


Abstract
Defining target sites for antisense oligonucleotides in highly structured RNA is a non-trivial exercise that has received much attention. Here we describe a novel and simple method to generate a library composed of all 20mer oligoribonucleotides that are sense- and antisense to any given sequence or genome and apply the method to the highly structured HIV-1 leader RNA. Oligoribonucleotides that interact strongly with folded HIV-1 RNA and potentially inhibit its dimerization were identified through iterative rounds of affinity selection by native gel electrophoresis. We identified five distinct regions in the HIV-1 RNA that were particularly prone to antisense annealing and a structural comparison between these sites suggested that the 3′-end of the antisense RNA preferentially interacts with single-stranded loops in the target RNA, whereas the 5′-end binds within double-stranded regions. The selected RNA species and corresponding DNA oligonucleotides were assayed for HIV-1 RNA binding, ability to block reverse transcription and/or potential to interfere with dimerization. All the selected oligonucleotides bound rapidly and strongly to the HIV-1 leader RNA in vitro and one oligonucleotide was capable of disrupting RNA dimers efficiently. The library selection methodology we describe here is rapid, inexpensive and generally applicable to any other RNA or RNP complex. The length of the oligonucleotide in the library is similar to antisense molecules generally applied in vivo and therefore likely to define targets relevant for HIV-1 therapy.
INTRODUCTION
Binding of antisense oligonucleotides to complementary targets in an mRNA can potentially abolish the expression of the encoded protein by degrading the message and/or interfering with ribosome function. This principle has been used extensively as a laboratory tool to elucidate gene function and in treatment of animal and human diseases (reviewed in 1). A critical limitation of antisense applications is the accessibility of the target. Naturally occurring RNA molecules are associated with proteins and folded into secondary and tertiary structures that may explain the variable efficiency of the antisense application (2,3). In the absence of any detailed structural information of the target RNA, antisense design has traditionally been based on the principle of trial and error. This can often be a cumbersome process considering that only an estimated 2–5% of randomly selected sites represent competent antisense targets (reviewed in 2). More recently, the application of small interfering RNAs (siRNAs), has been developed as a highly potent approach known as RNA interference (RNAi), that can lower gene expression in mammalian cells with an unprecedented efficiency and specificity (4). Although it was initially suggested that the siRNA approach was less sensitive to RNA structure in the target, it was recently demonstrated that the efficiency of RNAi-mediated ‘knock down’ is also influenced by RNA structure (5,6). This may be of particular concern if the targets are located within the 5′- and 3′-UTR of a mRNA or viral RNAs, which often are considerably structured.
A number of theoretical and practical approaches to determine accessible regions in RNA have been described (2,7). One approach, that is used extensively, is the mapping of nucleotides that are accessible to chemical reagents or nucleases (8). However, this information has proven inadequate for rational antisense design. Other more sophisticated approaches, that have been developed to globally search for effective antisense targets, include the mapping of hybrids formed between the mRNA and random DNA oligonucleotides using ribonuclease H (RNase H) (9–12), or alternatively annealing the labelled mRNA to an array of a complete set of antisense oligonucleotides (13). Although both techniques have proven useful for selection of functional antisense targets there are also limitations (see Discussion). Here we describe a novel method for selecting oligonucleotides from a complete library consisting of 20-nucleotide long sense- and antisense RNAs that are able to bind to target RNA under variable selective conditions. This technique is building on the genomic SELEX concept where natural sequences are used as a source for library construction (14).
The method was applied to a 355-nucleotide fragment of the HIV-1 genome that encompasses the highly structured 5′-UTR. Several regulatory elements that are instrumental at distinct steps of the retroviral replication cycle are located in this region: elements that control gene expression including the trans-activation response (TAR) element, that binds the transcription activator, Tat; the poly(A) hairpin that contains the suppressed 5′-poly(A) site; the major splice donor site 1 (SD1), and the initiation site for Gag translation. Sites that are functional at viral assembly steps in the viral replication cycle include the packaging signal (PSI) that specifies the encapsidation of the RNA genome, and the dimerization initiation site (DIS) that mediates the initial intermolecular contact in the dimeric genome, and the primer binding site (PBS) that binds the tRNALys3 used to prime reverse transcription (15). It has been demonstrated that at least two alternative conformers of the HIV-1 B-subtype leader RNA exist in vitro: a dimerization-competent branched multiple hairpin (BMH) conformation where the poly(A) and DIS hairpin structures are formed and a dimerization-incompetent long-distance interaction (LDI) conformation in which the poly(A) and the DIS sequences are involved in an alternative helix (reviewed in 16). The high level of sequence conservation and the notion that a large part the 5′-UTR is present in both genomic and sub-genomic HIV-1 RNA make it an attractive target for antisense and siRNA design. Here we identify five regions in the highly structured HIV-1 leader, which are feasible targets for antisense oligonucleotide binding.
MATERIALS AND METHODS
Constructs
The pUC18-LAI plasmid, containing a +1–355 nucleotide fragment of the HIV-1 genome sequence of the LAI isolate behind a T7 promoter was used throughout the selection assay. In the reverse transcriptase experiment we used an extended HIV-1 construct pUC18-LAI-extended containing the first 444 nucleotides of HIV-1 LAI. Both constructs have been described earlier (17).
