionophoretic activity (i.e., membrane transporter) as observed in human erythrocyte membranes.3 While several resin glycoside natural products have been synthesized,4 no member of merremoside family has succumbed to total synthesis.5 Despite their interesting biological activities, no detailed structure activity relationship (SAR) has been carried out.
Figure 1.
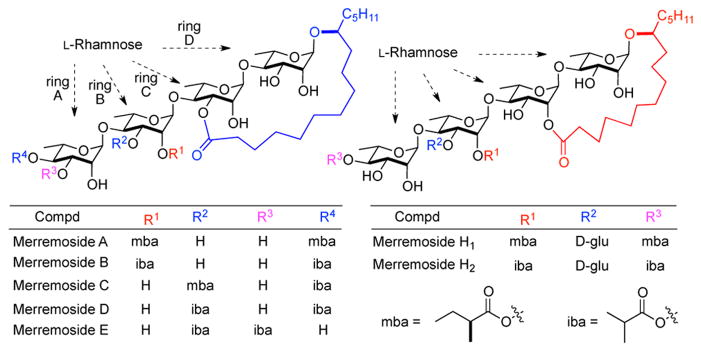
Structures of merremoside family of resin glycosides
To gain a better understanding of the promising and diverse biological activities associated with this novel set of natural products, we became interested in a synthesis led SAR-study of the merremosides. In this vein, we targeted for synthesis merremoside D. Intrigued by the possibility that enantiomeric analogues of these target compounds would possess the ion transport properties, yet would lack the same target protein interactions, we decided to develop a de novo asymmetric approach to the merremosides. We have demonstrated that a de novo approach to carbohydrates6 can be used for the assembly and medicinal chemistry study of oligosaccharides. 7, 8 The approach combines the use of asymmetric synthesis of pyranone glycosyl donor 5, a Pd(0)-catalyzed glycosylation and post-glycosylation transformation, which allow the enone functionality of the pyranones to serve as atom-less protecting groups for the C-2 to C-4 triol portion of the target rhamno-pyranose. Because the route uses asymmetric synthesis, it offers equal efficiency to access to various all D-, all L- and mixed D/L-isomer.9 Herein, we disclose our successful efforts at the synthesis of the purported structure of merremoside D.
Retrosynthetically we envisioned that construction of all the stereocenters in the merremoside D 1 could be accomplished from achiral starting materials utilizing our de novo approach to carbohydrates (Scheme 1). The tetrasaccharide 2 would be constructed by a convergent glycosylation between macrolactone disaccharide 9 and the imidate disaccharide 3, with a C-2 chloroacetate serving as an anomeric directing group. The desired C-3 esterification on 3 would be obtained by tin-mediated regioselective esterification of diol 4, which in turn would be obtained from pyranone 5. The macrolactone disaccharide 9 would be obtained from diol-acid 8, bis-rhamnose unit of which would be obtained from the same building block pyranone 5. This key building block pyranone 5 would be obtained from acetylfuran 6 in three steps. 7a,b A highly stereoselective Pd-catalyzed glycosylation would install all the desire 1,4-α-linkage in the tetra-rhamnose backbone. The aglycon 7 (jalapinolic ester) would be obtained from alkynone 10, using a modified asymmetric variant of a route reported by Heathcock.10 Thus as devised, all the chiral centers in 1 would have their absolute stereochemistry resulting from the Noyori asymmetric reduction of furan 6 and ynone 10.11
Scheme 1.