To construct the library 20 µg of pUC18-LAI was treated with 2 units of DNase I (2 units/µl, Ambion) for 8 min in 100 µl digestion buffer (250 mM Tris–HCl pH 7.6, 2.5 mM DTT, 125 mM NaCl2, 5 mM CaCl2, 50 mM MnCl2). The reaction was terminated by adding 10 µl 50 mM EDTA, followed by phenol extraction and ethanol precipitation. The ends of the fragments were filled-out with Klenow and the 5′-ends were phosphorylated using 2 units of T4 polynucleotide kinase (10.000 units/ml, New England Biolabs), followed by phenol extraction and precipitation. 200 pmol of a 5′-linker DNA cassette was made by annealing two oligonucleotides containing a T7 promoter, a Tag A sequence and a restriction site for MmeI: (sense: 5′-AGCCTGCAGTAATACGACTCAC TATAGGGATCGCTTAGTCCGAC-3′ and antisense: 5′-P-GTCGGACTAAGCGATCCCTATAGTGAGTCGTATTACTGCAGGC-3′; MmeI site underlined). This linker was mixed with 20 µg of the DNase I-digested DNA and ligated in a 50 µl ligation buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 10 mM DTT, 1 mM ATP, 25 µg/ml BSA) containing 120 units of T4 DNA ligase. The reaction was incubated at 14°C overnight and the DNA was phenol extracted, precipitated and redissolved in 20 µl H2O. The ligated sample was digested with 6 units of MmeI (2 units/µl, Centrum Transferu Technologii-Poland) in a total volume of 30 µl in MmeI buffer (10 mM HEPES pH 8.0, 2.5 mM KAc, 5 mM MgAc, 2 mM DTT, 40 µM S-adenosylomethionine hydrochloride) for 1 h at 37°C.
The 60 base pairs digestion product, corresponding to the T7 promoter, Tag A and the 20 bp of HIV-1 sequence, was excised from a 6% native polyacrylamide gel and extracted in 300 µl TE buffer (10 mM Tris–HCl pH 7.5, 0.2 mM EDTA). The DNA was precipitated and dissolved in 20 µl TE buffer. The DNA was mixed with 200 pmol of a 3′-linker DNA cassette, Tag B, made by annealing two oligonucleotides (sense: 5′-P-CCCTTTAGTGAGGGTTAATTAAGCTTA AGG-3′ and antisense: 5′-CTTAAGCTTAATTAACCC TCACTAAAGGGNN-3′), and ligated in 40 µl ligation buffer containing 120 units of T4 DNA ligase. The sample was phenol extracted, precipitated and redissolved in 20 µl H2O. Two microlitres of the ligated DNA fragment library was PCR amplified using 25 µM of forward (T7-Tag A: 5′-TAATACGACTCACTATAGG-3′) and reverse primer (Tag B: 5′-ATTAACCCTCACTAAAGGG-3′) in a standard 50 µl PCR reaction using Taq polymerase. The amplified DNA was purified in a 6% polyacrylamide gel as described above. To verify the random distribution of sequences within the library, the PCR product was inserted into the pCR2.1-TOPO (Invitrogen), transformed into TOP10 cells (Invitrogen) by electroporation and sequenced.
RNA transcription
Large scale RNA transcription was performed by run-off transcription using 1 µg library PCR product in a 20 µl reaction with T7 buffer (80 mM HEPES–KOH pH 7.5, 25 mM MgCl2, 2 mM spermidine, 20 mM DTT), 7.5 mM rNTPs, 10 units/ml yeast inorganic PPase, 20 units Ribonuclease Inhibitor (Promega) and 2 units of T7 polymerase. The transcription was incubated at 37°C for 4 h followed by DNA digestion using 2 units of DNase I for 15 min at 37°C. Twenty microlitres of 95% formamide loading buffer was added and the sample was heated to 90°C for 2 min prior to loading on a 6% denaturing polyacrylamide gel. After separation the RNA bands were excised using UV-shadowing and eluted in 300 µl nuclease-free solution of 25% v/w phenol pH 6.6. The RNA was precipitated and dissolved to a concentration of 1 µM.
In vitro selection assay
One picomole leader RNA (+1–355) was 5′-end-labelled with [γ-32P]ATP as described previously (18) and redissolved in 5 µl RNase-free water. The RNA was incubated at 85°C for 5 min and left on ice for 2 min. The sample was mixed with 5 pmol of either DIS primer (19), CN1 antisense primer (20) or library oligonucleotides and incubated at 37°C for 30 min in dimerization buffer (50 mM Na-cacodylate pH 7.5, 250 mM KCl, 5 mM MgCl2) in a total volume of 20 µl. Samples were analyzed in a 6% native TBM gel (50 mM Tris-borate pH 8.3, 5 mM MgCl2) and autoradiographed for 4 h at 4°C. The monomeric band was excised and eluted in 300 µl nuclease-free solution of 25% v/w phenol pH 6.6. Bound oligonucleotides were collected and amplified using RT–PCR. Reverse transcription was allowed to proceed at 41°C for 30 min in RT buffer (50 mM Tris–HCl pH 8.4, 10 mM MgCl2, 2 mM DTT, 5.8 µl 2.5 mM dNTPs, 2 units AMV RT) in a total volume of 8 µl. The PCR reaction was performed as described above and the sample was purified in a 1.5% agarose gel followed by microspin GFX-matrix columns (Amersham Biosciences). This PCR product was used for T7 run-off transcription generating RNA for the next round of selection. After the third and fifth rounds 15 and 19 clones, respectively, were sequenced as described above. Large scale run-off transcriptions was performed from EcoRI-cleaved plasmid clones 17, 21, 24, 29 and 34 as described above. DISupstream–RNA and DISskripkin–RNA oligos were constructed similarly, but using DNA oligos with fixed sequence corresponding to positions 242–263 and 249–270, respectively, as template.
Dimerization inhibition and primer extension assay
The RNA transcripts and the DNA oligonucleotides corresponding to the sequences of the selected inserts were used to inhibit dimerization of the viral leader as described above. The amount of monomer and dimer RNA were quantified using a Bio-Rad phosphorimager. To analyze how efficient DIS oligonucleotides disrupted HIV-1 RNA dimers, 1 pmol [γ-32P]RNA (+1–355) was incubated in nuclease-free water at 85°C for 5 min in a total volume of 10 µl, then snap-cooled on ice for 5 min, followed by adding 10 µl of dimerization buffer and incubation of the sample at 37°C for 30 min. Five picomoles of DIS–DNA, or DIS–RNA were added in total volume of 25 µl. The samples were incubated at 37°C and 6 µl aliquots were taken after 0, 30, 120 and 210 min and placed on ice. The samples were analyzed on a 6% native 1× TBM gel.