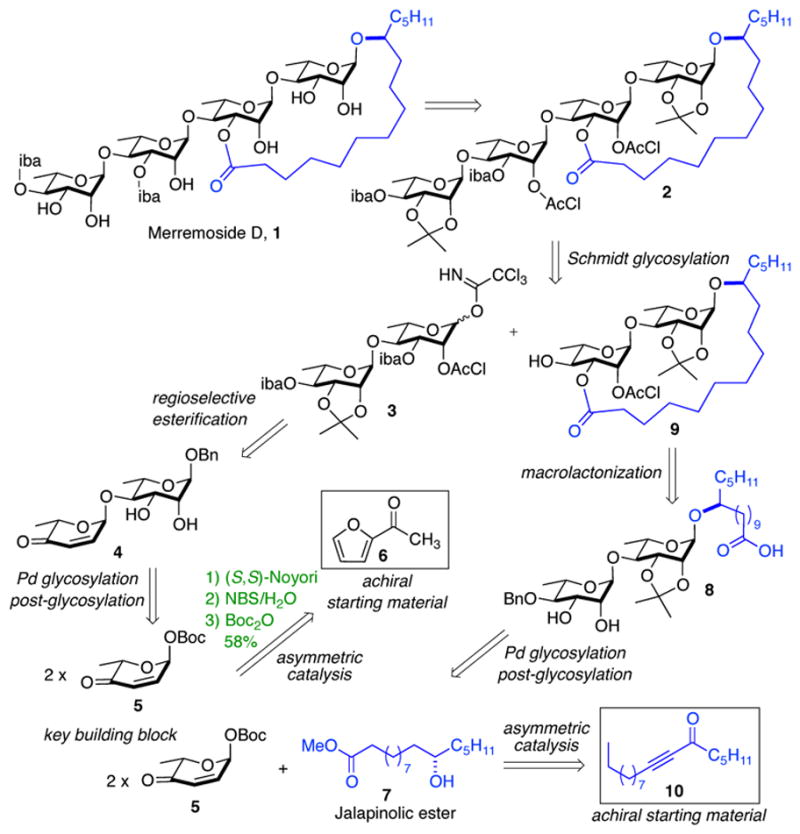
Retrosynthetic analysis: De Novo approach
The asymmetric approach to aglycon 7 began with the synthesis of alkynone 10 from undecyne 11 and hexanal 12 (Scheme 2).10 Enantioselective (S,S)-Noyori reduction of alkynone 10 gave alkynol 13 (89 % ee). 11a,b An alkyne-zipper reaction was used to isomerize the internal alkyne 13 into the terminal alkyne,12 which followed by hydroxyl protection, oxidative alkyne cleavage and esterification resulted the desired jalapinolic ester (i.e., 13 to 7). 10
Scheme 2.
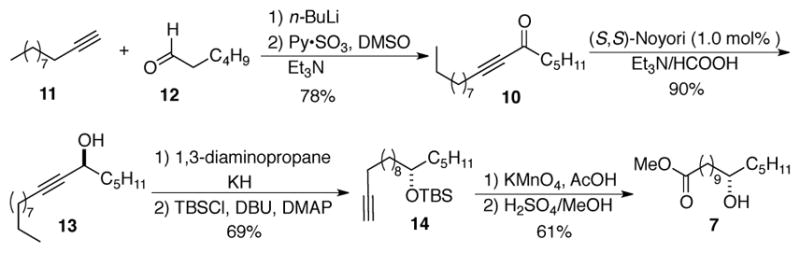
Synthesis of jalapinolic ester
The synthesis of the macrolactone disaccharide began with the de novo asymmetric synthesis of the key pyranone building block 5 (Scheme 3). The three step synthesis of 5 involves a Noyori reduction of acylfuran 611 followed by a subsequent Achmatowicz rearrangement 13 and diastereoselective tButyl carbonate (Boc)-protection of the axial anomeric alcohol. A Pd-catalyzed stereoselective glycosylation of 5 with jalapinolic ester 7 gave 15 (at this stage the minor diastereomer was removed by flash chromatography). The enone 15 was converted to glycosyl acceptor 18 via Luche reduction,14 Upjohn dihydroxylation15 and syn-diol protection. A second Pd-catalyzed glycosylation with pyranone 5 followed by enone reduction, gave allylic alcohol 19. Benzylation of the allylic alcohol followed by alkene dihydroxylation and a subsequent methyl ester saponification resulted in the diol-acid 8.