To perform the primer extension assay 1 pmol RNA spanning the first 444 nucleotides of the HIV-1 genome was mixed with 80 fmol 5′-end-labelled RT primer (5′-CCTTAACCGAATTTTTTCCC-3′) complementary to position 384–401 in the presence of 1 pmol of the different DNA or RNA oligonucleotides in a total volume of 6 µl annealing buffer (100 mM Tris–HCl pH 7.5, 400 mM KCl). The annealing mixture was incubated 2 min at 90°C followed by 20 min at 50°C. Reverse transcription and gel analysis was performed according to Damgaard et al. (21,22).
Oligonucleotide binding assay
A sample of 0.25 pmol of HIV-1 LAI RNA (not radioactive) was incubated in 12.5 µl water at 85°C for 5 min, followed by snap-cooling on ice for 5 min. The RNA sample was mixed with 1.25 pmol 5′-end-labelled RNA or DNA oligo and incubated in 50 µl dimerization buffer at 37°C. Eight microlitre aliquots were taken out at specified time points and placed on ice. The samples were loaded onto a 6 and 12% native 1× TBM polyacrylamide gel. The reactions were made in triplicate and bound radioactive oligo were quantified with a Bio-Rad phosphorimager.
RESULTS
Construction of a size-constrained RNA expression library from genomic HIV-1 DNA
A library of 20mer oligoribonucleotides containing sense- and antisense HIV-1 sequences was constructed as outlined in Figure 1. To restrict the length of the oligonucleotides we exploited the property of the restriction enzyme MmeI to cleave the DNA strands with an offset of 18 and 20 nucleotides from the recognition sequence. The presence of an upstream T7 promoter enabled run-off transcription and specific sequence tags flanking the HIV-1 RNA were used to facilitate RT–PCR amplification of the selected RNA oligonucleotides (Fig. 1). To restrict the selection towards the highly structured HIV-1 5′-UTR we used a plasmid containing the +1–355 region of HIV-1 LAI genome as input DNA. We found that it was important to use circular DNA as substrate to avoid a bias in the library sequences (data not shown). Since the plasmid backbone spans 2.7 kb we predict that only ∼10% of the RNAs in the library consist of sense or antisense HIV-1 sequences, and that the remaining 90% contain sequences derived from pUC18, which served as a selection control. To analyse the diversity of the library 24 clones were prepared and sequenced. As expected the sequences were randomly distributed throughout the plasmid in both the sense and antisense orientation (data not shown).
Figure 1.

Construction of the RNA library. A pUC18 plasmid containing the HIV-1(LAI) leader sequence (+1–355) was digested partially with DNase I to create random fragments. A double-stranded oligonucleotide, containing a T7 promoter, a specific tag sequence (Tag A) and a MmeI restriction site, was ligated to the fragments. After MmeI digestion the DNA fragments were ligated to a downstream linker, containing a random two-nucleotides 3′-overhang and another tag sequence (Tag B). The RNA library was synthesized by standard run-off transcription using T7 RNA polymerase.
Selection of antisense oligonucleotides that bind to HIV-1 RNA and inhibit dimerization
To select RNA species that bind and specifically inhibit dimerization of the HIV-1 leader we incubated 5 pmol of the library RNA with 1 pmol of 32P-labelled +1–355 HIV-1 RNA under conditions where >80% of the HIV-1 RNA forms dimer complexes in a native gel electrophoresis assay (Fig. 2, lane 1). RNA oligonucleotides, which both bind and specifically interfere with HIV-1 RNA dimerization, will presumably, remain bound to monomeric HIV-1 RNA during this assay. Therefore, monomeric HIV-1 RNA and the associated library RNAs was excised and extracted from the gel. After RT–PCR amplification using library-specific primers (T7-Tag A and Tag B; Fig. 1) a new pool of RNAs was transcribed and used for the next round of selection. After three rounds of selection the dimerization of the HIV-1 RNA was almost completely abolished in the presence of the selected library RNA (<5% dimerization; Fig. 2, lane 6). The selection was continued for a total of five rounds, but without any further increase in the inhibition of HIV-1 RNA dimerization (Fig. 2, lane 8). Two additional DNA oligonucleotides were included for comparison: CN1, which is complementary to nucleotides 123–150 and previously shown to disrupt the LDI conformer and stimulate dimerization of HIV-1 RNA (16) and DISskripkin, which is complementary to the distal part of the DIS stem–loop (positions 249–269; Fig. 3B) and previously shown to inhibit dimerization (Fig. 2, lanes 2 and 3, respectively) (19,23). To analyse the sequences of individual RNA in the pools, 15 and 19 clones from the third and fifth round of selection, respectively, were prepared and sequenced (Table 1). All sequences obtained were between 20 and 22 nucleotides long and complementary to six distinct regions in the HIV-1 leader, including the Poly(A), PBS, DIS, SD, PSI stem–loops, and at the U5-AUG stem (only found after the 5th round; Fig. 3A). Interestingly, with the exception of PSI, each region was represented by approximately the same number of clones in the selected pools suggesting that equimolar amounts of RNA oligonucleotides bound to HIV-1 RNA in a non-competitive fashion during the selection step. This also explains the appearance of multiple bands with slower mobility in the selection assay (Fig. 2; lanes 6–8) as compared to the smaller band shift produced with individual clones (see below; Fig. 4).
Figure 2.

Selection of RNA oligonucleotides that bind to the HIV-1 leader. Radiolabelled HIV-1 leader RNA (+1–355) was incubated under dimerization conditions alone (lane 1) or in the presence of control oligonucleotides, CN1, known to stimulate dimerization (lane 2), DISSkripkin, known to inhibit dimerization (lane 3), or the selected RNA pools after 1–5 rounds of selection (lanes 4–8). The positions of dimeric (D) and monomeric (M) RNA species are indicated.
Figure 3.


Position of the antisense oligonucleotides used in this study. (A) The selected target sites superimposed on the BMH secondary structure of the HIV-1 HXB2 subtype leader drawn according to Damgaard et al. (21). Thick lines denote the sequences complementary to the selected RNA oligonucleotides after five rounds of selection. The thin line denotes the sequence complementary to the PSI-specific oligonucleotides that was obtained frequently after the third round of selection, but only once after the fifth round (Table 1). The major structure elements are denoted (see text for details). (B) The annealing sites for DISupstream–, DISskripkin– and the selected DIS–oligonucleotides indicated by dotted, stippled and full lines respectively. (C) The annealing sites for PBSnat– and PBS–oligonucleotides.
Table 1. Sequences of the oligonucleotides obtained after three and five rounds of selection (denoted in the HIV-1 sense strand direction).
| Nr. | Regiona | 3rd Round of selection 5′ – 3′b | Position | Nr. | Regiona | 5th Round of selection 5′– 3′ | Position |
|---|---|---|---|---|---|---|---|
| 1 | Poly(A) | AAAGCUUGCCUUGAGUGCU | 76/94 | 16 | Poly(A) | ccAAAGCUUGCCUUGAGUG | 76/92 |
| 2 | AAUAAAGCUUGCCUUGAGUGC | 73/93 | 17 | * | AAGCUUGCCUUGAGUGCUUCA | 77/97 | |
| 3 | PBS | AAAGCGAAAGGGAAACCAGA | 203/222 | 18 | AAUAAAGCUUGCCUUGAGUG | 73/92 | |
| 4 | guggUGAAAGCGAAAGGG | 195/214 | 19 | AUAAAGCUUGCCUUGAGUGCUUCAG | 73/98 | ||
| 5 | AAAGCGAAAGGGAAACCAGA | 203/222 | 20 | PBS | GAAAGCGAAAGGGAAACCAGA | 202/222 | |
| 6 | CGAAAGGGAAACCAGAGGA | 207/225 | 21 | * | AAAGCGAAAGGGAAACCAGA | 203/222 | |
| 7 | DIS | AGCGCGCACGGCAAGAGGCG | 256/275 | 22 | AAAGCGAAAGGGAAACCAGAG | 203/223 | |
| 8 | AAGCGCGCACGGCAAGAGGCG | 255/275 | 23 | DIS | CUGAAGCGCGCACGGuAAGA | 252/271 | |
| 9 | AAGCGuGCACGGCAAGAGGCG | 255/275 | 24 | * | AAGCGCGCACGGGAAGAGGCG | 255/275 | |
| 10 | AAGCGCGCACGGCAAGAGGCG | 255/275 | 25 | AAGCGCGCACGGcAAGAGGCG | 255/275 | ||
| 11 | SD | AGaGGCGGCGACUGGUGAGU | 276/295 | 26 | AAGCGCGCACGGcAAGAGGuG | 255/275 | |
| 12 | AGGGGCGGCGACUGGUGAGUA | 276/296 | 27 | SD | AGGGGCGGCGACUGGUGAGUA | 276/296 | |
| 13 | PSI | AAUUUUGACUAGCGGAGGCU | 304/324 | 28 | AGGG-CGGCGACUGGUGAGUA | 276/296 | |
| 14 | AAUUgUGACUAGuCGGAGG | 304/322 | 29 | * | AcuGGCGGCGACUGGUGAGUA | 276/296 | |
| 15 | AAUUUUGACUAGCaGAGGCU | 304/324 | 30 | PSI | AAAcUUUGACUAGCGGAGGCU | 303/322 | |
| 31 | 5U-AUG | GAAGGAGAGAGAUGGGUGCG | 325/344 | ||||
| 32 | CUAGAAGGAGAGAGAUGGG | 322/340 | |||||
| 33 | AGAGAGAUGGGUGCGAGAG | 330/348 | |||||
| 34 | * | AGAAGGAGAGAGAUGGGUGCG | 224/344 |
aThe sequences are grouped according to the matching target region in the HIV-1. Asterisks mark the clones that were selected for further studies.
bThe sense sequence of the variable insert between the Mme1 site and Tag B is shown. Sequences that are identical to the HIV-1 sequence are indicated by capital letters; sequences that diverge from the HIV-1 sequence are indicated by non-capital letters, deletions relative to the HIV-1 sequence are indicated by –.
Figure 4.

HIV-1 leader RNA dimerization assay. HIV-1 leader RNA (+1–355) was incubated under dimerization conditions in the absence (Control), in the presence of oligonucleotide CN1 (lane 2), DISskripkin–DNA (lane 3) or selected antisense DNA and RNA oligonucleotides as denoted above (see Table 2 for sequences). The dimerization efficiency, indicated below, was calculated as [dimer]/[dimer]+[monomer] from three independent experiments. The positions of dimeric (D) and monomeric (M) RNA species are indicated.
Functional characterization of the selected oligonucleotides
To prove the applicability of the selection procedure one clone was selected from each of the five classes dominating the pool after five rounds of selection and their binding properties and effect on dimerization were investigated (Table 2, Fig. 4). The increase in the mobility shift observed upon RNA oligonucleotide binding compared to DNA binding can be explained by the increase in the mass of the RNA oligos from the tag sequences flanking the antisense portion. In addition, DNA oligonucleotides corresponding to the same sequences were synthesized and included in the analysis (Table 2, Fig. 4).
Table 2. The selected antisense oligonucleotides and their functional characteristics.
| Name of oligo | Sequence (5′ to 3′) | Binding HIV RNAa | Inhibition of dimera | Pausing of RTb |
|---|---|---|---|---|
| Poly(A)–DNA | AAG CAC TCA AGG CAA GCT TTA | - | - | (+) |
| PBS–DNA | CTC TGG TTT CCC TTT CGC TTT | ++ | - | ++ |
| DIS–DNA | CGC CTC TTG CCG TGC GCG CTT | +++ | +++ | ++ |
| SD–DNA | TAC TCA CCA GTC GCC GCC | + | - | ++ |
| 5U-AUG–DNA | CGC ACC CAT CTC TCT CCT T | + | - | (+) |
| Poly(A)–RNA | UGA AGC ACU CAA GGC AAG CUU | ++ | - | ++ |
| PBS–RNA | UCU GGU UUC CCU UUC GCU UU | +++ | - | +++ |
| DIS–RNA | CGC CUC UUG CCG UGC GCG CUU | ++++ | +++ | ++++ |
| SD–RNA | UAC UCA CCA GUC GCC GCC A | ++ | + | +++ |
| 5U-AUG–RNA | CGC ACC CAU CUC UCU CCU UCU | ++ | - | + |
All five selected RNA oligonucleotides bound strongly to the HIV-1 RNA since a distinct mobility shift was observed in all cases (Fig. 4, lanes 9–13). A binding experiment where the individual RNA oligonucleotides were added successively to the same binding reaction showed that all five RNAs can bind independently of each other to the HIV-1 leader RNA resulting in a progressively slower mobility (data not shown). Only DNA or RNA oligonucleotides complementary to the DIS region caused a nearly complete (>95%) inhibition of dimerization that was comparable to the inhibition obtained by DISskripkin–DNA (Fig. 4, lanes 3, 6 and 11), whereas the RNA oligonucleotide complementary to the SD region inhibited RNA dimerization partially with ∼30% (Fig. 4, lane 12). Of the DNA oligonucleotides it appears that only PBS–DNA, DIS–DNA and AUG–DNA were able to bind, presumably due to the lower stability of DNA–RNA duplexes as compared to RNA–RNA duplexes. From these data we conclude that it is primarily the RNA species antisense to the DIS region that are responsible for the observed inhibition of dimerization during the selection process whereas the other oligonucleotides were selected due to their binding affinity towards monomeric RNA.