Scheme 3.
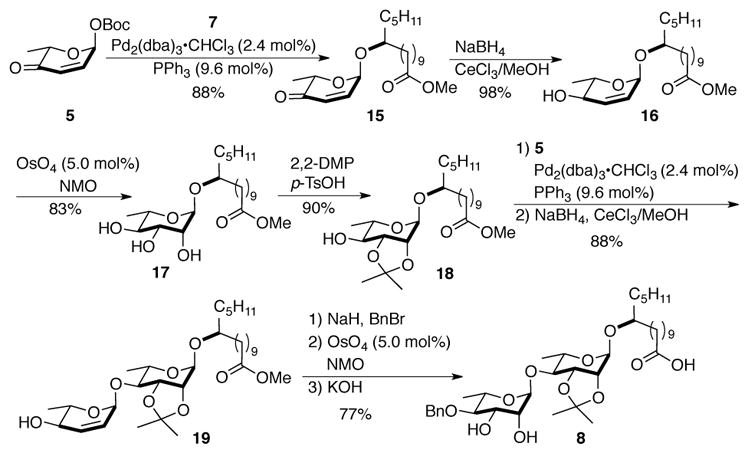
Synthesis of macrolactone disaccharide
With the macrolactone precursor 8 in hand, we investigated the macrolactonization utilizing conditions reported by Yang et al.5 Macrolactonization of 8 under Corey-Nicolaou conditions 16 gave a mixture of regioisomeric products 20 and 21 in a 4.7:1 ratio (Scheme 4). Extensive NMR analysis of the macrolactones confirmed that the major product was a C-2′ macrolactone 20 and the minor product was a C-3′ macrolactone 21. This observation was opposite to what was reported by Yang et al. (see SI).5
Scheme 4.
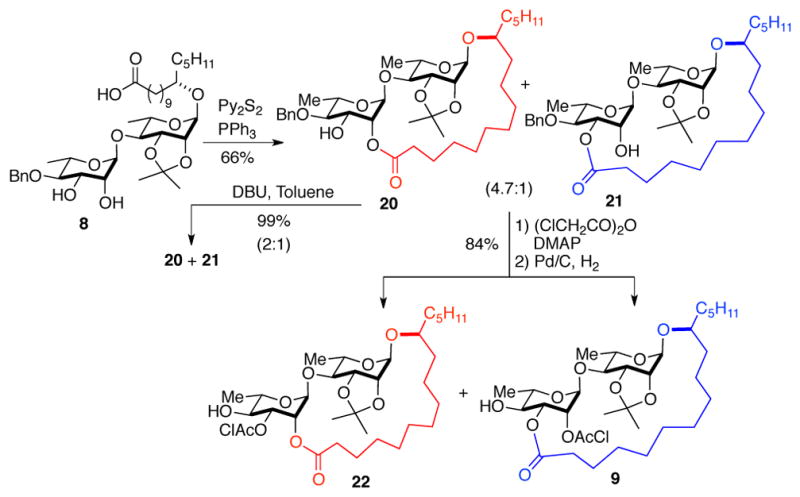
Macrolactonization/synthesis of glycosyl acceptors
To our delight, we found that the major C-2′ regioisomeric macrolactone 20 could be isomerized to the C-3′ macrolactone 21. Under the isomerization conditions (1 eq. DBU/toluene), the two macrolactones 20 and 21 reached a thermodynamic equilibrium (2:1) in 12 hr. The remaining hydroxyl group on the macrolactones (20 and 21) was protected as chloroacetic ester and the benzylether was removed to form the glycosyl acceptors (20 to 22 and 21 to 9) respectively.
The donor disaccharide portion was also prepared using our de novo asymmetric strategy. At the outset, we hoped to be able to carry the C-2/C-3 alkene functionality throughout the synthesis, thus serving as an atom-less protecting group (Scheme 5).7e The pyranone 5 was converted to protected rhamno-pyranosides 23a/b in four steps (Pd-catalyzed glycosylation, Luche reduction, Upjohn dihydroxylation and acetonation).17,18 A subsequent Pd-catalyzed glycosylation with pyranone 5 followed by acetonide removal gave syn-diol 4/24. This set the platform for the regioselective introduction of the required C-3 iba-ester, which was accomplished via a tin-mediated esterification with isobutyryl chloride (ibaCl).19 The C-2 hydroxyl of 25a/b was then protected as chloroacetic ester. Subsequent Luche reduction/esterification gave allylic esters (26a/b). At this stage disaccharide 26b was converted into our desired disaccharide donor 28 with the key C-2/C-3 double bond (DDQ; Cl3CCN/NaH). Unfortunately we were not able to find a promoter for glycosylation that did not cleave the allylic glycosidic bond in 28. Thus suggesting a limit to the use of C-2/3-alkenes in traditional glycosylations.
Scheme 5.
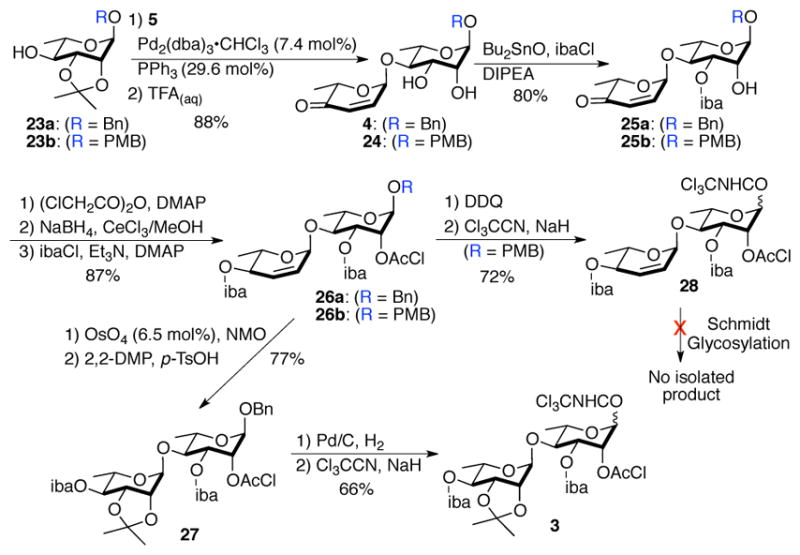
Glycosyl donor synthesis/attempted glycosylation
To overcome this problem, glycosyl donor 3 was synthesized from 26a by functionalizing the double bond and anomeric activation (OsO4/NMO; 2,2-DMP/H+; Pd/C, H2; Cl3CCN/NaH). As expected, the anomeric disaccharide bond in 3 was stable under Schmidt’s glycosylation conditions.20 Thus exposing a 2:1 mixture of disaccharide glycosyl donor 3 and disaccharide glycosyl acceptor 9 to 12% TMSOTf (CF3SO3Si(CH3)3) afforded tetrasaccharide 2 in 71% with complete α-selectivity via the anchimeric assistance of the C-2 chloroacetate (Scheme 6). Deprotection of the tetrasaccharide 2 began with the removal of both acetonide groups to furnish tetraol 29. Finally the two chloroacetates in 29 were removed to provide merremoside D (1) with thiourea without any deleterious effects to the macrolactone and the ester substituents. While the physio-chemical properties (specific rotation, melting point and HRMS) of the synthetic 1 match that reported for the natural material, unfortunately the comparison of the spectral data was more complicated (see SI). The structural identity of synthetic 1 was confirmed by detailed 1D and 2D NMR analysis (See, SI). The 1H and 13C NMR spectra of the synthetic merremoside D (1) were acquired in CDCl3, pyridine-d5, and pyridine-d5/D2O (5:1) and used for comparison with the limited NMR data reported for the isolated merremoside D. For instance, only 21 signals were reported for the 1H NMR (pyridine-d5/D2O) and 7 signals for the 13C NMR data (pyridine-d5).