The binding of the selected oligonucleotides to HIV-1 RNA may also interfere with elongation by reverse transcriptase. To test this we measured the ability of the RNA and DNA oligonucleotides to pause reverse transcription in vitro (Fig. 5 and data not shown). Except for the AUG–RNA that only had a modest effect, all of the selected RNA oligonucleotides resulted in strong pausing of the reverse transcriptase at the expected positions (Fig. 5). When using the DNA versions of the same oligonucleotides only PBS–DNA, DIS–DNA and SD–DNA were able to pause reverse transcription efficiently, which is in accordance with the binding data (data not shown; Fig. 4).
Figure 5.
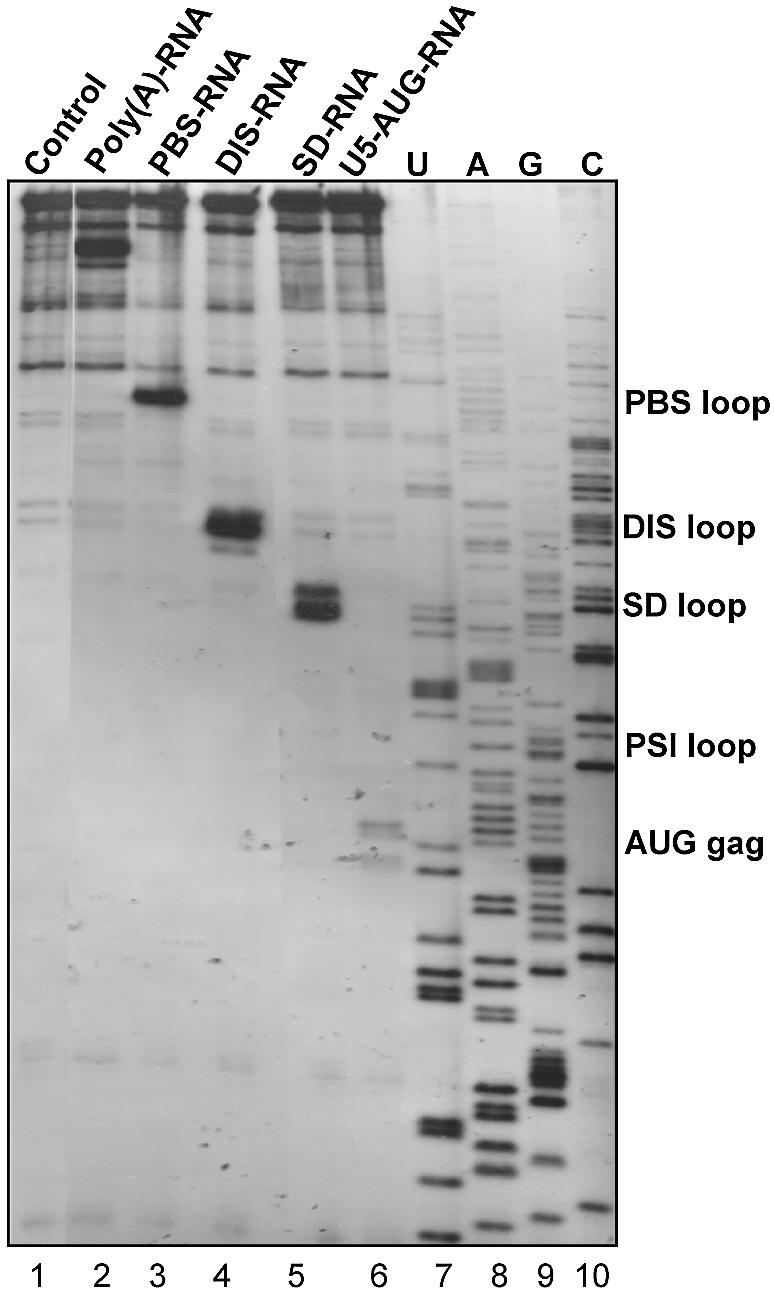
Pausing of reverse transcription by selected oligonucleotides. Binding of the selected oligonucleotides (Table 2) and their ability to block reverse transcription was investigated by a reverse transcription progressivity assay using an extended HIV-1 template (+1–444). A 5′-end-labelled DNA primer complementary to position 384–401 of the viral RNA was used to prime reverse transcription. A control without oligonucleotide (lane 1) and a sequencing ladder (lanes 7–10) are included.
Binding of oligonucleotides to the DIS hairpin and PBS loop
Interestingly, all selected DIS-specific antisense sequences that we selected were complementary to the distal loop and the 3′-side of the DIS helix suggesting a selective advantage for this mode of binding. To confirm this we compared the binding kinetics of the selected DIS–RNA clone with an RNA clone that was constructed to anneal either symmetrically to the DIS loop (position 249–269, DISskripkin–RNA; Fig. 3B) or to the 5′ side of the DIS hairpin (position 243–263; DISupstream–RNA; Fig. 3B). Unlabelled leader RNA was incubated with 32P-labelled RNA oligonucleotides and samples were taken at different time points and analysed by native gel electrophoresis (Fig. 6A). Notably, the selected DIS–RNA bound ∼2 to 3 times more strongly to the mRNA than the DISskripkin– and DISupstream–RNAs after 8 min of incubation (Fig. 6A). This difference in the binding kinetics most likely explains why only oligonucleotides that bind the 3′-part of the DIS stem–loop were obtained after three and five rounds of selection where the RNA were allowed to bind for 30 min, and demonstrates the sensitivity of the selection approach. The DISskripkin–RNA had a strong tendency to form dimers suggesting that this oligo forms a kissing-loop interaction with itself (Fig. 6A). This may explain its decreased annealing activity to the mRNA. A similar difference in binding kinetics was obtained when comparing DIS–DNA and DISskripkin–DNA oligoes (Fig. 6B).
Figure 6.



Binding analysis of the selected oligonucleotides to HIV-1 RNA. Non-labelled HIV-1 leader RNA (+1–355) was incubated in the presence of the indicated radiolabelled oligonucleotides that include (A) DISupstream–RNA (lanes 1–5), DIS–RNA (lanes 6–10) and DISSkripkin–RNA (lanes 11–15), (B) DISSkripkin –DNA (lanes 2–6) and DIS–DNA (lanes 7–11) and (C) PBSnat–DNA (lanes 2–6) and PBS–DNA (lanes 7–11). See Table 2 for sequences. The samples were incubated for the indicated time period (min) before being subjected to native gel electrophoresis. The relative binding efficiency of labelled oligonucleotide to the HIV-1 leader RNA was calculated from three independent experiments as the sum of counts in the monomer and dimer bands normalized in each experiment to the maximum binding observed at the last time point. (A) The bands corresponding monomeric and dimeric RNA oligos were investigated by running the same samples in a 12% polyacrylamide native gel (lower autoradiogram). Radiolabelled HIV-1 RNA was included as a marker in panels B and C. The positions of dimeric (D) and monomeric (M) RNA species are indicated.
Another intriguing observation was the identification of RNA oligonucleotides annealing to the 202–224 region in the PBS loop whereas no clones complementary to the natural primer binding site were obtained. To study the significance of this result we compared the binding kinetics of two DNA oligonucleotides complementary to either the natural PBS sequence (PBSnat–DNA; position 179–201; Fig. 3C) or to the selected sequence (PBS–DNA; 203–223; Fig. 3C). We found that the PBS oligonucleotide complementary to the selected site bound approximately two times faster at the early time points to the HIV-1 RNA than the oligonucleotide complementary to the natural PBS, further demonstrating the specificity of the selection protocol (Fig. 6C).
Next we investigated the ability of the DIS-specific oligonucleotides DIS–RNA and DIS–DNA, to disrupt preformed dimers into monomers (Fig. 7, lanes 2–9). Both DIS oligonucleotides were able to dissociate the HIV-1 dimer nearly to completion after 3 h, with the DIS–RNA being slightly more effective. This shows that the selected DIS oligonucleotide can bind the HIV-1 RNA and actively compete for dimer formation.
Figure 7.

Time course for the disruption of preformed HIV-1 dimer into RNA monomers in the presence of DIS-specific oligonucleotides. One picomole of radiolabelled HIV-1 leader RNA was dimerized and challenged with 5 pmol DIS–RNA or DIS–DNA for 0, 30, 120 or 240 min at 37°C before native gel electrophoresis. The dimerization efficiency was calculated as [dimer]/[dimer]+[monomer] from three independent experiments.
DISCUSSION
We have developed a simple and inexpensive methodology to construct a complete 20mer antisense and sense RNA library from genomic or cDNA sequences and applied it in a search for accessible target sites within the HIV-1 leader RNA. The diversity of the library in this study is limited to ∼6000 different RNA molecules (corresponding to all 20mer sequences in both strands of a 3 kb plasmid), but it can potentially accommodate up to 1012 different clones (or higher if scaled up). It is therefore feasible to make RNA libraries with up to 100-fold coverage of all possible 20mer sequences in the human genome in one reaction.
A number of library-based methodologies for finding effective antisense targets in RNAs have previously been described. However, in contrast to our approach that uses a library of RNA oligoes, previous methods are only applicable to DNA libraries. In one approach a randomized DNA–oligonucleotide library was synthesized and annealed to the mRNA. Upon RNase H digestion the cleavage site, which marks the binding site of antisense DNA, was mapped by primer extension (7,10,11). In an alternative approach, randomized DNA–oligonucleotides were annealed to the mRNA and used as reverse transcription primers to produce cDNAs that, upon PCR amplification, were cloned and characterized (24). There are three major limitations using the above mentioned methods: firstly, the binding of the short 10–12mer randomized DNA oligonucleotides may not reflect the binding efficiency of perfectly matched longer oligonucleotides that are generally applied in antisense experiments. Secondly, marking the binding site requires a subsequent enzymatic step (RNase H cleavage or reverse transcription) that also may be influenced by sequence and local structures in the RNA. Thirdly, in the RNase H-based approach only the RNase cleavage site is mapped whereas the sequence of the actual antisense DNA oligo is not characterized. These problems are circumvented in the array-based method (2,3,13,25), but this approach is considerably more expensive, non-suitable for RNA libraries and not attractive if a large number of genes or whole genomes are to be investigated. Also, based on its design this approach can only compare binding and not functional parameters like, for instance, inhibition of RNA dimerization.
The stringency of the selection approach described in this report is reflected by the observation that all clones present in the pool were complementary to the same five or six regions in the HIV-1 RNA after only three rounds of selection, in spite of the fact that >90% of the original library contained non-HIV-1 plasmid sequences. In addition the selected sequences in most regions were generally not identical but within two to three nucleotides from each other, implying that they did not derive from a common ancestor RNA in the library. Notably, the selection did not result in a single antisense winner RNA after an extensive number of selections. We believe that, due to the 5-fold excess of library RNA relative to HIV-1 RNA, non-competitive binding of approximately five non-overlapping RNA oligos was obtained after three and five rounds of selection. By lowering the library to target RNA ratio we suspect that fewer antisense targets will be selected after multiple selections. Hence, this method will not provide a complete list of all accessible sites in the target RNA, but rather an adjustable number of the most accessible ones.