Scheme 6.
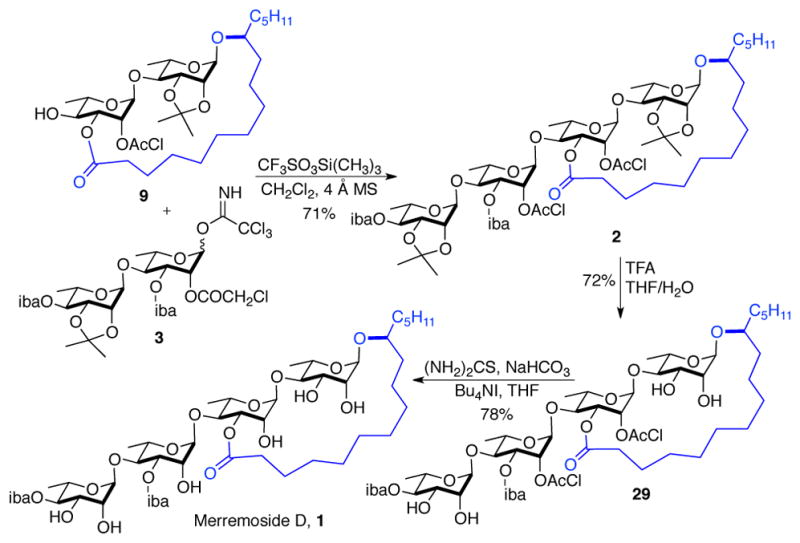
Total synthesis of merremoside D
While it is likely that these two materials are the same, the ill-defined ratio of the pyridine-d5/D2O mixture used to record the 1H NMR spectra of the natural material rendered it difficult to match the chemical shifts between the two samples with absolute certainty (e.g., depending on D2O/H2O concentration the Dppm for various signals varied from ≤ 0.78 ppm to 0.55 ppm in pyridine-d5). For instance, seven of the 21 signals had Dmax greater than 0.1 ppm with three of those being greater than 0.2 ppm. Similar spectral inconsistencies due to concentration/solvent effects have been observed in related oligosaccharides.21 Our efforts to find the identical solvent ratio used in the isolation were further confounded by the observation of ester migration/hydrolysis in synthetic 1 upon standing in pyridine-d5/D2O (see SI). In contrast to the chemical shifts, there was excellent agreement between the 1H-1H coupling constants for the 14 multiplets reported for natural merremoside D and the multiplets for synthetic 1, regardless of the ratio of pyridine-d5/D2O mixture. Similarly, there was significantly better agreement between the limited 13C NMR data (seven signals) reported for natural 1 in pyridine-d5 and that found for synthetic 1 (see SI). For instance, five of the seven signals were within 0.4 ppm and two are within 0.7 ppm, which is consistent with the known effects associated with small amounts of D2O on carbohydrates.1a,3,21
In conclusion, the first total synthesis of the purported structure of merremoside D was achieved in 22 longest linear steps with a 3% overall yield. The route demonstrates the power of a de novo asymmetric approach to a stereochemically complex (21 stereocenters) oligosaccharide natural product. The approach provided sufficient quantity of material (29 mg) for both structural and biological evaluation, enabling the screening against an array of organisms. In addition, the approach exposes some practical limitations of the use of atom-less protecting groups (i.e., C-2/3 alkene) with traditional glycosylation technology, in contrast to the Pd-catalyzed glycosylation. Detailed NMR analysis was used to confirm the structural identity of the synthetic material, which was consistent with the data reported for the natural 1. However, the lack of complete and reliable 1H and 13C NMR data precludes a conclusive confirmation of the structural assignment for merremoside D. Further efforts to elucidate the full biological structure activity relationships of the merremoside family of natural products is ongoing.