None of the selected target sites could have been predicted rationally from the proposed secondary structure model underscoring the importance of an experimental approach (Fig. 3) (21). For instance, all the selected DIS-specific antisense RNAs were complementary to the 3′-side of the DIS stem, whereas none of the RNA oligonucleotides resembled the antisense sequence of the oligo described by Skripkin et al. (19), which binds the distal loop symmetrically. The significance of this difference was tested experimentally by showing that the DIS–RNA oligo with the selected sequence binds to the HIV-1 RNA significantly faster than both the DISskripkin–RNA and DISupstream –RNA (Figs 3B and 6A). Over multiple rounds of selection this difference is most likely sufficient to enrich the DIS–RNA to a level where other RNA species that are antisense to other regions of the DIS hairpin escape detection.
Another strong binding site was identified in the major central loop of the PBS stem. Interestingly, this site did not overlap with the PBS used naturally by tRNA to prime reverse transcription, which one may have expected to be highly accessible (see Fig. 3C). By direct comparison we found that an oligonucleotide complementary to the selected site in the PBS region exhibited superior binding kinetics when compared to an oligo matching the natural primer binding site. One explanation for this observation may be that the binding of the natural tRNAlys primer is stimulated by the HIV-1 reverse transcriptase enzyme or structural elements within the tRNA itself in order to function as a primer in vivo. Several studies describing additional contact points between the tRNA and the RNA template, that are crucial to the reverse transcription process are in agreement with this explanation (26–28).
A structural comparison of the selected sites may enable us to rationally design antisense oligonucleotides in other systems. Interestingly, all six accessible sites in the HIV-1 RNA selected after three or five rounds were structurally similar: the 3′-part of the antisense RNA preferentially binds within putative single-stranded regions, whereas the 5′-part interacts with regions that are base paired. This suggests that antisense oligonucleotide prefers to initiate the binding process by annealing the 3′-end to a single-stranded region and then displace helical segments with its 5′-end. It is possible that asymmetric binding to a stem–loop structure lowers the likelihood of forming inhibitory self-structures in the antisense RNA molecule, which has been shown to have a profound impact on the binding kinetics (29,30). The observation that the DISskripkin–RNA has a strong tendency to form dimers supports this notion. Binding of oligonucleotides to regions in RNA that are predominantly double-stranded has previously been reported to confer a strong antisense response in vivo (31).
Only two of the selected RNA oligos, namely DIS–RNA and the SD–RNA, had any effect on dimerization of HIV-1 RNA. The DIS–RNA essentially blocked dimerization probably by competing for the kissing-loop interaction between the palindromic sequences in the DIS loop. The partial inhibition by the SD–RNA may be a result of its 3′-end binding closely to the highly conserved AGG loop (position 271–273), which has been implicated in dimerization (32–34). Binding of the SD–RNA may also affect the structure of the proximal part of the DIS stem and thereby interfere with the presentation of the distal-loop sequence for kissing-loop interactions.
The high level of conservation and functional importance in all HIV-1 mRNA species and genomic RNA make the 5′-leader the most desirable therapeutical target for oligonucleotide-based drugs. Through a relatively simple selection procedure we have screened the entire leader and identified five sites that are particularly accessible for the annealing of 20mer antisense oligonucleotides. This information will provide an important basis for the design of both antisense and siRNA molecules that can act as efficient inhibitors of viral replication in the future.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Rita Rosendahl for excellent technical assistance and Ray Brown for critical reading of the manuscript. The work was supported in parts by grants from the Danish Medical and Natural Research Councils, and the Karen Elise Jensen Foundation, NOVO’s Found, the Danish AIDS Foundation, and the Carlsberg Foundation. C.K.D. and E.S.A. were supported by University of Aarhus.
REFERENCES
- 1.Kurreck J. (2003) Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem., 270, 1628–1644. [DOI] [PubMed] [Google Scholar]
- 2.Sohail M. and Southern,E.M. (2000) Selecting optimal antisense reagents. Adv. Drug Deliv. Rev., 44, 23–34. [DOI] [PubMed] [Google Scholar]
- 3.Sohail M. and Southern,E.M. (2000) Hybridization of antisense reagents to RNA. Curr. Opin. Mol. Ther., 2, 264–271. [PubMed] [Google Scholar]
- 4.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 5.Bohula E.A., Salisbury,A.J., Sohail,M., Playford,M.P., Riedemann,J., Southern,E.M. and Macaulay,V.M. (2003) The efficacy of small interfering RNAs targeted to the type 1 insulin-like growth factor receptor (IGF1R) is influenced by secondary structure in the IGF1R transcript. J. Biol. Chem., 278, 15991–15997. [DOI] [PubMed] [Google Scholar]
- 6.Kretschmer-KazemiFar R. and Sczakiel,G. (2003) The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucleic Acids Res., 31, 4417–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho S.P., Britton,D.H., Bao,Y. and Scully,M.S. (2000) RNA mapping: selection of potent oligonucleotide sequences for antisense experiments. Methods Enzymol., 314, 168–183. [DOI] [PubMed] [Google Scholar]
- 8.Ehresmann C., Baudin,F., Mougel,M., Romby,P., Ebel,J.P. and Ehresmann,B. (1987) Probing the structure of RNAs in solution. Nucleic Acids Res., 15, 9109–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birikh K.R., Berlin,Y.A., Soreq,H. and Eckstein,F. (1997) Probing accessible sites for ribozymes on human acetylcholinesterase RNA. RNA, 3, 429–437. [PMC free article] [PubMed] [Google Scholar]