Supplementary Material
Acknowledgments
We are grateful to the NIH (GM088839) and NSF (CHE-0749451) for their generous support of our research program.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a) Kitagawa I, Baek NI, Kawashima K, Yokokawa Y, Yoshikawa M, Ohashi K, Shibuya H. Chem Pharm Bull. 1996;44:1680. doi: 10.1248/cpb.44.1680. [DOI] [PubMed] [Google Scholar]; b) Kitagawa I, Baek NI, Yokokawa Y, Yoshikawa M, Ohashi K, Shibuya H. Chem Pharm Bull. 1996;44:1693. doi: 10.1248/cpb.44.1693. [DOI] [PubMed] [Google Scholar]
- 2.Merremia mammosa is an herbal cancer medicines, for its use. see: http://www.khazanahherbal.com/product.php?category=21&product_id=111.
- 3.Kitagawa I, Ohashi K, Kawanishi H, Shibuya H, Shinkai H, Akedo H. Chem Pharm Bull. 1989;37:1679. doi: 10.1248/cpb.37.1679. [DOI] [PubMed] [Google Scholar]
- 4.Arias MB, Miranda RP, Heathcock CH. J Org Chem. 2004;69:4567. doi: 10.1021/jo030244c.Zhu SY, Huang JS, Zheng SS, Zhu K, Yang JS. Org Lett. 2013;15:4154. doi: 10.1021/ol4020255.Lu SF, O’yang QQ, Guo ZW, Yu B. Angew Chem Int Ed. 1997;36:2344.Fürstner A, Nagano T. J Am Chem Soc. 2007;129:1906. doi: 10.1021/ja068901g.Postema MHD, TenDyke K, Cutter J, Kuznetov G, Xu Q. Org Lett. 2009;11:1417. doi: 10.1021/ol900086b.For a review see: Fürstner A. Eur J Org Chem. 2004:943.
- 5.Zhu XM, He LL, Yang GL, Lei M, Chen SS, Yang JS. Synlett. 2006;20:3510. [Google Scholar]
- 6.(a) Ko SY, Lee AWM, Masamune S, Reed LA, Sharpless KB. Science. 1983;220:949–951. doi: 10.1126/science.220.4600.949. [DOI] [PubMed] [Google Scholar]; (b) Northrup AB, MacMillan DWC. Science. 2004;305:1752–1755. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]; (c) Ahmed MdM, Berry BP, Hunter TJ, Tomcik DJ, O’Doherty GA. Org Lett. 2005;7:745–748. doi: 10.1021/ol050044i. [DOI] [PubMed] [Google Scholar]
- 7.(a) Babu RS, O’Doherty GA. J Am Chem Soc. 2003;125:12406. doi: 10.1021/ja037097k. [DOI] [PubMed] [Google Scholar]; (b) Babu RS, Zhou M, O’Doherty GA. J Am Chem Soc. 2004;126:3428. doi: 10.1021/ja039400n. [DOI] [PubMed] [Google Scholar]; (c) Babu RS, O’Doherty GA. J Carb Chem. 2005;24:169. [Google Scholar]; (d) Guo H, O’Doherty GA. Angew Chem Int Ed. 2007;46:5206. doi: 10.1002/anie.200701354. [DOI] [PubMed] [Google Scholar]; (e) Guo H, O’Doherty GA. J Org Chem. 2008;73:5211. doi: 10.1021/jo800691v. [DOI] [PubMed] [Google Scholar]
- 8.(a) Wang HYL, Wu B, Zhang Q, Kang SW, Rojanasakul Y, O’Doherty GA. ACS Med Chem Lett. 2011;2:259. doi: 10.1021/ml100291n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang HYL, Rojanasakul Y, O’Doherty GA. ACS Med Chem Lett. 2011;2:264. doi: 10.1021/ml100290d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Iyer A, Zhou M, Azad N, Elbaz H, Wang L, Rogalsky DK, Rojanasakul Y, O’Doherty GA, Langenhan JM. ACS Med Chem Lett. 2010;1:326. doi: 10.1021/ml1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang HYL, Xin W, Zhou M, Stueckle TA, Rojanasakul Y, O’Doherty GA. ACS Med Chem Lett. 2011;2:73. doi: 10.1021/ml100219d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Babu RS, Chen Q, Kang SW, Zhou M, O’Doherty GA. J Am Chem Soc. 2012;134:11952–11955. doi: 10.1021/ja305321e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Guppi SR, O’Doherty GA. J Org Chem. 2007;72:4966. doi: 10.1021/jo070326r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Guo H, O’Doherty GA. J Org Chem. 2008;73:5211. doi: 10.1021/jo800691v. [DOI] [PubMed] [Google Scholar]
- 10.Larson DP, Heathcock CH. J Org Chem. 1997;62:8406. doi: 10.1021/jo971413u. [DOI] [PubMed] [Google Scholar]
- 11.(a) Noyori R, Ohkuma T, Kitamura M. J Am Chem Soc. 1987;109:5856. [Google Scholar]; (b) Borisova SA, Guppi SR, Kim HJ, Wu B, Penn JH, Liu H-w, O’Doherty GA. Org Lett. 2010;12:5150. doi: 10.1021/ol102144g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown CA, Yarnashita A. J Am Chem Soc. 1975;97:891.Kimmel T, Becker D. J Org Chem. 1984;49:2494.For synthetic application of this, see: Hoye RC, Baigorria AS, Danielson ME, Pragman AA, Rajapakse HA. J Org Chem. 1999;64:2450.Li M, O’Doherty GA. Org Lett. 2006;8:6087. doi: 10.1021/ol062595u.
- 13.Achmatowicz O, Bukowski P, Szechner B, Zwierzchowska Z, Zamojski A. Tetrahedron. 1971;27:1973. [Google Scholar]
- 14.Luche JL. J Am Chem Soc. 1978;100:2226. [Google Scholar]
- 15.VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973. [Google Scholar]
- 16.Corey EJ, Nicolaou KC. J Am Chem Soc. 1974;96:5614. [Google Scholar]
- 17.(a) Hinds JW, McKenna SB, Sharif EU, Wang HL, Akhmedov NG, O’Doherty GA. ChemMedChem. 2013;8:63. doi: 10.1002/cmdc.201200465. [DOI] [PubMed] [Google Scholar]; (b) Shan M, Sharif EU, O’Doherty GA. Angew Chem Int Ed. 2010;49:9492. doi: 10.1002/anie.201005329. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sharif EU, O’Doherty GA. Eur J Org Chem. 2012:2095. doi: 10.1002/ejoc.201101609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bedini E, Parrilli M, Unverzagt C. Tetrahedron Lett. 2002;43:8879. [Google Scholar]
- 19.David S, Hanessian S. Tetrahedron. 1985;41:663. [Google Scholar]
- 20.Schmidt RR, Michel J. Angew Chem Int Ed Engl. 1980;19:731. [Google Scholar]
- 21.a) Nagano T, Pospísil J, Chollet G, Schulthoff S, Hickmann V, Moulin E, Herrmann J, Müller R, Fürstner A. Chem Eur J. 2009;15:9697. doi: 10.1002/chem.200901449. [DOI] [PubMed] [Google Scholar]; b) Molinaro A, De Castro C, Lanzetta R, Manzo E, Parrilli M. J Am Chem Soc. 2001;123:12605. doi: 10.1021/ja016471i. [DOI] [PubMed] [Google Scholar]; c) Bekiroglu S, Sandström A, Kenne L, Sandström C. Org Biomol Chem. 2004;2:200. doi: 10.1039/b311852e. [DOI] [PubMed] [Google Scholar]; d) Kondoh A, Arlt A, Gabor B, Fürstner A. Chem Eur J. 2013;19:7731. doi: 10.1002/chem.201300827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.