- 10.Ho S.P., Bao,Y., Lesher,T., Malhotra,R., Ma,L.Y., Fluharty,S.J. and Sakai,R.R. (1998) Mapping of RNA accessible sites for antisense experiments with oligonucleotide libraries. Nat. Biotechnol., 16, 59–63. [DOI] [PubMed] [Google Scholar]
- 11.Ho S.P., Britton,D.H., Stone,B.A., Behrens,D.L., Leffet,L.M., Hobbs,F.W., Miller,J.A. and Trainor,G.L. (1996) Potent antisense oligonucleotides to the human multidrug resistance-1 mRNA are rationally selected by mapping RNA-accessible sites with oligonucleotide libraries. Nucleic Acids Res., 24, 1901–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lima W.F., Brown-Driver,V., Fox,M., Hanecak,R. and Bruice,T.W. (1997) Combinatorial screening and rational optimization for hybridization to folded hepatitis C virus RNA of oligonucleotides with biological antisense activity. J. Biol. Chem., 272, 626–638. [PubMed] [Google Scholar]
- 13.Milner N., Mir,K.U. and Southern,E.M. (1997) Selecting effective antisense reagents on combinatorial oligonucleotide arrays. Nat. Biotechnol., 15, 537–541. [DOI] [PubMed] [Google Scholar]
- 14.Dunn J.J., McCorkle,S.R., Praissman,L.A., Hind,G., Van Der Lelie,D., Bahou,W.F., Gnatenko,D.V. and Krause,M.K. (2002) Genomic signature tags (GSTs): a system for profiling genomic DNA. Genome Res., 12, 1756–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berkhout B. (2000) Multiple biological roles associated with the repeat(R) region of the HIV-1 RNA genome. Adv. Pharmacol., 48, 29–73. [DOI] [PubMed] [Google Scholar]
- 16.Huthoff H. and Berkhout,B. (2001) Two alternating structures of the HIV-1 leader RNA. RNA, 7, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen E.S., Jeeninga,R.E., Damgaard,C.K., Berkhout,B. and Kjems,J. (2003) Dimerization and template switching in the 5′ untranslated region between various subtypes of human immunodeficiency virus type 1. J. Virol., 77, 3020–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kjems J.E. and Christiansen,J. (1998) Analysis of RNA–protein complexes in vitro. In van der Vliet,P.C. (ed.), Laboratory Techniques in Biochemistry and Molecular Biology, vol. 26, pp. 43–46. Elsevier Science B.V., Amsterdam. [Google Scholar]
- 19.Skripkin E., Paillart,J.C., Marquet,R., Blumenfeld,M., Ehresmann,B. and Ehresmann,C. (1996) Mechanisms of inhibition of in vitro dimerization of HIV type I RNA by sense and antisense oligonucleotides. J. Biol. Chem., 271, 28812–28817. [DOI] [PubMed] [Google Scholar]
- 20.Berkhout B. and van Wamel,J.L. (2000) The leader of the HIV-1 RNA genome forms a compactly folded tertiary structure. RNA, 6, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Damgaard C.K., Andersen,E.S., Knudsen,B., Gorodkin,J. and Kjems,J. (2004) RNA interactions in the 5′ region of the HIV-1 genome. J. Mol. Biol., 336, 369–379. [DOI] [PubMed] [Google Scholar]
- 22.Damgaard C.K., Dyhr-Mikkelsen,H. and Kjems,J. (1998) Mapping the RNA binding sites for human immunodeficiency virus type-1 gag and NC proteins within the complete HIV-1 and -2 untranslated leader regions. Nucleic Acids Res., 26, 3667–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huthoff H. and Berkhout,B. (2002) Multiple secondary structure rearrangements during HIV-1 RNA dimerization. Biochemistry, 41, 10439–10445. [DOI] [PubMed] [Google Scholar]
- 24.Allawi H.T., Dong,F., Ip,H.S., Neri,B.P. and Lyamichev,V.I. (2001) Mapping of RNA accessible sites by extension of random oligonucleotide libraries with reverse transcriptase. RNA, 7, 314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Southern E.M., Case-Green,S.C., Elder,J.K., Johnson,M., Mir,K.U., Wang,L. and Williams,J.C. (1994) Arrays of complementary oligonucleotides for analysing the hybridisation behaviour of nucleic acids. Nucleic Acids Res., 22, 1368–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beerens N. and Berkhout,B. (2002) The tRNA primer activation signal in the human immunodeficiency virus type 1 genome is important for initiation and processive elongation of reverse transcription. J. Virol., 76, 2329–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brule F., Marquet,R., Rong,L., Wainberg,M.A., Roques,B.P., Le Grice,S.F., Ehresmann,B. and Ehresmann,C. (2002) Structural and functional properties of the HIV-1 RNA–tRNA(Lys)3 primer complex annealed by the nucleocapsid protein: comparison with the heat-annealed complex. RNA, 8, 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isel C., Westhof,E., Massire,C., Le Grice,S.F., Ehresmann,B., Ehresmann,C. and Marquet,R. (1999) Structural basis for the specificity of the initiation of HIV-1 reverse transcription. EMBO J., 18, 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patzel V., Steidl,U., Kronenwett,R., Haas,R. and Sczakiel,G. (1999) A theoretical approach to select effective antisense oligodeoxyribonucleotides at high statistical probability. Nucleic Acids Res., 27, 4328–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patzel V. and Sczakiel,G. (1998) Theoretical design of antisense RNA structures substantially improves annealing kinetics and efficacy in human cells. Nat. Biotechnol., 16, 64–68. [DOI] [PubMed] [Google Scholar]
- 31.Laptev A.V., Lu,Z., Colige,A. and Prockop,D.J. (1994) Specific inhibition of expression of a human collagen gene (COL1A1) with modified antisense oligonucleotides. The most effective target sites are clustered in double-stranded regions of the predicted secondary structure for the mRNA. Biochemistry, 33, 11033–11039. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi K.I., Baba,S., Chattopadhyay,P., Koyanagi,Y., Yamamoto,N., Takaku,H. and Kawai,G. (2000) Structural requirement for the two-step dimerization of human immunodeficiency virus type 1 genome. RNA, 6, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clever J.L. and Parslow,T.G. (1997) Mutant human immunodeficiency virus type 1 genomes with defects in RNA dimerization or encapsidation. J. Virol., 71, 3407–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greatorex J., Gallego,J., Varani,G. and Lever,A. (2002) Structure and stability of wild-type and mutant RNA internal loops from the SL-1 domain of the HIV-1 packaging signal. J. Mol. Biol., 322, 543–557. [DOI] [PubMed] [Google Scholar]